

Abstract
Drug-induced adaptations in the prefrontal cortex (PFC) contribute to several core aspects of addictive behaviors, but the underlying neuronal processes remain essentially unknown. Here, we demonstrate that repeated in vivo exposure to cocaine persistently reduces the voltage-gated K+ current (VGKC) in PFC pyramidal neurons, resulting in enhanced membrane excitability. Analysis of dopamine D1-class receptor (D1R)-mediated modulation of VGKC indicates that, despite the absence of direct D1R stimulation, downstream D1 signaling (the cAMP/protein kinase A pathway) is increased during withdrawal from chronic cocaine treatment and plays a central role in the drug-induced membrane plasticity in PFC. This long-lasting, cocaine-induced plasticity of membrane excitability in PFC pyramidal neurons may contribute to the impaired decision making and drug craving that characterize cocaine withdrawal.
Keywords: cocaine, prefrontal cortex, dopamine, K+ current, plasticity, adenylate cyclase
Introduction
Repeated exposure to drugs of abuse produces long-lasting neuronal adaptations that mediate the development and maintenance of addictive behaviors. Drug-induced adaptations do not occur globally, but within specific critical brain circuits, among which is the mesocorticolimbic dopamine (DA) system. This consists of the ventral tegmental area (VTA), the nucleus accumbens (NAc), and the prefrontal cortex (PFC), which receives a dense DA innervation from the VTA and sends glutamate efferents to the VTA and NAc. Given that the primary pharmacological effect of cocaine is to increase extracellular DA by blocking the DA reuptake transporter, it is conceivable that enhanced actions of DA in PFC after cocaine administration trigger important neuronal adaptations that are crucial to the development of cocaine addiction. Indeed, optimized DA modulation in the PFC is required for several cognitive processes, such as working memory, decision making, and goal planning, impairments of which contribute to addiction (Jentsch et al., 2000), whereas drug-induced DA-dependent neuronal adaptations and responses in the PFC contribute to the primary reinforcing effects of cocaine, development of cocaine locomotion sensitization, drug-elicited conditioned-place preference, cocaine self-administration, and reinstatement of cocaine seeking and relapse (Tzschentke, 2001).
Long-lasting changes in intrinsic membrane properties have recently received increased attention as a key mechanism underlying some forms of experience-dependent plasticity (Zhang and Linden, 2003). Because DA reversibly modulates the intrinsic membrane excitability of PFC neurons by affecting several ionic conductances (Yang and Seamans, 1996; Maurice et al., 2001; Dong and White, 2003; Dong et al., 2004a), it is conceivable that such short-term modulation of PFC neurons may, after repeated cocaine administration, evolve into a long-term adaptation. A previous study demonstrated that ID, the major component of the whole-cell voltage-gated K+ current (VGKC) (with relatively slow inactivation) is suppressed during D1R/cAMP/PKA (D1-class receptor/cAMP/protein kinase A) cascade stimulation, whereas two other types of currents, IA (with rapid inactivation) and IK (with very slow inactivation), are very small relative to ID and insensitive to DA D1 signaling (Dong and White, 2003). Here, we report that, via DA D1 signaling, repeated cocaine treatment induces a reduction of whole-cell VGKC (mainly ID), resulting in a persistent increase of membrane excitability in PFC neurons. This cocaine-induced, long-lasting neural adaptation may contribute to the behavioral sequelae and cognitive impairments that characterize the addicted state (Jentsch et al., 2000).
Materials and Methods
Animals and treatment. After a 1 week acclamation to the vivarium, male Sprague Dawley rats (4-5 weeks of age) were randomly distributed to two groups that received an intraperitoneal injection of saline (1.0 ml/kg) or (-)cocaine HCl (15.0 mg/kg) once per day for 5 consecutive days. The electrophysiology experiments were conducted on the third day after cessation of injections.
