

Abstract
To study the efficacy of moxifloxacin treatment for tuberculosis, we utilized a novel cartridge system to simulate in vivo pharmacokinetics. We found this system to be a robust method for modeling in vivo pharmacokinetics and present data supporting the utility of intermittent moxifloxacin treatment as a component of antituberculosis chemotherapy.
Tuberculosis (TB) continues to be a leading cause of morbidity and mortality worldwide. The current treatment for active TB requires a lengthy course of medication, adherence to which is often limited by clinical, social, financial, and behavioral factors. Although supervised chemotherapy is recommended by the World Health Organization, it remains to be completely implemented, particularly in resource-poor countries. The cost of supervision frequently exceeds the cost of the drugs themselves. If a more intermittent treatment regimen could be developed, supervision costs would be reduced. The maximum interval between doses for an antibiotic is limited both by the in vivo half-life (t1/2) of the drug and by the postantibiotic effect (PAE), the continued inhibition of bacterial replication after an antibiotic has been withdrawn (4, 12). Moxifloxacin (MXF) is an 8-methoxy quinolone with in vitro and in vivo activities against Mycobacterium tuberculosis (9, 11, 22).
In this study, we examined the bactericidal effects and PAEs of MXF against M. tuberculosis using a novel cartridge system designed to closely simulate in vivo parameters for pharmacokinetics (PK) and pharmacodynamics (PD) (10). The cartridge system (Fig. 1A) consists of a hollow-fiber cartridge (BioVest International, Minneapolis, Minn.), a central reservoir, an input reservoir, an output (waste) reservoir, a fast (perfusion) pump, a slow (dilution) pump, and a dosing pump (to simulate oral dosing). The cartridge (Fig. 1B) consists of semipermeable capillaries (70-kDa molecular mass cutoff) which divide the cartridge into an intracapillary compartment (ICC), for perfusion of the growth medium, and an extracapillary compartment (ECC), in which bacteria can be inoculated. The perfusion pump circulates medium between the central reservoir and the cartridge. The input reservoir contains medium that is delivered to the central reservoir by the dilution pump. Excess medium is bled from the central reservoir to the waste reservoir in such a way that the volume of medium in the system (central reservoir, tubing, ICC, and ECC) is maintained at a constant 52 ml. The dosing pump delivers drug to the central reservoir at a programmed rate for a programmed interval of time. The flow rate from the input reservoir to the central reservoir determines the drug t1/2 in the system.
FIG. 1.

(A) Schematic diagram of the PK-PD system used in this study. See the text for a complete description of the various components. (B) Capillary cartridge used for perfusion of the bacterial culture. Shown clamped at either end are tubes connected to the ICC, through which the culture medium is circulated. At the top of the cartridge are ports for inoculation and aspiration of the ECC, in which the bacteria reside.
To simulate the PK parameters of a 400-mg dose of MXF in humans (maximum concentration of the drug in serum [Cmax], 3.1 to 4.5 μg/ml; area under the concentration-time curve, 31 to 48 μg · h/ml; t1/2, 12 to 13 h [12]), the required dose of MXF administered to the cartridge system was calculated to be 100 μg. This dosage takes into account 50% protein binding in human serum (12) and thus was designed to achieve a Cmax of 1.6 to 2.3 μg/ml, equivalent to the free-drug Cmax in human serum. However, it should be noted that we could find no experimental evidence to either support or invalidate the assumption that the degree of protein binding impacts the clinical efficacy of the quinolones. We set up separate dosing regimens for each of the five cartridges: (i) no drug given; (ii) 100 μg given at time zero; (iii) 100 μg given at time zero followed by 100 μg at 168 h; (iv) 100 μg given at time zero and 96 and 192 h; and (v) 33 μg given at time zero and 96 and 192 h. In all cases, drug was administered by the dosing pump in a total volume of 1 ml over 15 min.
A model describing the PK properties of the cartridge system is shown below:
 |
where X1 is the mass of drug (in micrograms) at any time (t) in the infusion pump, X2 is the mass of drug at time t in the system, Q is the zero-order rate constant of administration of the drug, and kelim is the first-order elimination rate constant. Since in our studies the drug was administered over the course of 15 min, the value of Q is 4 h−1 (there are four 15-min intervals per h). From basic PK, the t1/2 in hours of the drug in the system is defined by
 |
(1) |
where kelim is the rate constant of elimination of the drug from the system. Solving for the desired t1/2 of 12 h gives a kelim of 0.05775 h−1. Again, from pharmacokinetics, drug clearance (CL) from the system is defined by
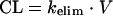 |
(2) |
where V is the volume of distribution. Thus, a kelim of 0.05775 h−1 and a V of 52 ml (see the description of the system above) gives a clearance rate of 3.003 ml/h. In order to achieve a t1/2 of 12 h, the flow rate of the dilution pump was therefore set at 3.003 ml/h. Assuming that distribution is instantaneous, the amount of drug in the system during the infusion of a dose can be described by the following differential equation:
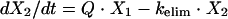 |
(3) |
which, when subjected to the LaPlace transformation (18), becomes
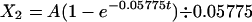 |
(4) |
where A is four times the dose of drug (in micrograms) and X2 is the mass of drug (in micrograms) in the system at t (in hours). Following the 15-min infusion, X2 is given by
 |
(5) |
where Xpost is the mass of drug (in micrograms) in the system following the infusion (i.e., equation 4 is solved for a t of 0.25 h).
