


Abstract
Nucleic acids are molecules of choice for both established and emerging nanoscale technologies. These technologies benefit from large functional densities of ‘DNA processing elements’ that can be readily manufactured. To achieve the desired functionality, polynucleotide sequences are currently designed by a process that involves tedious and laborious filtering of potential candidates against a series of requirements and parameters. Here, we present a complete novel methodology for the rapid rational design of large sets of DNA sequences. This method allows for the direct implementation of very complex and detailed requirements for the generated sequences, thus avoiding ‘brute force’ filtering. At the same time, these sequences have narrow distributions of melting temperatures. The molecular part of the design process can be done without computer assistance, using an efficient ‘human engineering’ approach by drawing a single blueprint graph that represents all generated sequences. Moreover, the method eliminates the necessity for extensive thermodynamic calculations. Melting temperature can be calculated only once (or not at all). In addition, the isostability of the sequences is independent of the selection of a particular set of thermodynamic parameters. Applications are presented for DNA sequence designs for microarrays, universal microarray zip sequences and electron transfer experiments.
INTRODUCTION
Nucleic acids can be synthetically ‘programmed’ by designing their primary sequence to form pre-defined structures that are easily detectable on the molecular level (1). Moreover, base-pairing by hydrogen bonding between complementary bases (2) is the basic and well-known mechanism by which the final molecular state is related to the nucleic acid sequence. Higher level corrections to this basic principle, such as stabilizing contributions of base stacking, destabilization by mismatches (3–12), or probabilistic and kinetic aspects of secondary structure formation (13–17) are also well characterized. Nucleic acids are thus molecules of choice for both established and emerging nanoscale technologies. These technologies benefit from large functional densities of ‘DNA processing elements’ that can be readily manufactured. DNA-based devices are used in many diverse applications. They include biological DNA microarrays for gene expression analyses (18–20), chips for gene sequencing by hybridization (21) and medical diagnostic applications, such as branched DNA chips (22,23), single nucleotide polymorphism (SNP) detection microarrays (18,24–27), chips for phylogenetic studies (28), for transcriptome arrays (29) and even for tracking the temporal ordering of events in biological systems (30). Technical applications of DNAs include nanoscale self-assembly systems (31–36), molecular transport devices (33), components for molecular computing (switches, logical units and programmable molecular systems for massively parallel computing) (32,37–63) and ‘molecular motors’ (64). To achieve the desired functionality in all these technological applications, polynucleotides are currently designed by tedious and laborious filtering of potential candidates against a series of requirements and parameters. This ‘brute force’ process is used to ensure that the selected molecules hybridize optimally under pre-defined experimental conditions (29,65,66). Among these requirements, a narrow distribution of melting temperatures (Tms) of the selected sequences is standard for all applications. For example, to achieve a better uniformity during hybridization on microarrays, it is more important to have a narrow Tm distribution rather than a uniform oligonucleotide length. Therefore, some approaches for microarray oligomer design suggest that adjusting the arrayed sequence length by one or a few nucleotides would result in the narrowest possible Tm range (13).
Thermodynamic methods (2–4,10) provide a general relationship between the DNA sequence composition and its stability. In their current form, these methods can be routinely used to calculate the Tm of a given polynucleotide sequence. Thus, any DNA design in which isostability is desirable is currently a very inefficient process because it requires that the sequence is actually generated before its Tm can be predicted. This can be done by a brute-force approach where the computer generates all possible oligomers, calculates their Tm and keeps only those with the desired stability. As the number of possible sequences increases by six orders of magnitude for every 10 bp added, the search through the complete set of possible sequences becomes very soon unrealistic. To bypass this problem, a manageable set of candidate sequences is generated randomly as the input for stability filtering (1,65). The stringency of the isostability criterion defines the extent of inevitable sequence rejections in this step. Moreover, the designer has very limited control over the candidate sequence features. This leads to the rejections of additional isostable sequences in subsequent design steps, which further contribute to the overall ineffectiveness of the process.
Thus, for optimized sequence design, the formulae predicting Tm should be mathematically inverted into a form that would use the desired Tm as input and generate the polynucleotide sequences as output. However, this problem is not solvable until now. In this contribution, we present a specific solution of the inversion problem. This solution constitutes a major progress for the broad range of applications which are not forced to start from a given natural set of sequences (as is for example required for SNP microarrays). At the same time, we show, by an illustrative example, that the benefits of having an effective solution to the Tm-inversion problem also solve the probe design issues for natural sequences. Our solution to the Tm-inversion problem is a general, mathematically complete and most importantly practical method for the direct rational design of large sets of isostable DNA sequences. Formulation of this method stems from discrete mathematics that has provided numerous tools for solving DNA structural problems (67,68). We show that encoding of DNA sequences by Eulerian graphs contains not only relevant thermodynamic information for our goal but is also instrumental in providing algorithmic and molecular insight into basic aspects of isostability of nucleic acid duplexes.
Eulerian graphs are mathematical objects that directly reflect the most important structural feature of a DNA molecule—the linearity and connectivity of its unbranched polymer chain. There are two variants of Eulerian graphs that encode DNA sequences. Pevzner et al. (69) introduced the DNA sequence encoding in the form of a de Bruijn graph, in which the algorithmic determination of the Eulerian path solves the problem of assembly of DNA sequences from fragments. Zhang and Waterman (70) recently used the multisequence variant of the de Bruijn graph for improving the accuracy of multisequence alignment. Chechetkin (71) used the de Bruijn graph to analyze the translational stability of the genetic code. Vertices in the de Bruijn graph are structurally defined by subsegments of the encoded sequence. Therefore, DNA polymers with different lengths will have different de Bruijn graphs.
Alternatively, the Eulerian graph with four vertices representing the four nucleobases was introduced as a descriptor of DNA polymers (2). In this encoding, the number of vertices is independent of the polynucleotide length, a fact that is beneficial for our application here. A simplified form of this graph was used to characterize the similarity of DNA sequences (72). We recently used this graph to analyze the efficiency of RNA interference (73). In that application, the decomposition of the complete DNA graph into a finite set of subgraphs (so called basic cycles) was introduced. Here, we show how this decomposition of DNA graphs can be used to directly design isostable sequences without the necessity to actually calculate the Tm. At the same time, general insight into the ‘anatomy’ of the primary structure composition of isostable sequences is gained and used to extend the capabilities of the design method.
MATERIALS AND METHODS
Thermodynamic interpretation of Eulerian graph representation of DNA sequence
We will start our analysis with the nearest-neighbor (nn) level of thermodynamic interpretation of DNA Eulerian graphs. Generalization to higher levels of thermodynamic models is possible and will be shown with the applications in the next section. Within the framework of nn-approximation, the thermodynamic model for calculating enthalpy ΔHduplex of a DNA oligomer from its sequence can be formally described as the following functional:

Here, the second bracket, [TD], collects all thermodynamic constants and experimental parameters (ΔSx are sequence-dependent, nucleation and duplex formation entropies, respectively; Txy is the average melting temperature of AT or CG base pairs; δHxy/x′y′ are stacking energies for neighboring bases xy; ΔG0 is the standard free energy; C is the duplex concentration; [Na+] defines the buffer ionic strength; and T0 is the system temperature). In practice, once the parameter selection and the experimental conditions are optimized, the terms in [TD] do not vary from sequence to sequence but are constant. The first bracket contains information about the DNA sequence (Nxy are the numbers of AT and CG base pairs, and Nxy/x′y′ are the numbers and types of respective nearest-neighbor interactions).
Next, we show how encoding of a DNA sequence by the Eluerian graph Γ can be related to Equation 1. In general DNA-graph Γ, the vertices (nodes) correspond to ordered n-tuples of bases (n = 1, 2, …, t), and the edges correspond to sugar-phosphate backbone connections between them. The encoded DNA sequence is in mathematical jargon represented as an Eulerian path in Γ. Existing theory of Eulerian graphs, namely the theorems related to paths (74), show that expressing a given ‘mother’ DNA sequence by Γ immediately produces the representation of the potentially very large set of ‘daughter’ sequences. Next, we analyze what is unique about these ‘daughter’ DNA sequences. Let us consider the simplest generators of ‘daughter’ sequences—DNA graphs able to encode sequence information up to the nearest-neighbor level. The corresponding Eulerian graphs Γnn have four vertices, each labeled with a single base A, T, C or G. The method of representing a given DNA sequence by this four-vertex Eulerian graph Γnn is shown in Figure 1A [also see (2,73) for other examples]. The resulting graph Γnn can then be uniquely characterized by its adjacency matrix A(Γnn) (Figure 1A, bottom). Figure 1B shows the correspondence between matrix elements of A(Γnn) and values of Nxy's in the first term of Equation 1. With this projection, can be re-written as
Figure 1.