Electrophysiology. Cell/slice preparation and patch-clamp recordings were conducted as reported previously (Dong and White, 2003). Briefly, rats were anesthetized and decapitated. Brains were quickly removed, blocked, and sliced. For voltage-clamp recordings from dissociated neurons, the coronal slices (400 μm) were incubated for 1-4 h at room temperature in a sodium bicarbonate-buffered Earle's balanced salt solution. The deep layers (V and VI) of medial PFC (mPFC) were isolated and then placed into an oxygenated, HEPES-buffered HBSS containing 1.5 mg/ml protease (type XIV) at 35°C for 30 min. The tissue was then triturated with a graded series of fire-polished Pasteur pipettes. The cell suspension was transferred to the recording chamber. The intracellular recording solution was as follows (in mm): 70 K2SO4, 60 N-methylglucamine (NMG+), 30 HEPES, 5 BAPTA, 12 phosphocreatine, 3 Na2ATP, 0.2 Na3GTP, 2 MgCl2, and 0.5 CaCl2, pH 7.2, with KOH (275 mOsm/L). The normal extracellular recording solution contained the following (in mm): 140 Na-isethionate, 10 HEPES, 12 glucose, 17.5 sucrose, 1-4 KCl, 4 MgCl2, and 0.001 TTX, pH 7.35, with NaOH/HCl (300 mOsm/L). In some preparations, when both outward and inward K+ currents were recorded at the same time, the concentration of K+ in the extracellular solution was increased to 20 mm, and the NMG+ was reduced to keep the optimal osmolarity. Extracellular recording solutions were applied via one of a series of glass capillaries (inner diameter, ∼250 μm) in which gravity-fed flow was regulated by electronic valves (BioLogic, Claix, France). Recordings were obtained with an Axon Instruments (Foster City, CA) 200A patch-clamp amplifier, controlled, and monitored with a personal computer running pClamp (version 8.1) with a 125 kHz interface (Axon Instruments). Electrode resistances were ∼1-4 MΩ in bath. Series resistance was compensated (70-80%) and periodically monitored. Recordings were restricted to neurons with pyramidal soma and small remnants of the apical dendrites. Current density of VGKC or inwardly rectifying K+ current (IRKC) was calculated as (whole-cell current)/(whole-cell capacitance). Recordings were performed at room temperature (22-24°C). The liquid junction potential (∼2 mV) was not compensated. Leak subtraction was not applied.
For whole-cell current-clamp recordings in PFC slice, the PFC brain slices were anchored in the recording chamber and perfused by oxygenated artificial CSF (34°C). The internal solution contained the following (in mm): 120 K+-gluconate, 10 HEPES, 0.1 EGTA, 20 KCl, 2 MgCl2, 3 Na2ATP, and 0.3 Na2GTP. Whole-cell current-clamp recordings were performed using an SEC-05L NPI amplifier (ALA Scientific Instruments, Westbury, NY). Action potentials were generated by injection of step-depolarizing current pulses. Whole-cell pipette series resistance was <20 MΩ, and bridge was compensated. Only cells with a resting membrane potential of less than -60 mV, stable baseline responses, and evoked spikes >0 mV were used for analysis. All of the reagents were obtained from Sigma (St. Louis, MO), except for ATP and GTP (Boehringer Mannheim, Indianapolis, IN) and BAPTA (Calbiochem, La Jolla, CA).
PKA assay. The mPFC and motor cortex (MC) were respectively dissected, combined, lysed in hypotonic buffer [10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 1 mm DTT, 50 μm FK506 (tacrolimus), and 10 μm microcystin], and supplemented with Complete Protease Inhibitor tablets (Roche Diagnostics). PKA activity in 2 μg of each sample was determined by the PepTag PKA assay (Promega, Madison, WI). Positive controls contained 50 ng of purified PKA catalytic subunit (Promega), whereas negative controls contained a 2 μg sample plus 50 μm protein kinase inhibitor (Calbiochem, San Diego, CA). The PepTag assay uses the Leu-Arg-Arg-Ala-Ser-Leu-Gly (Kemptide) peptide substrate tagged with a fluorescent dye. During phosphorylation, the net charge of this peptide changes and alters the direction of peptide migration when run on an agarose gel. Activity was detected by the amount of phosphorylated substrate migrating toward the anode. This assay, starting from animal treatment, was repeated four times, and the data were combined for analysis.