In this study, the growth medium was Middlebrook 7H9 broth with 10% oleic acid-albumin-dextrose-catalase supplement (both from BD Diagnostic Systems, Sparks, Md.), 0.5% glycerol (Mallinckrodt Baker, Phillipsburg, N.J.), and 0.05% Tween 80 (Sigma-Aldrich, St. Louis, Mo.). A total of 105 CFU of M. tuberculosis H37Ra were inoculated into the ECCs of each of five cartridge systems and allowed to equilibrate overnight in a 37°C, 5% CO2 incubator. Gas exchange was provided by a 0.2-μm-pore-size filter venting the central reservoir. The five cartridge systems were run in parallel.
The culture in the untreated control cartridge demonstrated steady exponential growth, with a doubling time of approximately 33 h for the first 300 h of the experiment, at which point the number of viable bacteria stabilized at between 107 and 108 CFU (Fig. 2A). This demonstrated the feasibility of establishing bacterial populations within the cartridge. In the cartridges receiving 100-μg doses of MXF, there was a significant decline in the number of viable bacilli (Fig. 2B to D). The CFU counts in the cartridge receiving 33-μg doses of MXF did not vary significantly during the course of the experiment (Fig. 2E). These results demonstrate the bactericidal effects of MXF in the environment within the cartridge.
FIG. 2.


Effects of different courses of MXF treatment on M. tuberculosis H37Ra as a function of time. An inoculum of 105 CFU of H37Ra was placed into each of five cartridges and continuously perfused with medium. The cultures were exposed to no drug (A), one 100-μg pulse of MXF (B), two 100-μg doses 7 days apart (C), three 100-μg doses every 4 days (D), or three 33-μg doses every 4 days (E). Bacterial titers were determined by plate counts of samples taken from the cartridges at regular intervals. The calculated concentration of MXF in the cartridge is shown by dotted curves, which are scaled to the right vertical axes. Note that sampling from the cartridges whose data are shown in panels B and C was terminated at 300 h due to contamination observed in the central reservoir of the system.
Of particular interest was the PAE, which, for the purposes of this study, was defined as the time required for the CFU count in a treated cartridge to increase 1 log10 unit above the nadir count observed after dosing. The schedule for intermittent chemotherapy is constrained by the duration of the PAE. Specifically, dosing should be spaced such that no resumption of bacterial multiplication occurs between doses (5). Thus, while a single 100-μg dose in the cartridge system demonstrated a PAE of 195 h (8.1 days), it is clear that bacterial multiplication resumed within 140 h (5.8 days) after administration of the drug (Fig. 2B). In addition, once bacterial growth resumed, the culture exhibited a doubling time nearly equal to that of the control (approximately 37 h, compared to 33 h for the untreated culture). Even weekly dosing (Fig. 2C) allowed some bacterial replication to occur. In contrast, dosing with 100 μg of MXF every 4 days resulted in a continual decline in bacterial counts (Fig. 2D). Dosing with only 33 μg of MXF every 4 days resulted in neither a significant decline nor an increase in bacterial load (Fig. 2E). Based on the results of this in vitro study, and with the assumption that the same PK properties would occur in humans, 400-mg doses of MXF could be spaced 3 to 4 days apart without allowing a significant rebound of bacterial multiplication. However, spacing doses 1 week apart is not advised, as an appreciable amount of bacterial growth was seen to occur (Fig. 2C).