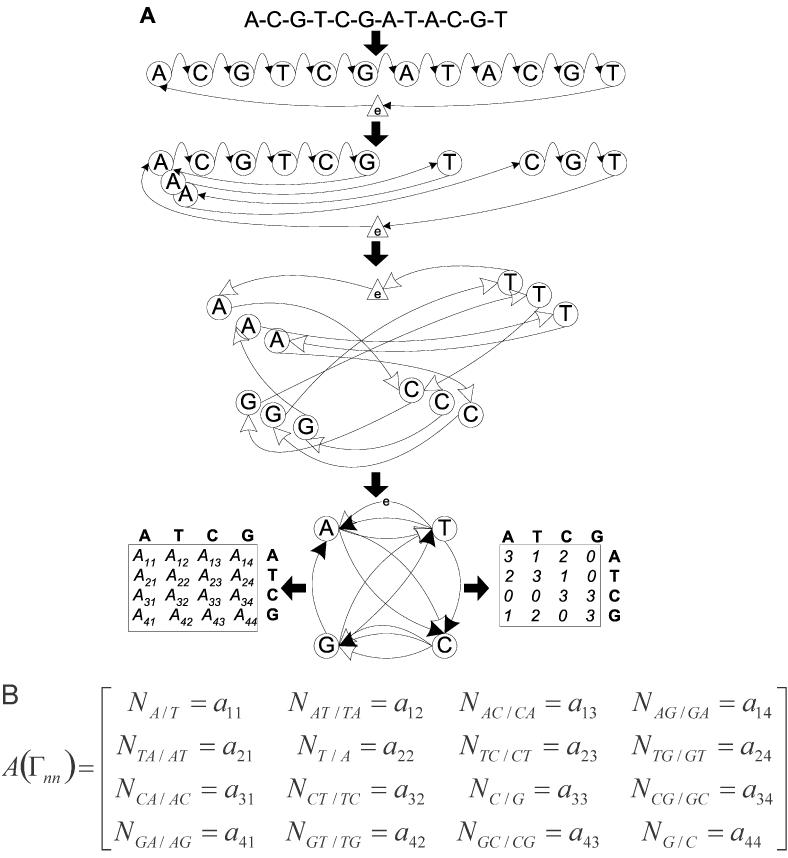
Schematic representation of transformation of a given DNA sequence into Eulerian graph Γnn and its adjacency matrix A(Γnn). (A) Graphical representation of the transformation showing annealing of identically labeled vertices into final form of Γnn. Construction of adjacency matrix A(Γnn) follows established mathematical rules. (B) Correspondence of matrix elements of A(Γnn) and entries in the first term of Equation 1.
![]()
In summary, Equation 1 shows that all DNA sequences of the same length that have the same values of NAT, NCG and {Nxy/x′y′} are isostable on a nn-level. We will name these sequences ‘contextually isostable’ to distinguish them from DNA duplexes that differ in their [NAT, NCG, {Nxy/x′y′}] terms, but accidentally have Tms that are similar. Equation 2s hows that all ‘daughter’ n-mer sequences represented by one Eulerian graph Γnn have the same values of NAT, NCG and {Nxy/x′y′}. Thus every sequence from the complete isostable set of ‘daughter’ sequences, represented by various Eulerian paths in the same graph Γnn, is contextually isostable both to the ‘mother’ and to all other ‘daughter’ sequences. For a given oligomer length, graph Γnn can therefore be interpreted as a parameter-independent encoding of both primary sequences and the common thermodynamic stability for all contextually isostable DNA polymers that exist (on the nearest-neighbor approximation level).
Next we show that by extending the above analysis, Equation 2 transforms from being a trivial copy of Equation 1 into the foundation of the sequence design algorithms that we describe here. Thermodynamic interpretation of the DNA Eulerian graph Γnn shows that the problem of inverting thermodynamic formula for Tm into a generator of contextually isostable DNA ‘daughter’ sequences of a given length is solved by finding all Eulerian paths in the graph Γnn of the ‘mother’ oligomer. What is the molecular background for this claim? The numerical representation of graph Γnn by its adjacency matrix A(Γnn) might resemble the dinucleotide frequency profile of a (mother) DNA sequence. Taken out of the context of the matrix scheme, the elements aij are indeed just that—the components of this profile. Nevertheless, this interpretation improperly reduces the informational content of Γnn and A(Γnn) to a level that prevents any attempt to rationally design contextually isostable DNA sequences. The additional molecular information that is uniquely captured by the DNA graph Γnn describes details of dinucleotide connectivity in the ‘mother’ sequence. This contextual sequence feature is represented by the network of edges connecting the Γnn vertices. Molecularly, a DNA graph Γnn is Eulerian because the oligonucleotide sequence is a linear unbranched polymer (see Figure 1). In a mathematical sense, the Eulerian graph Γnn is defined by exclusive topology of its edges, that is to say the number of incoming and outgoing arrows in each vertex is the same. The combination of these molecular and mathematical realizations of a specific structural situation in DNA in Γnn enables quantitative capturing of the dinucleotide connectivity: it is necessary to order the dinucleotide frequencies into an unambiguous matrix form of A(Γnn). More importantly, this result also explains why not all combinations of dinucleotide frequencies can describe an actual DNA sequence. The reason is that the diagonal and off-diagonal elements of the corresponding adjacency matrix A(Γnn) are inter-related to satisfy the condition of the Eulerianity of Γnn. This is explained below and summarized in Equation 3 (74).
Molecularly, the diagonal elements A(Γnn) enumerate the four bases in the sequence. The off-diagonal elements of A(Γnn) enumerate the number of 5′ neighbors (lower left triangle of the matrix) and 3′ neighbors (upper right triangle) of any base in the sequence. In the linear unbranched polymer, we obtain the same result if we count the bases directly or by counting their 5′ or 3′ neighbors. Thus, for the subset of DNA sequence without a-plets of identical bases, the off-diagonal elements in a given row and column should sum to the diagonal element of A(Γnn). If, on the other hand the neighbor of a given base is the same base, it is reflected by an increase in the corresponding diagonal element in A(Γnn). Thus, the final topological feature of a general DNA sequence, i.e. the numbers of ∼XX∼ motifs in it, is enumerated by the differences
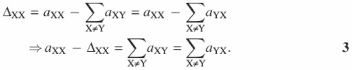
This also represents the mathematical formulation of the above discussed relationship between elements of the adjacency matrix A(Γnn). Moreover, it completely defines all relationships between the variables in terms [Nc] of Equation 1. A concrete example in Figure 2 demonstrates how these relationships are applied in a ‘pen-and-paper’ variant of our rational DNA sequence design.
Figure 2.
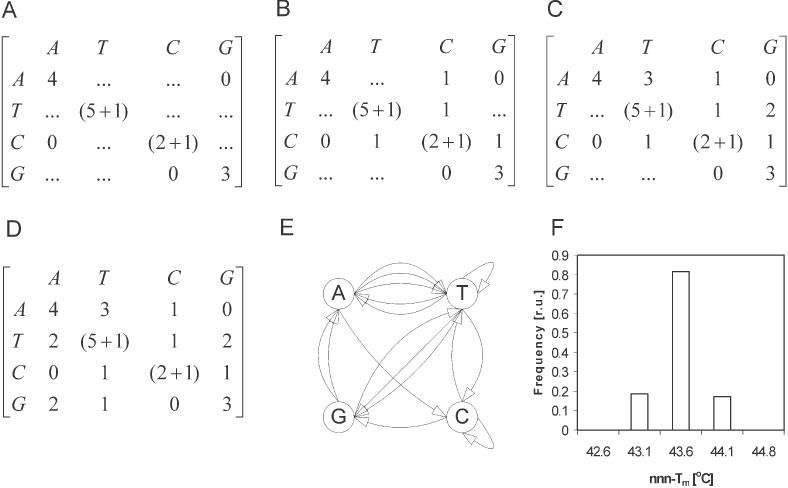
Example of construction of adjacency matrix A(Γnn) and corresponding Eulerian graph Γnn using the set of structural requirements defined in the text. (A) Initial form of matrix A(Γnn) after implementation of requirements for length, nucleobase composition, presence of only one ∼TT∼ and ∼CC∼ motif and absence of ∼CA∼ and ∼AG∼ stacking interactions in all generated contextually isostable sequences. (B) First intermediate form of the adjacency matrix A(Γnn) after relations between diagonal and off-diagonal elements were used to fill the C-row and column. (C) Second intermediate form of A(Γnn) with elements aAT and aTG determined by the rules of Equation 3 (see text for details). (D) Final form of the A(Γnn) with remaining off-diagonal elements calculated from the previous intermediate form. (E) Eulerian graph Γnn defined by A(Γnn) (F) Distribution of Tm predicted for contextually nn-isostable sequences generated from the graph E using nnn-level of thermodynamic formulation of Equation 1w ith parameters from (2).
First, let us define our goal. Say that we need a set of 16mer that is contextually nn-isostable. Next, each sequence should contain 25% A, the (A+T)/(C+G) and C/G ratios in these sequences should be 1:1.7 and 1:1, respectively. No sequence in the set should contain ∼AA∼, ∼GG∼, ∼GC∼, ∼AG∼ and ∼CA∼ arrangement of bases. Moreover, ∼TT∼ and ∼CC∼ motifs can appear only once in every sequence. Even though these base composition requirements are rather complex, we show that they can be unambiguously used to easily determine (by hand!) the adjacency matrix A(Γnn) and thus the corresponding graph Γnn. (i) The required base composition and the common length of the desired sequences defines the main diagonal of A(Γnn). (ii) The restrictions on 2-tuple motifs in the sequence define zero off-diagonal elements in the A(Γnn) (see Figure 2A). In the next step, we use the mathematical restrictions (Equation 3) on elements of the adjacency matrix for Eulerian graphs to determine the remaining off-diagonal elements of A(Γnn). An alternative formulation of these relations is that the off-diagonal elements of A(Γnn) can only be integer partitions of diagonal elements, reduced by the number of base repeats. (iii) The decomposition of aTT and aCC diagonal matrix elements into (5+1) and (2+1) reflects the required presence of ∼TT∼ and ∼CC∼ in the sequence just once. We can now fill in the C-row and column of A(Γnn). There are two integer partitions of 2 = (2+0) and 2 = (1+1). We select (1+1), because having an additional zero would increase the scope of input restrictions listed above. The resulting intermediate form of A(Γnn) is shown in Figure 2B. Following systematically the sum and partition rules further allows to identify the elements aTG = 2 and aAT = 3 (see Figure 2C). This in turn defines elements aTA = 2, aGA = 2 and aGT = 1. This completes the adjacency matrix A(Γnn) (see Figure 2D). The corresponding graph Γnn is shown in Figure 2E. The enumeration formula (2) reveals that this graph represents ∼104 contextually isostable sequences which all obey the composition and sequence topology restrictions formulated upon input. Brute-force search for sequences with these specific properties would require very detailed scanning of 4 × 109 candidates.