Data analysis. More than 70% of the data were collected and analyzed without knowledge of the treatment that the animals had received. There were no differences in the results from blinded and nonblinded experiments, and, therefore, the results were combined. Statistica (StatSoft, Tulsa, OK) was used for most of the statistical analysis. Origin (Microcal Software, Northampton, MA) was used to plot the current traces and graphs. Student's t tests were used in all of the comparisons. Data are presented as mean ± SE.
Results
To begin to characterize cocaine-induced adaptations in the PFC, we acutely dissociated deep layer (V and VI) mPFC neurons from animals 3 d after cessation of repeated administration of either cocaine or saline and examined the whole-cell IRKC and VGKC. A previous study demonstrated that ID contributed most of the whole-cell VGKC (IA is not obvious in most PFC neurons, and IK contributes <10% of the whole-cell current), and, therefore, we operationally defined the whole-cell VGKC as ID (Dong and White, 2003). A hyperpolarization voltage step (200 ms, -100 mV) from the holding potential of -50 mV elicited IRKC, and a subsequent depolarization step (500 ms, 10 mV) elicited whole-cell VGKC. In PFC pyramidal neurons, the IRKC density (measured at the end of the current) was not significantly affected (saline, 22.71 ± 5.83 pA/pF, n = 14; cocaine, 19.18 ± 2.46 pA/pF, n = 26) (Fig. 1A), whereas the early component of ID density (measured at 50 ms from current initiation) was significantly decreased by cocaine treatment (saline, 175.58 ± 16.95 pA/pF, n = 14; cocaine, 115.26 ± 16.10 pA/pF, n = 15; p < 0.01) (Fig. 1A). In contrast, in motor cortex pyramidal neurons prepared from the same brain slices, neither the IRKC density (saline, 25.91 ± 4.93 pA/pF, n = 9; cocaine, 24.79 ± 2.79 pA/pF, n = 9) nor the early ID density (saline, 166.41 ± 14.09 pA/pF, n = 14; cocaine, 159.98 ± 12.76 pA/pF, n = 15) (Fig. 1B) was altered by cocaine treatment. It has been demonstrated that there are two components in ID: the early component, with an inactivation τ of 220 ms, and the late component, with an inactivation τ of 3.4 s (Dong and White, 2003). In contrast to the early component, the density of the late component (measured at 400 ms from current initiation) in PFC neurons was only minimally affected by cocaine treatment (saline, 112.2 ± 10.4 pA/pF, n = 14; cocaine, 96.4 ± 7.5 pA/pF, n = 26, p = 0.06) (Fig. 1), indicating that chronic cocaine treatment preferentially suppresses the early component of ID.
Figure 1.

Repeated cocaine treatment reduces the VGKC and increases membrane excitability in PFC pyramidal neurons. A1, Examples of IRKC and VGKC in PFC pyramidal neurons from animals receiving cocaine (COC) and saline (SAL) pretreatments. Voltage protocol used to sequentially elicit IRKC and VGKC is shown above the traces. The arrows indicate the time points at which IRKC (downward arrows) and VGKC (upward arrows) were measured. A2, Summary of current densities indicates that VGKC, but not IRKC, is significantly reduced in PFC neurons from animals receiving cocaine when compared with saline pretreatments (*p < 0.05 in this and all of the subsequent figures). B1, Examples of IRKC and VGKC in motor cortex (Motor Ctx) pyramidal neurons from animals receiving cocaine and saline pretreatments. B2, Summary of current densities indicates that both VGKC and IRKC in motor cortex neurons are not significantly affected by chronic cocaine administration. C, Examples of action potentials elicited by a constant current injection (200 pA, 500 ms) in PFC neurons from animals receiving saline or cocaine pretreatment. D, Summary of the number of action potentials elicited by constant current injection in cocaine-pretreated and saline-pretreated PFC neurons.