To demonstrate the quality of the contextual isostability of the daughter DNA sequences defined by this graph, we generated the sequences defined above and calculated their Tm with the next-nearest-neighbor (nnn-level) thermodynamic formula. The distribution of calculated Tm using this higher-level model is shown in Figure 2F. Such a calculation emulates what might be observed experimentally. The result is very satisfactory: the total variation of predicted melting temperatures for all daughter sequences is only ±0.6°C. Of course, only one identical Tm would be calculated for all 104 DNAs if the nn-level of thermodynamic model is used.
The construction of the DNA graph Γnn can be done de novo as in Figure 2 or using the given ‘mother’ DNA sequence as is shown in Figure 1. Although it is intellectually pleasing to represent thousands of Avogadro's numbers of isostable DNA sequences by a simple sketch or a 16-element mathematical descriptor, it is only the first part of the solution leading to practical applications. Next, we need to solve the problem of how to generate the actual DNA sequences once their graph representation Γnn is known. This step is seemingly formal and more suited for those interested in the developments of algorithms. Methods for finding all Eulerian paths in Γnn are indeed well established and are shown to run in polynomial time (69,70,74). Nevertheless, we present here a novel specific solution for Γnn representation that provides deep molecular insight into what are the unique features of contextually isostable sequences. The details of software implementation of this algorithm and the link to its website are presented as the Supplementary Material on the NAR website. More importantly, the results of this analysis directly answer the next question about DNA sequences that are contextually isostable on higher levels of thermodynamic approximation (next-nearest-neighbor—nnn—or generally n…. n level).
To gain the needed molecular insight, we will use our previous result which shows that every DNA graph Γnn can be decomposed into a finite set of subgraphs—cycles γk,(k = 1, …, 24) (73).
To re-iterate, cycles γk are Eulerian graphs with only one in- and one out-edge incident to all vertices (73). Molecularly, they represent segments of the analyzed oligonucleotide sequence. It is important that for Γnn representation of any DNA of any length, there are only 24 γk cycles (73). In our approach, the graph Γnn is first decomposed into these basic cycles using the adjacency matrix representation (73). Formally, the decomposition of Γnn can be written as Γnn = ∑kckγk (ck is the multiplicity of occurrence of a given cycle γk in Γnn). The cycles from the resulting set are then used as building blocks to generate the isostable daughter sequences. To this end, we systematically anneal the cycles γk via superimposing identically labeled vertices. The process is demonstrated by a simple example in Figure 3.
Figure 3.
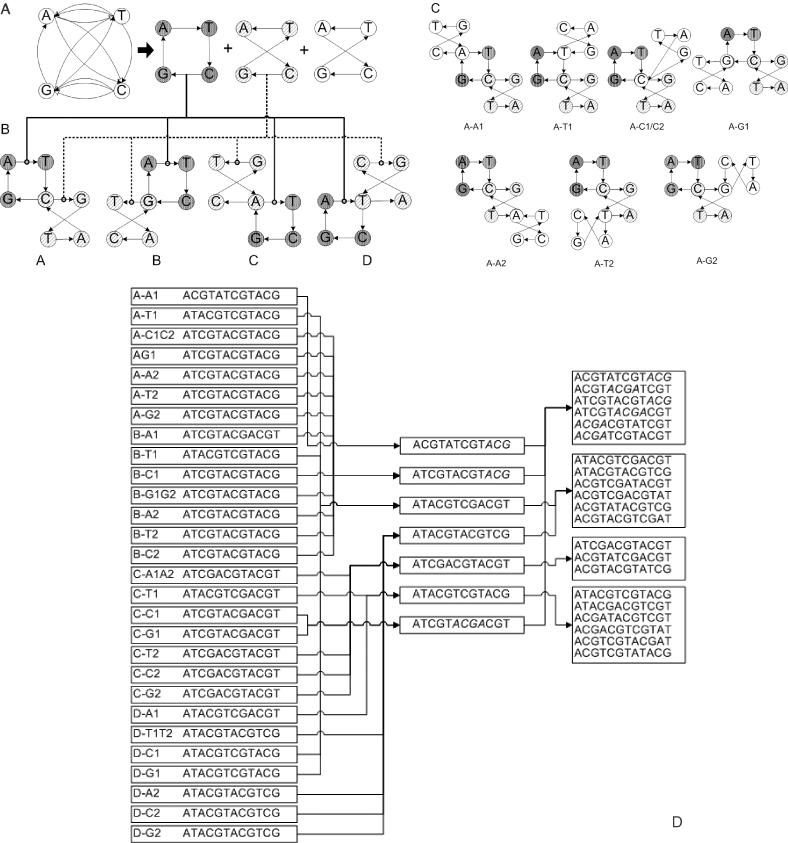
Schematic example of our algorithm that generates sets of contextually isostable DNA sequences from basic cycles γk defined by decomposition of the Eulerian graph Γnn. (A) Given its connectivity, the original graph Γnn is decomposed of into three basic cycle subgraphs γa + 2γb. (B) The first level of building template structures T from γk is demonstrated. All four possible ways of annealing one basic cycle γa to another basic cycle γb via identical vertices are shown (A–D). (C) Completion of the template structures T by annealing the second basic cycle γb to intermediate structure A from level B. Final template A-A1 means that the intermediate structure A is connected to the second γbvia the upper left A-vertex. Final template A-A2 means that the intermediate structure A is connected to the second γb via the lower right A-vertex and so forth. The remaining templates [derived from (B) to (D)] are omitted for clarity. (D) Left panel: The complete set of sequences as read from all final templates, starting at base A. Central panel: Selection of a unique sequence set from the complete list. Reduction in the number of sequences is the consequence of many identities among templates, which, in turn, is a consequence of the symmetry of graph Γnn. This redundancy is only generated due to the ‘manual’ nature of the construction for demonstration purposes. However, in the final software implementation, these redundancies are avoided. Right panel: the final set of contextually isostable sequences generated by systematic permutation of the permutable segments.
Using this concrete example, we can show how this approach provides insight into the common molecular properties of the contextually isostable DNA sequences. In Figure 3, Γnn = γa + 2γb and the input into annealing operation are three cycles γa,γb,γb from panel A. By two levels of annealing them with each other, we generate a series of new Eulerian graphs T, with a representative subset shown in panel C. Each of these graphs T encodes one possible network of annealing connections that is allowed given the input set of cycles. We will call this scheme the template. The vertices in template graphs can be divided into two topologically distinct categories. The first kind of vertex has only one incoming and one outgoing edge (d+ = d− = 1). The second kind of vertex has more than one incoming and outgoing edge (d+ = d− > 1). These second kind vertices form ‘nodes’ where segments consisting of only vertices of the first kind start and end. By selecting a starting vertex, the DNA sequence So can be generated by following the oriented edges in T and recording the nucleobase labels of vertices visited along the path. The consequence is that the template T has a branched topology. The branches represent the segments, which start and end in a node in a T graph.
In plain language, this analysis reveals how the sequence of any DNA molecule from a set of contextually isostable oligomers is severely constrained in base composition and neighbor combinations. These constraints follow the relative weights of the terms in Equation 1. Thus, with constant length, first we need to preserve the base composition to keep the number of duplex hydrogen bonding unchanged. Second, for each base, we need to preserve the largest fraction of its stacking π–π interactions. This means that the nearest-neighbors of each base should be kept constant in the resulting ‘daughter’ linear polymers. Therefore, any ‘daughter’ DNA from an isostable set should be a permutation of certain blocks. By analogy, a ‘mother’ DNA polymer can be represented by a set of ‘balls’ connected by strings. We now ‘throw them in the air’ and cut the strings in few specific places such that no matter how the pieces re-shuffle upon landing, each base has the same nearest-neighbors as before. We demonstrated above that finding the Eulerian paths in the graph Γnn via its decomposition into cycles does exactly that (i.e. finds the places where to cut) in a direct way: the nodes in a T graph are the sites at which the string of the ‘connected balls thrown in the air’ has to be cut to generate a contextually isostable set of ‘daughter’ sequences. This is because arbitrary permutations of these segments generate new ‘daughter’ DNA sequences Sp that are contextually isostable with the original ‘mother’ molecule So (see Figure 3C). This is the gain in information that DNA graphs provide over the dinucleotide profile. We cannot simply take the pre-defined number of dinucleotides and combine them arbitrarily into a linear polymer to preserve its stability. Our result shows that arbitrarily permutable units that provide contextually isostable daughter sequences are blocks of bases, represented by the branches in the template graph T. Generalization of this result (see below) shows what is a general form of graphs Γnn up to Γn, …, n representing DNA sequences that are contextually isostable on a higher than nearest-neighbor approximation level.
Beyond the nearest-neighbor—higher levels of contextual isostability of DNA sequences
The topology of the template graph T and the final steps of the above described algorithm open the way for generating contextually isostable sequences that will have identical numbers of not only nearest-neighbor (nn), but also next-nearest-neighbor (nnn) and even higher (x times n) stacking interactions along their nucleobase sequence. The generalization requires only modified definition of the node labels in the template graphs. If instead of single base labels we use oriented 2-tuple (oriented means that the ∼AT∼ label is different from the ∼TA∼ label), then the topology of permutable segments will be XY-…-XY and the sequences [Sp] generated by systematic rearrangement of them will be contextually isostable at the nnn-level of thermodynamic approximation. In general, by labeling template graph nodes with oriented (x−1)-tuples of nucleobases, the sequences [Sp] will be contextually isostable at the (x times n)-level of thermodynamic approximation.
This method can be used as a standalone algorithm for generating contextually isostable sequences of the desired level of thermodynamic detail. However, it can be equally efficiently used as continuation of the rational design strategy described above. An example of this approach is shown in Figure 4. Assume that the oligomer ATGACATGATCATCA is a member of a contextually isostable set of sequences. Because the definition of the node in any template graph is that its in-degree equals its out-degree which has to be larger than one, we can systematically scan the oligomer sequence for repeated occurrences of x-tuples. At each step, all identical x-tuples, if present, are joined into one node of the template graph T*. The rest of the sequence defines the loops in the template graph that, in turn, are invariable blocks to be systematically permuted. For the given ‘mother’ sequence, this generates all possible sets [Sp]X,…,Y of contextually isostable sequences at the (x+1) times n-approximation level. For the oligomer in our example, the representative set of these higher-level template graphs is shown in Figure 4. Also shown is the combined list of sequences generated from all T*'s by systematically rearranging their permutable segments.
Figure 4.
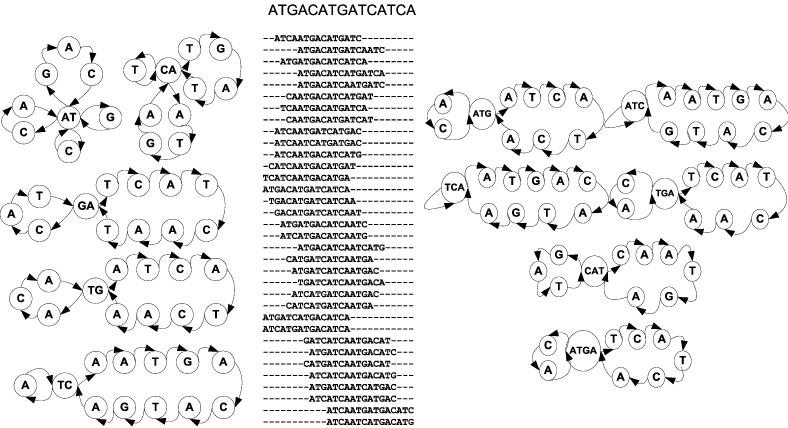
Schematic representation of how to generate the set of sequences that are contextually isostable at the next-nearest-neighbor level. The ‘mother’ oligomer ATGACATGATCATCA is converted into template graphs T* by a systematic search for identically oriented 2-, 3- and 4-tuples. If more than one n-tuple is found in the sequence, they are combined into the node vertex of T*. This operation defines permutable segments. Their systematic permutation yields the contextually isostable ‘daughter’ oligomers, shown in the central panel. For comparison, multiple alignment of the resulting sequences using ClustalW is shown.
Although theoretically one could increase the x-tuple label of the T* node up to half of the seed sequence length, for practical purposes we limit it to four nucleobases. This is because the probability of finding multiple occurrences of x-tuples in the sequence of a given length decreases rapidly with increasing x. At the same time, the contribution of the corresponding x times n neighbor interactions to the thermodynamic stability become negligible. We attach a Maplet application (designed with Maple v. 9.03; Maplesoft, Waterloo Maple Inc.) together with the source code and operating instructions as Supplementary Material.
RESULTS AND DISCUSSION
To demonstrate various practical aspects of our approach, several applications are presented. The first practical question to be answered is if the sets of contextually isostable sequences are large enough to provide sufficient numbers of sequences for further functional selection. For nn-approximation and Γnn graphs, this question can be easily answered. In our previous paper (2), we modified the Aardene–Ehrenfest theorem (74) for DNA graphs Γnn. This provides a formula that enumerates all Eulerian paths (representing contextually isostable ‘daughter’ DNA sequences) in graph Γnn. Consider the ‘mother’ sequence CGTACAAGCCCTGGACGATCACTCACAATGACGAAATCATAGCCTCGATGATCCGACGTAAA. Converting it into graph Γnn and applying the enumeration formula from (2), we find that the number ND of ‘daughter’ sequences that are contextually isostable (on the nn-level) with this oligomer is ∼2.6 × 1027. This can be used as a reference for quantitative scoring of the gain of our approach. ND is only a fraction (1.1 × 10−6%) of the complete set of 2.1 × 1037 62mers that would need to be generated and characterized by calculating Tm in a complete brute-force search for isostable sequences. At the same time, it is a number large enough to ensure that there are plenty of sequences to be screened for practical applications. At the same time, however, this result reveals the other side of the coin: we see that even the sets of contextually isostable sequences might be too large for complete processing.
Sequence design to study electron transfer in DNA molecules
The next example is derived from the field of molecular electronics and relates to the study of electron transfer in DNA chains. It is known that ∼GG∼ motifs in a DNA sequence function as traps for radical cations that can be generated photochemically by irradiating a DNA duplex complexed with light absorbing antraquinone derivatives. The relative distances of ∼GG∼ motifs and the kind of bases between them determine the hoping rate of the radical cation (75–77). To study the details of the transfer mechanism, idealized oligomers [(A)nGG]m and [(T)nGG]m with variable distances between the ∼GG∼ motifs were studied recently (77). In practice, it might be desirable to design more complex oligomers in which the ∼GG∼ traps have variable distances but are placed within sequences that preserve the base stacking interactions throughout the whole polymer. We generated a series of contextually nn-isostable ‘daughter’ 60mers using the ‘mother’ sequence from (76). This seed sequence was converted into graph Γnn, which was in turn decomposed into basic cycles γk. We then used our algorithm to generate a set of 693 824 contextually isostable sequences from these base cycles. Their Tm's (predicted for the sake of stringency by a nnn-level thermodynamic model) were within ±1°C, and 97% of them were within ±0.5°C. Because of our construction from graph Γnn representing the seed sequence, all these sequences contain exactly four ∼GG∼ motifs at various positions. This demonstrates the design superiority of our approach: if further restrictions are imposed (e.g. the regularity of distances between the GG-traps), it is practically certain that the desired sequences will be found. This is demonstrated in Table 1 that contains oligomers [(GG)Xm(GG)Ym(GG)] with regular distances m ranging from 8 to 14, which were selected from this contextually isostable set. The generation of this sequence pool took ∼30 s on a 2.66 GHz PC with 512 KB RAM. A text editor was then used to select the oligomers shown in Table 1. The quantitative gain of using our method over a conventional sequence-design application can be estimated as follows. There are ∼1.4 × 1036 60mers—an intractably large number of sequences for any processing and storage. We therefore use 107 as the realistic number of sequences that can be processed. An alternative strategy to our approach would be to place the four ∼GG∼ motifs randomly along the sequence and then fill-in the gaps in each oligomer from the pool of remaining bases of the ‘mother’ sequence to generate up to 107 daughter oligomers. This would be then followed by a (relatively complex) search for sequences having the same dinucleotide connections and frequency profile as the mother sequence. Finally, the oligomers with the desired distances between the GG-traps would be selected. Instead of this tedious process, our graph-based approach incorporates directly the goal of the design: every generated daughter sequence has the ∼GG∼ traps in the same neighbor context. It also guarantees finding the solution and eliminates completely the most complex part of the alternative design. A smaller and thus more tractable dataset with no irrelevant sequences is an additional (but important) benefit of our approach.
Table 1. Example of the sequence design to study electron transfer in DNA molecules.
| Oligomer | Sequence |
|---|---|
| TTTGGCACAGTAGTTCCCAGGTAAAAAAAGACATTGGCGCTGAACTGTCTTGGACGCGTA | |
| [(GG)X14(GG)Y14(GG)] | CTGACTGGATTGCACGCGCCCAGGTCACATTGCTGTAAGGACGTAGTAAAAAAAGGTTTT |
| CGCCAATGTACGGTAGTTCTGAACAGGTTTTTCGCCACTGGAGTAAAAAAGATGGCACTG | |
| [(GG)X12(GG)Y12(GG)] | GTAAGGTTTTTCCCAGATGGCGCAACTGTCACGGCTGTAGATGCACGGTACTTGAAAAAA |
| AGACTGTGCAATCATGGCGTAACGCAAGGTTTTTCTAAAGGTGAACTGTTAGGAACGCCC | |
| [(GG)X10(GG)Y10(GG)] | AGTAGTGGATGCACCTCTGGCATGAAACACGGTTAGTAAAAAGGTTTTTCTGAACGCGCC |
| CTTGAACATGCAGCAGTATGGTAACGCGCGGTTTTCCCAGGTAAAAAGTGGAACTGACTT | |
| [(GG)X8(GG)Y8(GG)] | AGTCGCAGGTAAAAAAAGGTTTCTGTAGGATTGCATTGGCACTTGACTGAACGTACGCCC |
| TGCATAACGCGTTGTGAACTCAGCATGGTTTTTCGGCCAGTAGGTAAAAAGGACCTGAAC | |
| [(GG)X6(GG)Y6(GG)] | AGTAACAGCAATGTGAACAATGGCTTCTTGGACAAAAGGTGACTAGGTTTTCGCGCCCGT |
The presence of the relevant motif, (GG)Xm(GG)Ym(GG), was directly incorporated into the generating graph Γnn: it contained three and only three ‘loops’ on the G-vertex. Length, nucleobase composition and all nearest-neighbor interactions (up to the terminal residues) are identical within the set and are the same as for the seed sequence studied in (76). See text for details.
Design of permutable blocks for the synthesis of contextually isostable zip-code sequences
In the next example, we demonstrate another novel aspect of working with contextually isostable sequences. The essential technological part of universal microarrays (78,79) are so called zip-code sequences that, besides lacking any sequence similarity, need to be isostable. This allows for reactions on chip microarrays to be carried out at a single temperature, resulting in stringent and rapid hybridization. For technical reasons, zip-code sequences are sometimes synthesized from shorter blocks (e.g. 24mer from six 4mers). Our approach provides a tool for the rational design of such blocks and for tuning their properties to optimize the zip-sequence quality. Although a design based on thermodynamic properties cannot directly lead to such a sequence diversity that prevents cross-hybridization, our approach allows to incorporating some sequence features that overcome this inadequacy. For example, the periodicity of zip-code sequences constructed from blocks of equal length facilitates energetic stabilization of unrelated sequences when target and probe strands are shifted by multiples of the block lengths. To minimize this effect, blocks of unequal length can be used. The template graph in Figure 5A is a prototype for designing such blocks. To demonstrate the flexibility of our approach, we omit nucleobase labeling of template graph vertices within the permutable blocks. This can be done because only the topology of the template graph T* determines the contextual isostability of the generated sequences. Sequences synthesized from these blocks by systematic permutation will be contextually isostable, irrespective of the actual base composition of the permutable segments. This provides the opportunity to tune the thermodynamic properties of the zip-code sequences without changing the overall design. Two levels of Tm modulations are possible. First, the node base X can be replaced and the composition of the remaining parts of T* are conserved. Selecting X = A or X = T will provide zip-code sequences with lower Tm, selecting X = C or X = G will provide sequences with higher Tm. This replacement might have an impact on the overall similarity or dissimilarity of the zip-code sequences. Thus, the second possibility is to permute systematically all bases in all positions (e.g. A→G, T→C, C→A and G→T). The new sequences will again be contextually isostable, although their Tm will be different from that of the parent set. The dissimilarity of the respective sequences will be at least formally identical to that of the parent set. Here is the example of this design strategy: two eight-block sets were derived from the same template graph T* using two alternative associations of vertices with nucleobases. The template graphs T*1 and T*2 are shown in Figure 5B. The resulting permutable segments were systematically combined into a set of 40 320 37-meric zip-code sequences. Tm predictions, calculated within the nnn-approximation for these two sets are shown in Table 2.