One predictable functional consequence of the cocaine-induced reduction in ID is an overall enhancement of membrane excitability in PFC neurons. To test this prediction, we made whole-cell current-clamp recordings from PFC pyramidal neurons in brain slices and measured the number of action potentials generated by a positive current injection from a normalized resting membrane potential (-65 mV). More action potentials were elicited by this constant current injection (0.2 nA, 500 ms) in neurons from animals pretreated with cocaine than from saline control neurons (saline, 4.17 ± 0.53 spikes, n = 12; cocaine, 6.08 ± 0.44 spikes, n = 12; p < 0.01) (Fig. 1C), a result consistent with the idea that chronic cocaine treatment persistently elevates intrinsic membrane excitability in PFC neurons by reducing VGKC (ID).
A critical question is whether this enhancement of membrane excitability could also be triggered by acute cocaine treatment. To address this concern, we first acutely applied cocaine (1 μm) to the PFC slice. Ten minutes of direct cocaine perfusion did not alter action potential firing evoked by a constant current step (net change, 0.20 ± 0.22; n = 5; p = 0.37; paired t test). We next examined the elicited action potential firing from animals receiving only one injection of cocaine, followed by a 24 h withdrawal. This short-term cocaine administration also did not alter the action potential firing in PFC neurons (again evoked by a 200 pA, 500 ms step from Vm of -65 mV; saline, 4.08 ± 0.52 spikes, n = 13; cocaine, 3.82 ± 0.51 spikes, n = 11; p = 0.72; t test). These results suggest that the enhancement in the membrane excitability of PFC neurons is a characteristic neuronal adaptation induced by chronic, but not acute, cocaine administration.
A major structural difference between PFC and motor cortex in rodents is that PFC receives dense DA projections but motor cortex does not (Carr et al., 1999), suggesting a potential role of DA in this cocaine-induced adaptation. Indeed, DA, via activation of the D1R/cAMP/PKA signaling pathway, inhibits VGKC (ID) in PFC neurons (Dong and White, 2003). Thus, it is possible that cocaine treatment may persistently upregulate DA D1 signaling, resulting in the observed reduction in ID. To test this hypothesis, we compared the D1R-mediated modulation of VGKC in PFC neurons from saline-versus cocaine-pretreated rats. Consistent with previous findings (Dong and White, 2003), stimulation of D1Rs with 1 μm SKF81297 [(+-)-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1M-3-benzazepine], a selective DA D1R agonist, suppressed ID (measured at 50 ms from the current initiation) in PFC neurons obtained from saline-pretreated animals, an effect that was significantly reduced by cocaine pretreatment (saline, 19.60 ± 1.83%, n = 8; cocaine, 7.43 ± 1.46%, n = 7; p < 0.05) (Fig. 2A).
Figure 2.