Figure 5.
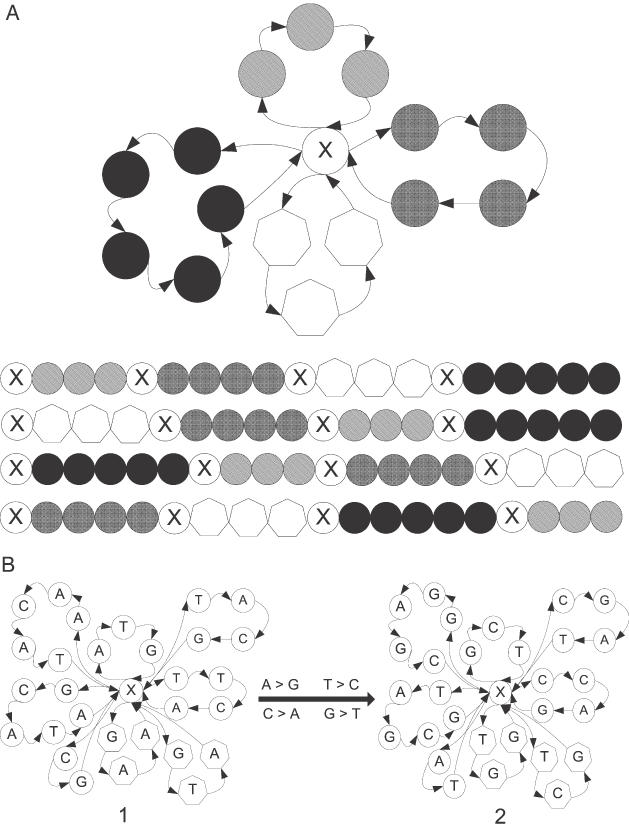
Design of permutable blocks for the synthesis of contextually isostable zip-code sequences for universal microarrays. (A) Demonstration of the topology of template graph T* (top) and several arrangements of the permutable segments in the final zip-code templates. The permutable segments have different lengths to minimize cross-hybridization that derives from the periodicity of the zip sequences. The different shading of the template vertices demonstrates that contextual isostability is a property derived from the template graph topology, rather than from the concrete nucleobase identities. Vertices in the permutable blocks can be arbitrarily filled with any nucleobase. As long as these vertex assignments remain unchanged throughout the permutation, the resulting set of sequences will be contextually isostable. This increases the overall effectivity of the design: Once the satisfactory (sub)set of contextually isostable zip-sequences is selected, its common thermodynamic properties can be fine-tuned by systematic replacement of the nucleobases in the oligomers. (B) An example of how to adjust the Tm for contextually nn-isostable 37mers, generated from eight permutable segments that are defined by the common template T*. The assignments of nucleobases to vertices in the right template (set 2) were generated by systematic replacements of A→G, T→C, C→A and G→T in the left template (set 1). The resulting block permutation yielded 40 320 sequences. Tm predictions were calculated for these two series using all four selections of node base X = A, T, C and G. See Table 2 for results.
Table 2. Range of melting temperatures for the set of 40 320 zip-code sequences constructed from the T* graphs in Figure 5B.
| Set | Base X in the node | |||
|---|---|---|---|---|
| A | T | C | G | |
| Tm (°C) | Tm (°C) | Tm (°C) | Tm (°C) | |
| 1 | 65.1 ± 0.3 | 65.4 ± 0.3 | 75.6 ± 0.5 | 73.7 ± 0.5 |
| 2 | 72.5 ± 0.5 | 73.6 ± 0.4 | 83.2 ± 0.7 | 83.0 ± 0.6 |
Data are shown for all selections of node base X. Tms were predicted using the nnn-level of the thermodynamic model to increase stringency of the isothermality test.
Analysis of design feasibility for universal expression profiling microarrays
In the next example, we show how the Eulerian graph representation of the DNA sequence can help in answering basic questions related to practical aspects of microarray design. A recent proof of principle describes a novel hexamer-based universal microarray platform using a single hybridization step to expression profile a complete transcriptome (80). A total cDNA is processed with two different restriction endonucleases and two adaptor ligations to generate equal length DNA fragments containing gene-identifying hexamer sequences. These fragments were detected by a complete set of all possible 4096 (46) hexameric tags printed on the microarray. Final ligation to the arrays yields thousands of 14mer sequence tags. However, before this system could be applied for expression analysis, 153 parameters needed to be experimentally determined using 2136 synthetic DNA oligomers to reveal the complexity of cross-hybridizations and enable quantitative analysis of the array (80,81). The largest numbers of these parameters are related to evaluating the impact of mismatches at individual positions and in the context of its 5′ and 3′ nearest-neighbors within the hexameric tag.
Here, the theoretical ability to design a set of isostable sequences with identical next-nearest-neighbor interactions might reduce the number of parameters required because every potential mismatch will occur in the same context (has the same 5′ and 3′ neighbors). Hence, the experimental effort to design such a universal microarray might be reduced. To demonstrate this, we have to hypothesize that we can design a sufficient number of contextually isostable oligomers for such a microarray and still ensure a complete representation of the transcriptome.
Our graph-based approach provides a direct way to derive the length and topology of such sequences. In the previous section, we have shown that sequences characterized by the same subset of identical nnn-interactions can be constructed from permutable segments defined in Figure 6. Our formalism also allows to determining how many permutable segments are needed to obtain the required number of such sequences. There are only 720 permutations (factorial of 6) of six blocks and 5040 permutations (7!) of seven blocks. This enumeration defines the general topology of the generating graph (Figure 6). As a result, we see that minimal the length of an oligomer with these special properties is 21 bases (this corresponds to one base per graph ‘leaf’ in Figure 5), resulting in the sequence template b1XY b2XY b3XY b4XY b5XY b6XY b7XY. Obviously, this length and design will result in marked cross-hybridization due to high homology among the oligomers (at least 81%). To avoid cross-hybridization, we can search for the next possible shortest topology. It is the 24mer b11XY b21 b22XY b31XY b41 b42XY b51XY b61 b62XY b71XY with graph ‘leafs’ that alternate between one and two bases. In this second design topology, the selection of bxy bx(y+1) permutable blocks can be made to minimize the overall homology (e.g. bxy bx(y+1) ≠ XY, etc.). Moreover, the length difference in the permutable blocks will help to break the unwanted periodicity of the XY segments that is unavoidable for the above 21mer design.
Figure 6.
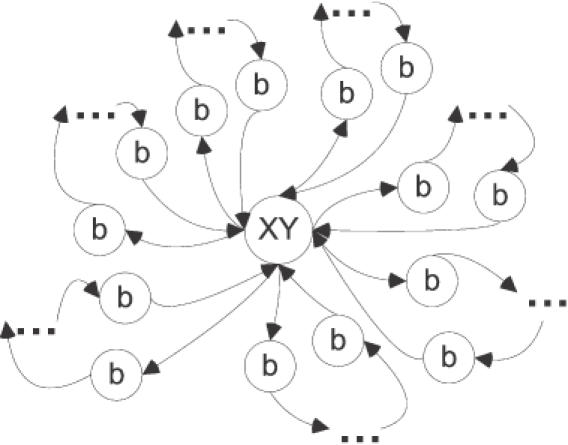
Graph-based design of generalized permutable blocks for the generation of at least 4000 contextually isostable sequences with a given subset of next-nearest-neighbor (nnn-) interactions. This graph facilitates the feasibility analysis for the use of such sequences in n-mer-based universal microarrays for expression profiling (80). See text for details.
In summary, the graph-based analysis provides a reasonable basis for the evaluation of the original hypothesis. In this example, we can conclude that the deduced minimal length of 24mers prevents the realistic chance of finding a complete coverage of the transcriptome.
Optimal selection of targeted regions of natural genes
In this application, we take advantage of the fact that our fast graph-based algorithm generates large numbers of contextually isostable ‘daughter’ DNA oligomers, which inherit not only the stability but also dominant sequence features from their ‘mother’ molecule. We used this generating step in an alternative strategy that optimizes the selection of target sequences for gene expression microarrays. We compare the properties of the final set of targets to the commercial probes from Affymetrix YG98 yeast microarray. The basic idea of our novel approach is to select first the optimal (isostable and non-homologous) ‘mother’ sequences from the target genes. Each ‘mother’ sequence is subsequently used to generate a set of contextually isostable ‘daughter’ sequences, for which we search for their ‘partners’ in the genes. Our analysis of isostability via graphs Γnn provides an additional characteristic (‘potency’) of every ‘mother’ molecule that is obtained by calculating the number of DNA-paths in its graph Γnn. The combination of unique properties of novel, graph-based descriptors is used to streamline the selection process. Another advantage of this design strategy is its scalability. Every step of the algorithm is deterministic, which supports a parallel implementation on multi-processor platforms for large design projects.
The reference set of yeast sequences for this illustrative example was selected from 5885 yeast open reading frame (ORF) entries in the SGD database (82) by searching for those ORF sequences that contained one or more targets for Affymetrix yeast S98 probes (or their reverse complements). A total of 935 oligomers were found in 49 yeast ORF sequences in the database. The distribution of their Tm, calculated within a nn-approximation, is shown in Figure 7A. This distribution spans 29°C and can be fitted by Gaussian function with half-width σ = 5.0°C. To characterize the homology of probe sequences, we used the following quantitative characteristics of pair wise similarities: a sequence of a probe i was slid over the complementary sequence of probe j, one base at a time. At each duplex configuration, the number of complementary base pairs was determined, together with the length of the longest contiguous segment of complementary base pairs. If either the number of complementary base pairs was >60% of the total probe length or the contiguous complementary segment was ≥50% of the probe length in any overlay configuration, the pair i–j was assigned 0 in a 935 × 935 sequence homology matrix [H]. If the probe pair was not homologous according to this criterion, the corresponding entry in [H] was set to 1. Then the fraction of rows in [H] that do not contain any 0 was calculated. This number is the low estimate of the fraction of mutually non-homologous probes in the set. (The actual number is the size of the largest clique in a graph defined by [H] (74). We did not attempt to use this algorithmically difficult descriptor here.)
Figure 7.
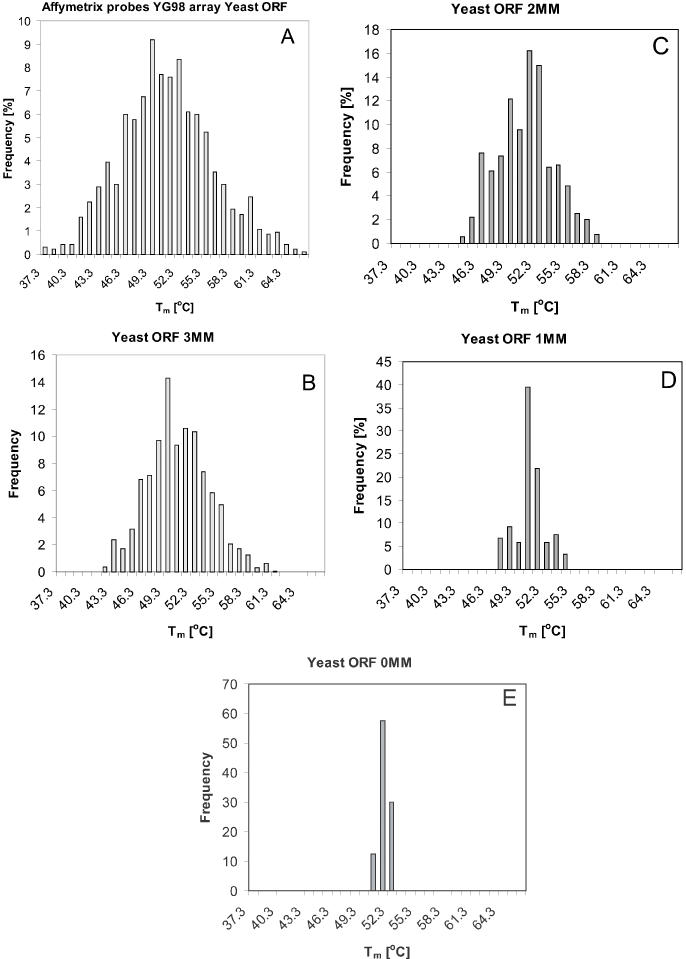
Histograms of Tm calculated on nn-level of approximation for sets of probes of 49 yeast ORFs. (A) Affymetrix probe set (935) from yeast S98 microarray. (B) Probes designed by our method with up to three mismatched bases between daughter and target sequences (853). (C) Probes designed with up to two mismatched bases between daughter and target sequences (795). (D) Probes designed with up to one mismatched base between daughter and target sequences (786). (E) Probes designed with no mismatched bases between daughter and target sequences (744).
Next, the same set of 49 ORF sequences was used as the input into our method. First, ‘mother’ sequences were selected as follows: candidate probe oligomers from all 49 ORF sequences were characterized by adjacency matrices A(Γnn) and the number NM of DNA paths in their graph Γnn. Only probes having NM > 50% of the maximum (NM)max for each ORF were considered further. To avoid the homology of ‘mother’ sequences, we eliminated duplicities of adjacency matrices A(Γnn) in the set. The Tm distribution was calculated for these most ‘potent’ and diverse ‘mother’ sequences. The iterative generation of ‘daughters’ started with the ‘mother’ and was characterized by the Tm in the center of the most-populated temperature interval. We used the algorithm described here (see Figure 3). In each set of ‘daughter’ sequences, we first eliminated occasional duplicities and sequence homologies >70% of the total probe length. In the following step, we searched through all ORF sequences for target regions, identical to any ‘daughter’ probe, allowing up to 3, 2, 1 and 0 mismatches per probe–target pair. The uniqueness of the probe to the particular target gene is controlled in this step: if for any given ‘daughter’ probe candidate we found two or more targets on different gene sequences, that probe is eliminated from the result. After all the ‘daughter’ sequences from the respective iteration step were screened, the ‘mother’ with Tm closest to the first one entered the iteration. This process continued until all ORF sequences from the set were covered by at least one probe or the pool of ‘mothers’ was depleted.
The partner sequences (i.e. not daughters, but the oligomers that might differ from them up to the selected number of mismatches) were then subjected to the same characterization of their quality as the reference Affymetrix probe set. Figure 7B–E and Table 3 show the quantitatively scored improvements in properties of the respective probe sets. The improvement both in isothermality (by a factor of 7) and in sequence uniqueness over the Affymetrix probe set is obvious. Moreover, the method allows simple and very effective control of the outcome. Because the iteration starts with a ‘mother’ sequence, which has known difference in Tm and the set of ‘daughter’ sequences is contextually isostable with it, the selection process can be interrupted at a pre-defined width of the predicted Tm distribution. An essential feature of this novel selection strategy is the improvement in the level of diversity between the probes in the set. The utilization of the graph-based generation of probe candidates enabled us to incorporate both isothermality and the non-cross-hybridization requirements directly into the design process. This is facilitated by the high level of sequence context information encoded in the graph characterization of the DNA. The contextual diversity of ‘mother’ sequences with different graphs Γnn is inherited by daughter candidates and that property is passed (with the level controlled by the fraction of allowed differences between daughter and target sequences) to the final outcome of the design process.
Table 3. Characterization of probe sets for 49 yeast ORF sequences.
| IDa | ΔTm (°C)b | σ(°C)c | [H]-quality (%)d |
|---|---|---|---|
| Affymetrix | 29 | 5.0 | 7.7 |
| 3 MM | 19 | 3.4 | 12.0 |
| 2 MM | 14 | 3.0 | 31.4 |
| 1 MM | 7 | 1.0 | 71.4 |
| 0 MM | 1.5 | 0.7 | 52.5 |
aProbe set identification—Affymetrix—probes from YG98 microarray, x MM—sets designed by method described in this paper, allowing x mismatches between ‘daughter’ and target sequence.
bTotal interval of melting temperatures calculated from probe sequences using nn-approximation.
cHalf-width of the Gaussian function fitted to the Tm histograms in Figure 7 (regression coefficients >0.9).
dCharacterization of the probe sequence homology using the [H] matrix (see text for details). The number is the fraction of the total number of probes that are non-homologous to any other probe in the set. The criterion for any pair of probes was that no more than 60% complementary bases and no continuous segment of complementary bases longer than 50% of the probe length were found.
CONCLUSIONS
Here, we present a novel method for the rational design of optimized DNA sequences for a wide range of technological applications. The advantages of our new Eulerian graph-based design concept can be summarized as follows: the sets of contextually isostable sequences exhibit extremely narrow ranges of melting temperatures, a requirement that is central to all applications. Furthermore, the mathematical tools of our graph-based method allow to imposing very complex and detailed requirements on the sequences to be generated. These requirements are then automatically satisfied without exception in every one of them. More importantly, the molecular part of the design process can be done without computer assistance, in a highly efficient engineering fashion—by drawing a ‘blueprint’ graph Γ, or, by partitioning the total numbers of each base into three components by writing the ‘crossword-puzzle’ adjacency matrix A(Γ) (following the rules in Equation 3). The efficiency of this stage stems from the fact that a single graph can represent enormous numbers of sequences with the desired properties. The method eliminates the necessity of extensive thermodynamic calculations. The melting temperature can be calculated only once or not at all; yet the isostability of the sequences is guaranteed and independent of the selection of a particular set of thermodynamic parameters.
One important question is how close the theoretical Tm of a contextually isostable sequence set is likely to be to the real Tms of a set of sequences selected for experimental validation from the identified ‘daughter’ sequence pool. Of course, because the method is based on Equation 1, it cannot predict a Tm closer than what current algorithms for Tm calculations allow. Correction of any disparity between a thermodynamic model and experimental stability of DNA duplexes—as was for example observed for oligomers on microarrays or metal nanoparticles (83)—can in principle be achieved in two ways. One is to determine new, more precise and for example microarray-specific thermodynamic parameters. The other is to fundamentally change the thermodynamic model itself by incorporating additional system descriptors (state functions) that go beyond the hydrogen bonding, nucleation, stacking, ionic strength, etc. Important advantage of our approach concerning parameters is the fact that our method is parameter-independent. The molecular identities of ‘daughters’ in our contextually isostable oligomer sets will never change with improved quality of thermodynamic parameters that might emerge from increasingly precise calorimetric and other experimental studies. Second, the narrowness of Tm distribution within this set is also constant. The only change that might be observed upon the changed parameters (see Equation 1) is the actual value of the predicted Tm. Concerning the thermodynamic model itself, it is of course difficult to anticipate the putative impact that a complete change in the assumptions and principles underlying Equation 1 would bring about. Nevertheless, if there is any theoretical possibility that novel thermodynamic contributors will be oligomer dependent, then the sequence context will again be important to predict their quantitative impact. To this effect, it is important to realize that our sequences are not only predicted to be thermodynamically isostable but are necessarily and under all circumstances contextually homogeneous. This last property is perhaps more relevant than the value of the calculated Tm itself, because it guarantees that other DNA properties that are contextually dependent will have the narrowest possible distributions for our set than for any other methods of selection would produce.