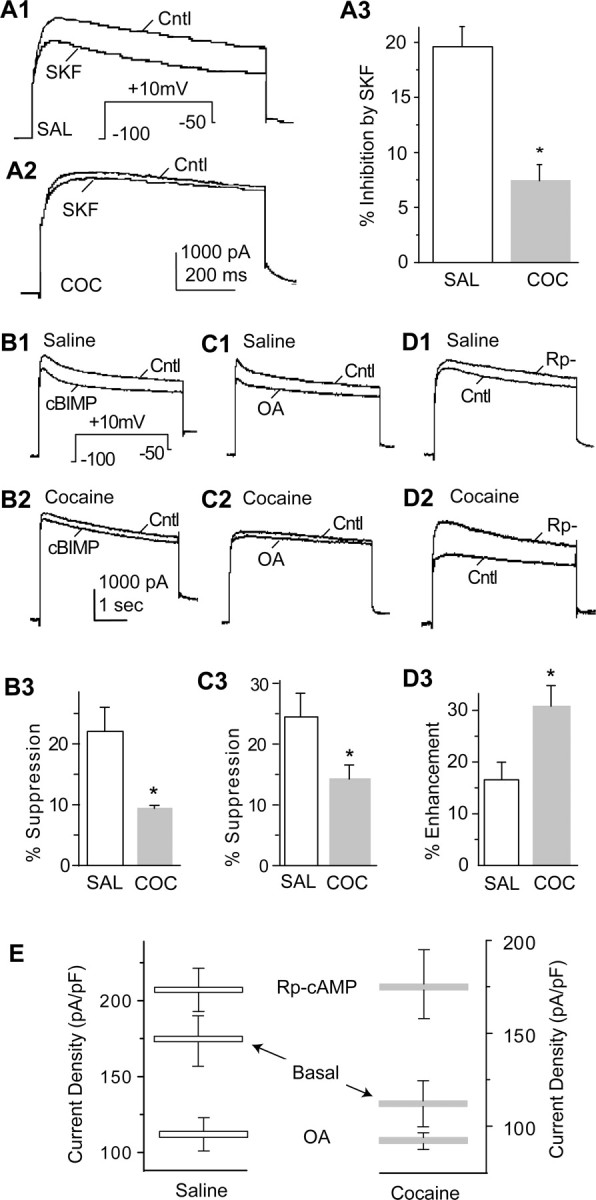
A central role of DA D1 receptor signaling in cocaine-induced neuroadaptations in PFC neurons. A1, A2, Examples of D1R-mediated suppression of VGKC in PFC neurons from animals receiving saline (SAL; A1) and cocaine (COC; A2) pretreatments. A3, Summary shows that the D1R-mediated suppression of VGKC in PFC neurons is diminished by cocaine pretreatment. B1, B2, Examples of effects of cBIMP on VGKC in PFC neurons from animals receiving saline (B1) and cocaine (B2) pretreatments. B3, Summary shows that the cBIMP-induced suppression of VGKC in PFC neurons is diminished by cocaine pretreatment. C1, C2, Examples of effects of OA on VGKC in PFC neurons from animals receiving saline (C1) and cocaine (C2) pretreatments. C3, Summary shows that OA-induced suppression of VGKC in PFC neurons is diminished by cocaine pretreatment. D1, D2, Examples of effects of Rp-cAMP on VGKC in PFC neurons from animals receiving saline (D1) and cocaine (D2) pretreatments. D3, Summary shows that the Rp-cAMP-induced enhancment of VGKC in PFC neurons is increased by cocaine pretreatment. E, Summary of current density of basal VGKC and modulated VGKC (by Rp-cAMP and OA) suggests that basal cAMP/PKA activity is upregulated by chronic cocaine pretreatment. Cntl, Control; SKF, SKF81297.
Although several mechanisms could account for the observed reduction of the D1R effect on ID in neurons from animals that had received cocaine, because of the overall reduction of ID in these PFC neurons, we proceeded to test the hypothesis that the chronic cocaine treatment caused a persistent elevation in D1R signaling, specifically an increase in the basal activity of the cAMP/PKA signaling pathway. If basal D1R/cAMP/PKA activity is already elevated in the PFC neurons from cocaine-pretreated animals, additional stimulation of this pathway should produce less effect, whereas inhibition of this pathway should result in a larger effect in cocaine-pretreated neurons. To directly activate PKA, we applied 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole-3′,5′-cyclic monophosphorothioate (cBIMP) (50 μm), a membrane-permeable PKA activator, and found that the inhibition of ID (measured at 50 ms from the current initiation) was significantly weaker in the PFC neurons from the cocaine group (saline, 22.07 ± 3.80%, n = 6; cocaine, 9.39 ± 0.51%, n = 8; p < 0.05) (Fig. 2B). We next examined the effects of inhibiting protein phosphatase activity with okadaic acid (OA) (20 μm), a manipulation that should increase basal PKA-mediated phosphorylation. Okadaic acid also inhibited ID to a significantly greater extent in the saline control neurons than in cocaine-pretreated neurons (saline, 24.48 ± 3.89%, n = 5; cocaine, 14.24 ± 2.31%, n = 6; p < 0.05) (Fig. 2C). We also performed the converse experiment of inhibiting cAMP/PKA activity using Rp-cAMP (100 μm). This caused an enhancement of ID amplitude, an increase that was significantly larger in cocaine-pretreated neurons than in control neurons (saline, 16.55 ± 3.36%, n = 6; cocaine, 30.77 ± 10.51%, n = 7; p < 0.05) (Fig. 2D). Together, these results (Fig. 2E) are consistent with the idea that the cocaine treatment caused a persistent elevation in cAMP/PKA activity and, as a consequence, reduced the overall VGKC.