For a biologically most relevant application—the selection of probes for microarray functional genomics applications—our graph-based approach supplements existing strategies with a streamlined alternative. The first level benefit of this approach is a direct consequence of the possibility to select probe sequence properties and their differences collectively on the level of (few) ‘mother’ oligomers. These features are inherited by the ‘daughter’ sequence sets, which are much smaller than those of probe candidates in various steps of existing methods. It is therefore easier and faster to eliminate sequences that violate requirements for uniqueness and non-cross-hybridization. This feature of our method is best quantified by the relative sizes of ‘cliques’ of mutually non-homologous probes in Table 3. In summary, graph-based generation of contextually isostable sequences opens the possibility to define a priori technological criteria for the microarray probes and to match these criteria optimally for a given set of natural gene sequences by choosing the characteristics of graphs representing the probe candidates.
Last but not least, the molecular insight into the ‘anatomy’ of contextual isostability that our approach provides opens new possibilities for detailed experimental and theoretical studies of relationships between a DNA sequence and its properties and functions.
The interface to the program that generates ‘daughter’ sequences using DNA graphs Γnn is made available on http://max2.physics.sunysb.edu/users/pancoska/sage.html.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Martin Rocek (Institute for Theoretical Physics, Stony Brook University) for insightful discussions and Chi Ming Hung (Institute for Theoretical Physics, Stony Brook University) for help with web-application. We also thank anonymous referees for constructive comments. Support for this project was provided by National Cancer Institute grants 2RO1CA0600664 and 1RO1CA93853 to U.M.M.
REFERENCES
- 1.Dirks R.M., Lin,M., Winfree,E. and Pierce,N.A. (2004) Paradigms for computational nucleic acid design. Nucleic Acids Res., 32, 1392–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benight A.S., Pancoska,P., Owczarzy,R., Vallone,P.M., Nesetril,J. and Riccelli,P.V. (2001) Calculating sequence-dependent melting stability of duplex DNA oligomers and multiplex sequence analysis by graphs. Methods Enzymol., 340, 165–192. [DOI] [PubMed] [Google Scholar]
- 3.SantaLucia J. Jr, Kierzek,R. and Turner,D.H. (1991) Stabilities of consecutive A.C, C.C, G.G, U.C, and U.U mismatches in RNA internal loops: evidence for stable hydrogen-bonded U.U and C.C.+ pairs. Biochemistry, 30, 8242–8251. [DOI] [PubMed] [Google Scholar]
- 4.SantaLucia J. Jr, Allawi,H.T. and Seneviratne,P.A. (1996) Improved nearest-neighbor parameters for predicting DNA duplex stability. Biochemistry, 35, 3555–3562. [DOI] [PubMed] [Google Scholar]
- 5.Xia T., SantaLucia,J.,Jr, Burkard,M.E., Kierzek,R., Schroeder,S.J., Jiao,X., Cox,C. and Turner,D.H. (1998) Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson–Crick base pairs. Biochemistry, 37, 14719–14735. [DOI] [PubMed] [Google Scholar]
- 6.Allawi H.T. and SantaLucia,J.,Jr (1998) Nearest-neighbor thermodynamics of internal A.C mismatches in DNA: sequence dependence and pH effects. Biochemistry, 37, 9435–9444. [DOI] [PubMed] [Google Scholar]
- 7.Arghavani M.B., SantaLucia,J.,Jr and Romano,L.J. (1998) Effect of mismatched complementary strands and 5′-change in sequence context on the thermodynamics and structure of benzo[a]pyrene-modified oligonucleotides. Biochemistry, 37, 8575–8583. [DOI] [PubMed] [Google Scholar]
- 8.Allawi H.T. and SantaLucia,J.,Jr (1998) Thermodynamics of internal C.T mismatches in DNA. Nucleic Acids Res., 26, 2694–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allawi H.T. and SantaLucia,J.,Jr (1998) Nearest neighbor thermodynamic parameters for internal G.A mismatches in DNA. Biochemistry, 37, 2170–2179. [DOI] [PubMed] [Google Scholar]
- 10.SantaLucia J. Jr (1998) A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA, 95, 1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peyret N., Seneviratne,P.A., Allawi,H.T. and SantaLucia,J.,Jr (1999) Nearest-neighbor thermodynamics and NMR of DNA sequences with internal A.A, C.C, G.G, and T.T mismatches. Biochemistry, 38, 3468–3477. [DOI] [PubMed] [Google Scholar]
- 12.Bommarito S., Peyret,N. and SantaLucia,J.,Jr (2000) Thermodynamic parameters for DNA sequences with dangling ends. Nucleic Acids Res., 28, 1929–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rouillard J.M., Zuker,M. and Gulari,E. (2003) OligoArray 2.0: design of oligonucleotide probes for DNA microarrays using a thermodynamic approach. Nucleic Acids Res., 31, 3057–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res., 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuker M. (2000) Calculating nucleic acid secondary structure. Curr. Opin. Struct. Biol., 10, 303–310. [DOI] [PubMed] [Google Scholar]
- 16.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- 17.Zuker M. and Jacobson,A.B. (1998) Using reliability information to annotate RNA secondary structures. RNA, 4, 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heller M.J. (2002) DNA microarray technology: devices, systems, and applications. Annu. Rev. Biomed. Eng., 4, 129–153. [DOI] [PubMed] [Google Scholar]
- 19.Hartuv E., Schmitt,A.O., Lange,J., Meier-Ewert,S., Lehrach,H. and Shamir,R. (2000) An algorithm for clustering cDNA fingerprints. Genomics, 66, 249–256. [DOI] [PubMed] [Google Scholar]
- 20.Kingsley M.T., Straub,T.A., Call,D.R., Daly,D.S., Wunschel,S.C. and Chandler,D.P. (2002) Fingerprinting closely related Xanthomonas pathovars with random nonamer oligonucleotide microarrays. Appl. Environ. Microbiol., 68, 6361–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fedrigo O. and Naylor,G. (2004) A gene-specific DNA sequencing chip for exploring molecular evolutionary change. Nucleic Acids Res., 32, 1208–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai C.Y., Yu,M.L., Chen,S.C., Lin,Z.Y., Hsieh,M.Y., Wang,L.Y., Tsai,J.F., Chuang,W.L. and Chang,W.Y. (2004) Clinical evaluation of the COBAS Amplicor HBV monitor test for measuring serum HBV DNA and comparison with the Quantiplex branched DNA signal amplification assay in Taiwan. J. Clin. Pathol., 57, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morishima C., Chung,M., Ng,K.W., Brambilla,D.J. and Gretch,D.R. (2004) Strengths and limitations of commercial tests for hepatitis C virus RNA quantification. J. Clin. Microbiol., 42, 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cutler D.J., Zwick,M.E., Carrasquillo,M.M., Yohn,C.T., Tobin,K.P., Kashuk,C., Mathews,D.J., Shah,N.A., Eichler,E.E., Warrington,J.A. et al. (2001) High-throughput variation detection and genotyping using microarrays. Genome Res., 11, 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding Y. (2002) Rational statistical design of antisense oligonucleotides for high throughput functional genomics and drug target validation. Statist. Sin., 12, 273–296. [Google Scholar]
- 26.Lee I., Dombkowski,A.A. and Athey,B.D. (2004) Guidelines for incorporating non-perfectly matched oligonucleotides into target-specific hybridization probes for a DNA microarray. Nucleic Acids Res., 32, 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ott J. and Hoh,J. (2003) Set association analysis of SNP case–control and microarray data. J. Comput. Biol., 10, 569–574. [DOI] [PubMed] [Google Scholar]
- 28.DeSantis T.Z., Dubosarskiy,I., Murray,S.R. and Andersen,G.L. (2003) Comprehensive aligned sequence construction for automated design of effective probes (CASCADE-P) using 16S rDNA. Bioinformatics, 19, 1461–1468. [DOI] [PubMed] [Google Scholar]
- 29.Nielsen H.B., Wernersson,R. and Knudsen,S. (2003) Design of oligonucleotides for microarrays and perspectives for design of multi-transcriptome arrays. Nucleic Acids Res., 31, 3491–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magwene P.M., Lizardi,P. and Kim,J. (2003) Reconstructing the temporal ordering of biological samples using microarray data. Bioinformatics, 19, 842–850. [DOI] [PubMed] [Google Scholar]
- 31.Chen J.H. and Seeman,N.C. (1991) Synthesis from DNA of a molecule with the connectivity of a cube. Nature, 350, 631–633. [DOI] [PubMed] [Google Scholar]
- 32.Conrad M. and Zauner,K.P. (1998) DNA as a vehicle for the self-assembly model of computing. Biosystems, 45, 59–66. [DOI] [PubMed] [Google Scholar]
- 33.Phadke R.S. (2001) Biomolecular electronics in the twenty-first century. Appl. Biochem. Biotechnol., 96, 269–276. [DOI] [PubMed] [Google Scholar]