To more directly assess the effect of chronic cocaine treatment on basal PKA activity in PFC, we used a biochemical assay involving phosphorylation of a peptide substrate of PKA (Kemptide). Tissue lysates from both the PFC and motor cortex were prepared from saline- and cocaine-pretreated animals. Consistent with our electrophysiological findings, PKA activity in PFC, but not motor cortex, was significantly increased by cocaine administration (PFC-saline, 17.31 ± 5.6; PFC-cocaine, 37.1 ± 9.4, p < 0.05, n = 4; motor cortex-saline, 20.8 ± 7.0; MC-cocaine, 31.4 ± 6.0, p > 0.1, n = 4) (Fig. 3).
Figure 3.

Chronic cocaine enhances basal PKA activity in PFC neurons. A, Examples of gels showing phosphorylation of peptide substrate resulting from PKA activity in tissue from PFC and MC obtained from saline (SAL)- and cocaine (COC)-pretreated animals. B, Summary shows that PKA activity within PFC, but not in MC, is significantly increased by cocaine pretreatment.
Discussion
As the backbone of corticomesolimbic glutamate transmission, the PFC seems to play a critical role in mediating several important components of addictive behaviors. For example, direct electrical stimulation of PFC leads to rewarding effects and sensitization to cocaine, whereas lesions of PFC or impairment of corticomesolimbic glutamate transmission prevent the development of cocaine sensitization (Li et al., 1997, 1999; Tzschentke, 2001). In addition, excitatory lesions of PFC or microperfusion of cocaine into PFC augments drug-taking behavior (Goeders and Smith, 1993; Weissenborn et al., 1997). Thus, a critical question in understanding the neuronal mechanisms of addiction is what neuronal adaptations occur in PFC neurons in response of drugs of abuse (Wolf, 1998; Hyman and Malenka, 2001). Although important progress has been made in delineating some of the morphological and molecular adaptations that occur in this structure (Robinson and Kolb, 1997, 1999; Shaw-Lutchman et al., 2003), little is known about how drugs modify the physiological properties of PFC neurons.
Here, we demonstrate that previous cocaine exposure reduces whole-cell VGKC (mainly ID) and enhances membrane excitability in PFC neurons. These cocaine-induced electrophysiological adaptations are persistent and remain several days after termination of cocaine treatment. A direct consequence of this cocaine-induced neuronal adaptation would be an increase of presynaptic glutamate release in the mesocorticolimbic DA system. In fact, an increased glutamate release has been observed in the VTA, the NAc, and the PFC after cocaine administration (Smith et al., 1995; Reid et al., 1997), and this increase, which can be blocked by inactivation of PFC, is temporally correlated with drug-seeking behaviors (Smith-Roe et al., 1999; McFarland et al., 2003). Furthermore, this adaptation in PFC neurons may also serve as a presynaptic trigger for the drug-induced long-term synaptic plasticity expressed at glutamatergic synapses in mesocorticolimbic DA system (Thomas et al., 2001; Dong et al., 2004b).
Our results indicate that cocaine-induced effects occur preferentially in the early component of ID. There are three types of currents expressed in PFC neurons, namely ID, IA, and IK (Dong and White, 2003). As done previously (Dong and White, 2003), in this study, we regard the whole-cell VGKC as being predominantly attributable to ID. IK contributes <10% of the whole-cell VGKC and does not inactivate under normal recording course (seconds); therefore, it was considered “background” or leak current (Dong and White, 2003). IA is also very small and, in most PFC neurons, is obscured by ID (Dong and White, 2003). Furthermore, it inactivates very rapidly (inactivation τ ∼12 ms), and we measured the early ID at 50 ms from current initiation, a time point at which most IA has already inactivated (Dong and White, 2003). Thus, although the possibility of cocaine-induced changes in IA and IK cannot be excluded, modulation of ID contributes most of the cocaine-induced alteration in whole-cell VGKC.
Consistent with this conclusion, PFC neurons from animals receiving the same cocaine treatment exhibited an increased input resistance and decreased outward rectification when membrane potential was depolarized, whereas no change in resting membrane potential was observed (Nasif et al., 2002). However, neuronal excitability and action potential firing in response to depolarization are influenced by many additional ionic conductances, such as Na+ and Ca2+ currents, and, thus, it will be important in future studies to examine how in vivo cocaine administration affects these currents in PFC neurons.
It has been suggested that the K+ channels presumably underlying VGKCs exist in a state of balance between D1R/cAMP/PKA-dependent phosphorylation, which downregulates the VGKC, and dephosphorylation, which upregulates the VGKC (Dong and White, 2003). Results from pharmacological experiments in which we manipulated the cAMP/PKA pathway suggest that the cocaine-induced, long-lasting neuronal adaptation in PFC neurons is attributable to the persistent upregulation of cAMP/PKA signaling. Such elevation of basal activity in the cAMP/PKA pathway would not only account for the smaller whole-cell VGKC that we observed in cocaine-pretreated neurons but also accounts for the diminished D1R modulation, the diminished effects of cBIMP and OA, and the enhanced action of Rp-cAMP.
Increased cAMP/PKA signaling (Shaw-Lutchman et al., 2003) also seems to contribute to long-lasting, cocaine-induced electrophysiological adaptations in NAc medium spiny neurons, although the effects involve decreases in Na+ and Ca2+ currents (Zhang et al., 1998, 2002), changes that will decrease excitability. Thus, although alterations in this intracellular signaling pathway may be a common biochemical adaptation to chronic cocaine throughout the mesocorticolimbic DA system, there are marked cell-type-specific differences in the physiological consequences. Given the importance of the PFC for making rational choices (Damasio, 1995; Jentsch et al., 1997), we suggest that the long-lasting cocaine-induced changes in PFC pyramidal neuron excitability reported here may contribute importantly to the compulsive and maladaptive decision making that characterizes addiction.
Footnotes
This work was supported by National Institutes of Health Grants DA12618, DA00456 (F.J.W.), and DA009264 (R.C.M.). We thank Kerstin Ford for excellent technical assistance.