- 34.Seeman N.C. (1991) Construction of three-dimensional stick figures from branched DNA. DNA Cell Biol., 10, 475–486. [DOI] [PubMed] [Google Scholar]
- 35.Seeman N.C. (1999) DNA engineering and its application to nanotechnology. Trends Biotechnol., 17, 437–443. [DOI] [PubMed] [Google Scholar]
- 36.Winfree E., Liu,F., Wenzler,L.A. and Seeman,N.C. (1998) Design and self-assembly of two-dimensional DNA crystals. Nature, 394, 539–544. [DOI] [PubMed] [Google Scholar]
- 37.Rozenberg G. and Spaink,H. (2003) DNA computing by blocking. Theoret. Comput. Sci., 292, 653–665. [Google Scholar]
- 38.Penchovsky R. and Ackermann,J. (2003) DNA library design for molecular computation. J. Comput. Biol., 10, 215–229. [DOI] [PubMed] [Google Scholar]
- 39.Livstone M.S., van Noort,D. and Landweber,L.F. (2003) Molecular computing revisited: a Moore's law? Trends Biotechnol., 21, 98–101. [DOI] [PubMed] [Google Scholar]
- 40.Braun-Sand S.B. and Wiest,O. (2003) Theoretical studies of mixed-valence transition metal complexes for molecular computing. J. Phys. Chem. A, 107, 285–291. [Google Scholar]
- 41.Benenson Y., Adar,R., Paz-Elizur,T., Livneh,Z. and Shapiro,E. (2003) DNA molecule provides a computing machine with both data and fuel. Proc. Natl Acad. Sci. USA, 100, 2191–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zimmermann K.H. (2002) Efficient DNA sticker algorithms for NP-complete graph problems. Comput. Phys. Commun., 144, 297–309. [Google Scholar]
- 43.Yin Z., Zhang,F. and Xu,J. (2002) A Chinese Postman Problem based on DNA computing. J. Chem. Inf. Comput. Sci., 42, 222–224. [DOI] [PubMed] [Google Scholar]
- 44.Wang S.Y. (2002) DNA computing of bipartite graphs for maximum matching. J. Math. Chem., 31, 271–279. [Google Scholar]
- 45.Tour J.M., Van Zandt,W.L., Husband,C.P., Husband,S.M., Wilson,L.S., Franzon,P.D. and Nackashi,D.P. (2002) Nanocell logic gates for molecular computing. IEEE Trans. Nanotechnol., 1, 100–109. [Google Scholar]
- 46.Rozenberg G. (2002) Models of molecular computing based on molecular reactions. New Generation Comput., 20, 237–249. [Google Scholar]
- 47.Normile D. (2002) Molecular computing—DNA-based computer takes aim at genes. Science, 295, 951. [DOI] [PubMed] [Google Scholar]
- 48.Liu Y.C., Xu,J., Pan,L.Q. and Wang,S.Y. (2002) DNA solution of a graph coloring problem. J. Chem. Inform. Comput. Sci., 42, 524–528. [DOI] [PubMed] [Google Scholar]
- 49.Gillmor S.D., Rugheimer,P.P. and Lagally,M.G. (2002) Computation with DNA on surfaces. Surface Sci., 500, 699–721. [Google Scholar]
- 50.Amos M., Paun,G., Rozenberg,G. and Salomaa,A.T. (2002) Topics in the theory of DNA computing. Theoret. Comput. Sci., 287, 3–38. [Google Scholar]
- 51.Paun G. (2001) From cells to computers: computing with membranes (P systems). Biosystems, 59, 139–158. [DOI] [PubMed] [Google Scholar]
- 52.Head T. (2001) Biomolecular realizations of a parallel architecture for solving combinatorial problems. New Generation Comput., 19, 301–312. [Google Scholar]
- 53.Freivalds R., Hromkovic,J., Paun,G. and Unger,W. (2001) Communication, molecular computing and randomized algorithms—Foreword. Theoret. Comput. Sci., 264, 1–2. [Google Scholar]
- 54.Wasiewicz P., Janczak,T., Mulawka,J.J. and Plucienniczak,A. (2000) The inference based on molecular computing. Cybernet. Syst., 31, 283–315. [Google Scholar]
- 55.Seife C. (2000) Molecular computing—RNA works out knight moves. Science, 287, 1182–1183. [DOI] [PubMed] [Google Scholar]
- 56.Sakamoto K., Gouzu,H., Komiya,K., Kiga,D., Yokoyama,S., Yokomori,T. and Hagiya,M. (2000) Molecular computation by DNA hairpin formation. Science, 288, 1223–1226. [DOI] [PubMed] [Google Scholar]
- 57.Ruben A.J. and Landweber,L.F. (2000) The past, present and future of molecular computing. Nature Rev. Mol. Cell Biol., 1, 69–72. [DOI] [PubMed] [Google Scholar]
- 58.Rotman D. (2000) Molecular computing. Technol. Rev., 103, 5258. [Google Scholar]
- 59.Head T., Rozenberg,G., Bladergroen,R.S., Breek,C.K., Lommerse,P.H. and Spaink,H.P. (2000) Computing with DNA by operating on plasmids. Biosystems, 57, 87–93. [DOI] [PubMed] [Google Scholar]
- 60.Hagiya M. (1999) Perspectives on molecular computing. New Generation Comput., 17, 131–151. [Google Scholar]
- 61.Garzon M.H. and Deaton,R.J. (1999) Biomolecular computing and programming. IEEE Trans. Evol. Comput., 3, 236–250. [Google Scholar]
- 62.Deaton R., Garzon,M., Rose,J., Franceschetti,D.R. and Stevens,S.E.,Jr (1999) DNA computing: a review. Fundamenta Informaticae, 35, 231–245. [Google Scholar]
- 63.Deaton R., Garzon,M., Murphy,R.C., Rose,J.A., Franceschetti,D.R. and Stevens,S.E. (1998) Reliability and efficiency of a DNA-based computation. Phys. Rev. Lett., 80, 417–420. [Google Scholar]
- 64.Yurke B., Turberfield,A.J., Mills,A.P.,Jr, Simmel,F.C. and Neumann,J.L. (2000) A DNA-fuelled molecular machine made of DNA. Nature, 406, 605–608. [DOI] [PubMed] [Google Scholar]
- 65.Deaton R., Kim,J.W. and Chen,J. (2003) Design and test of noncrosshybridizing oligonucleotide building blocks for DNA computers and nanostructures. Appl. Phys. Lett., 82, 1305–1307. [Google Scholar]
- 66.Matveeva O.V., Shabalina,S.A., Nemtsov,V.A., Tsodikov,A.D., Gesteland,R.F. and Atkins,J.F. (2003) Thermodynamic calculations and statistical correlations for oligo-probes design. Nucleic Acids Res., 31, 4211–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Waterman M.S. (1995) Introduction to Computational Biology. Chapman and Hall, London. [Google Scholar]
- 68.Blazewicz J., Hertz,A., Kobler,D. and de Werra,D. (1999) On some properties of DNA graphs. Discrete Appl. Math., 98, 1–19. [Google Scholar]
- 69.Pevzner P.A., Tang,H.X. and Waterman,M.S. (2001) An Eulerian path approach to DNA fragment assembly. Proc. Natl Acad. Sci. USA, 98, 9748–9753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y. and Waterman,M.S. (2003) An Eulerian path approach to global multiple alignment for DNA sequences. J. Comput. Biol., 10, 803–819. [DOI] [PubMed] [Google Scholar]
- 71.Chechetkin V.R. (2003) Block structure and stability of the genetic code. J. Theoret. Biol., 222, 177–188. [DOI] [PubMed] [Google Scholar]
- 72.Feng S. and Zhongxi,M. (2002) Similarity among nucleotide sequences. Acta Biotheoret., 50, 95–99. [DOI] [PubMed] [Google Scholar]
- 73.Pancoska P., Moravek,Z. and Moll,U.M. (2004) Efficient RNA interference depends on global context of the target sequence. Quantitative analysis of silencing efficiency using Eulerian graph representation of siRNA. Nucleic Acids Res., 32, 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nesetril J. (1998) Invitation to Discrete Mathematics. Oxford University Press, Oxford. [Google Scholar]
- 75.Lewis F.D., Liu,J., Zuo,X., Hayes,R.T. and Wasielewski,M.R. (2003) Dynamics and energetics of single-step hole transport in DNA hairpins. J. Am. Chem. Soc., 125, 4850–4861. [DOI] [PubMed] [Google Scholar]
- 76.Schuster G.B. (2000) Long-range charge transfer in DNA: transient structural distortions control the distance dependence. Acc. Chem. Res., 33, 253–260. [DOI] [PubMed] [Google Scholar]
- 77.Liu C.S., Hernandez,R. and Schuster,G.B. (2004) Mechanism for radical cation transport in duplex DNA oligonucleotides. J. Am. Chem. Soc., 126, 2877–2884. [DOI] [PubMed] [Google Scholar]
- 78.Favis R., Day,J., Gerry,N.P., Phelan,C., Narod,S. and Barany,F. (2000) Universal DNA array detection of small insertions and deletions in BRCA1 and BRCA2. Nat. Biotechnol., 18, 561–564. [DOI] [PubMed] [Google Scholar]
- 79.Gerry N.P., Witkowski,N.E., Day,J., Hammer,R.P., Barany,G. and Barany,F. (1999) Universal DNA microarray method for multiplex detection of low abundance point mutations. J. Mol. Biol., 292, 251–262. [DOI] [PubMed] [Google Scholar]
- 80.Roth M.E., Feng,L., McConnell,K.J., Schaffer,P.J., Guerra,C.E., Affourtit,J.P., Piper,K.R., Guccione,L., Hariharan,J., Ford,M.J. et al. (2004) Expression profiling using a hexamer-based universal microarray. Nat. Biotechnol., 22, 418–426. [DOI] [PubMed] [Google Scholar]
- 81.van Dam R.M. and Quake,S.R. (2002) Gene expression analysis with universal n-mer arrays. Genome Res., 12, 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Christie K.R., Weng,S., Balakrishnan,R., Costanzo,M.C., Dolinski,K., Dwight,S.S., Engel,S.R., Feierbach,B., Fisk,D.G., Hirschman,J.E. et al. (2004) Saccharomyces Genome Database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res., 32 (Database issue), D311–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jin R., Wu,G., Li,Z., Mirkin,C.A. and Schatz,G.C. (2003) What controls the melting properties of DNA-linked gold nanoparticle assemblies? J. Am. Chem. Soc., 125, 1643–1654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.