Correspondence should be addressed to Dr. Yan Dong, Merck Research Laboratories, P.O. Box 2000, RY80M-213, Rahway, NJ 07065. E-mail: yan_dong@merck.com.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250936-05$15.00/0
References
- Carr DB, O'Donnell P, Card JP, Sesack SR (1999) Dopamine terminals in the rat prefrontal cortex synapse on pyramidal cells that project to the nucleus accumbens. J Neurosci 19: 11049-11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damasio AR (1995) On some functions of the human prefrontal cortex. Ann NY Acad Sci 769: 241-251. [DOI] [PubMed] [Google Scholar]
- Dong Y, White FJ (2003) Dopamine D1-class receptors selectively modulate a slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J Neurosci 23: 2686-2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Cooper D, Nasif F, Hu XT, White FJ (2004a) Dopamine modulates inwardly rectifying potassium currents in medial prefrontal cortex pyramidal neurons. J Neurosci 24: 3077-3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, Malenka RC (2004b) Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(-/-) mice. Proc Natl Acad Sci USA 101: 14282-14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeders NE, Smith JE (1993) Intracranial cocaine self-administration into the medial prefrontal cortex increases dopamine turnover in the nucleus accumbens. J Pharmacol Exp Ther 265: 592-600. [PubMed] [Google Scholar]
- Hyman SE, Malenka RC (2001) Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci 2: 695-703. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Elsworth JD, Redmond Jr DE, Roth RH (1997) Phencyclidine increases forebrain monoamine metabolism in rats and monkeys: modulation by the isomers of HA966. J Neurosci 17: 1769-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH, Taylor JR (2000) Role for dopamine in the behavioral functions of the prefrontal corticostriatal system: implications for mental disorders and psychotropic drug action. Prog Brain Res 126: 433-453. [DOI] [PubMed] [Google Scholar]
- Li Y, Vartanian AJ, White FJ, Xue CJ, Wolf ME (1997) Effects of the AMPA receptor antagonist NBQX on the development and expression of behavioral sensitization to cocaine and amphetamine. Psychopharmacology (Berl) 134: 266-276. [DOI] [PubMed] [Google Scholar]
- Li Y, Hu XT, Berney TG, Vartanian AJ, Stine CD, Wolf ME, White FJ (1999) Both glutamate receptor antagonists and prefrontal cortex lesions prevent induction of cocaine sensitization and associated neuroadaptations. Synapse 34: 169-180. [DOI] [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ (2001) D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci 21: 2268-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW (2003) Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23: 3531-3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasif FJ, Hu X-T, White FJ (2002) Effects of repeated cocaine (COC) administration on high voltage activated (HVA)-Ca2+ potentials in medial prefrontal cortex pyramidal (MPFC) neurons. Soc Neurosci Abstr 28: 898.10. [Google Scholar]
- Reid MS, Hsu Jr K, Berger SP (1997) Cocaine and amphetamine preferentially stimulate glutamate release in the limbic system: studies on the involvement of dopamine. Synapse 27: 95-105. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B (1997) Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J Neurosci 17: 8491-8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Kolb B (1999) Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci 11: 1598-1604. [DOI] [PubMed] [Google Scholar]
- Shaw-Lutchman TZ, Impey S, Storm D, Nestler EJ (2003) Regulation of CRE-mediated transcription in mouse brain by amphetamine. Synapse 48: 10-17. [DOI] [PubMed] [Google Scholar]
- Smith JA, Mo Q, Guo H, Kunko PM, Robinson SE (1995) Cocaine increases extraneuronal levels of aspartate and glutamate in the nucleus accumbens. Brain Res 683: 264-269. [DOI] [PubMed] [Google Scholar]
- Smith-Roe SL, Sadeghian K, Kelley AE (1999) Spatial learning and performance in the radial arm maze is impaired after N-methyl-d-aspartate (NMDA) receptor blockade in striatal subregions. Behav Neurosci 113: 703-717. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001) Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4: 1217-1223. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM (2001) Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol 63: 241-320. [DOI] [PubMed] [Google Scholar]
- Weissenborn R, Robbins TW, Everitt BJ (1997) Effects of medial prefrontal or anterior cingulate cortex lesions on responding for cocaine under fixed-ratio and second-order schedules of reinforcement in rats. Psychopharmacology (Berl) 134: 242-257. [DOI] [PubMed] [Google Scholar]
- Wolf ME (1998) The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol 54: 679-720. [DOI] [PubMed] [Google Scholar]
- Yang CR, Seamans JK (1996) Dopamine D1 receptor actions in layers V-VI rat prefrontal cortex neurons in vitro: modulation of dendritic-somatic signal integration. J Neurosci 16: 1922-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ (2003) The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci 4: 885-900. [DOI] [PubMed] [Google Scholar]
- Zhang XF, Hu XT, White FJ (1998) Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J Neurosci 18: 488-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Cooper DC, White FJ (2002) Repeated cocaine treatment decreases whole-cell calcium current in rat nucleus accumbens neurons. J Pharmacol Exp Ther 301: 1119-1125. [DOI] [PubMed] [Google Scholar]