Several in vitro PK and PD models for testing antibiotic activity have been described previously (1, 2, 6, 7, 20, 21). In contrast to what occurs with conventional methods for antibiotic testing, bacteria in these models are exposed to drug concentrations that change over time to better simulate the in vivo situation. While these in vitro models have been used to assess the PK and PD parameters of antimicrobials, including fluoroquinolones, against other bacteria (8, 13-17), to our knowledge they have not been used to evaluate the PAE properties of fluoroquinolones against M. tuberculosis. By simulating PK parameters in humans, our in vitro PK-PD system determined the PAE of a single 400-mg dose of MXF on M. tuberculosis to be over 1 week. This result is compared to a PAE of greater than 15 days in a traditional static in vitro study of MXF against M. tuberculosis H37Rv after an exposure period of 24 h (A. S. Ginsburg et al., unpublished data). Recently, Chan et al. reported the in vitro PAE of a 2-μg/ml concentration of MXF against M. tuberculosis to be 0.3 h (3). However, this study used a radiometric assay to measure the cumulative growth of control and treated cultures, and the PAE was defined in a different manner, such that the results of that study and our study cannot be directly compared. Because the cartridge-based dynamic PK-PD system better simulates the PK properties of MXF in humans, the clinically relevant PAE is most likely better approximated in this system than in the traditional in vitro model. Our finding that bacillary growth resumes less than 1 week following a single 100-μg dose of MXF in the cartridge model is in good agreement with another study in which weekly doses of up to 400 mg of MXF per kg of body weight, a dose approximating the area under the concentration-time curve of a 400-mg dose in humans (19), had no effect on bacterial growth in infected mice. However, daily administration of 400 mg of MXF/kg resulted in a >5-log reduction in lung bacillary count compared to the count in untreated mice after 28 days of therapy (23). The cartridge-based in vitro system utilized in this study has wide-ranging potential in the study of antimicrobials, encompassing more than the study of a single drug-pathogen interaction. The design of this apparatus may facilitate the study of multidrug regimens, mixed infections, and the dynamics of selection for antibiotic-resistant mutants.
ADDENDUM IN PROOF
We refer the reader to a recently published article (T. Gumbo, A. Louie, M. R. Deziel, L. M. Parsons, M. Salfinger, and G. L. Drusano, J. Infect. Dis. 190:1642-1651, 2004) for the description of a similar in vitro system used to study the kinetics of moxifloxacin resistance in M. tuberculosis.
Acknowledgments
This work was supported by the Global Alliance for Tuberculosis Drug Discovery and NIAID grant R01 AI43846. S.C.W. was supported by NIAID grant F32 AI 50360.
REFERENCES
- 1.Blaser, J., B. B. Stone, M. C. Groner, and S. H. Zinner. 1987. Comparative study with enoxacin and netilmicin in a pharmacodynamic model to determine importance of ratio of antibiotic peak concentration to MIC for bactericidal activity and emergence of resistance. Antimicrob. Agents Chemother. 31:1054-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blaser, J., B. B. Stone, and S. H. Zinner. 1985. Two compartment kinetic model with multiple artificial capillary units. J. Antimicrob. Chemother. 15(Suppl. A):131-137. [DOI] [PubMed] [Google Scholar]
- 3.Chan, C. Y., C. Au-Yeang, W. W. Yew, C. C. Leung, and A. F. Cheng. 2004. In vitro postantibiotic effects of rifapentine, isoniazid, and moxifloxacin against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 48:340-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craig, W. A., and B. Vogelman. 1987. The postantibiotic effect. Ann. Intern. Med. 106:900-902. [DOI] [PubMed] [Google Scholar]
- 5.Dickinson, J. M., and D. A. Mitchison. 1968. In vitro and in vivo studies to assess the suitability of antituberculous drugs for use in intermittent chemotherapy regimens. Tubercle 49(Suppl.):66-70. [DOI] [PubMed] [Google Scholar]
- 6.Dudley, M. N., J. Blaser, D. Gilbert, and S. H. Zinner. 1990. Significance of “extravascular” protein binding for antimicrobial pharmacodynamics in an in vitro capillary model of infection. Antimicrob. Agents Chemother. 34:98-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dudley, M. N., and S. H. Zinner. 1991. Single daily dosing of amikacin in an in-vitro model. J. Antimicrob. Chemother. 27(Suppl. C):15-19. [DOI] [PubMed] [Google Scholar]
- 8.Garrison, M. W., K. Vance-Bryan, T. A. Larson, J. P. Toscano, and J. C. Rotschafer. 1990. Assessment of effects of protein binding on daptomycin and vancomycin killing of Staphylococcus aureus by using an in vitro pharmacodynamic model. Antimicrob. Agents Chemother. 34:1925-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ginsburg, A. S., J. H. Grosset, and W. R. Bishai. 2003. Fluoroquinolones, tuberculosis, and resistance. Lancet Infect. Dis. 3:432-442. [DOI] [PubMed] [Google Scholar]
- 10.Hamzeh, F. M., J. Lee, M. Huber, A. Shahkolahi, H. Farzadegan, G. Wang, and P. S. Lietman. 1997. Presented at the Tenth International Conference on Antiviral Research, Atlanta, Ga.
- 11.Hu, Y., A. R. Coates, and D. A. Mitchison. 2003. Sterilizing activities of fluoroquinolones against rifampin-tolerant populations of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 47:653-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim, M. K., and C. H. Nightingale. 2000. Pharmacokinetics and pharmacodynamics of the fluoroquinolones, p. 169-202. In V. T. Andriole (ed.), The quinolones, 3rd ed. Academic Press, San Diego, Calif.
- 13.Klepser, M. E., E. J. Ernst, C. R. Petzold, P. Rhomberg, and G. V. Doern. 2001. Comparative bactericidal activities of ciprofloxacin, clinafloxacin, grepafloxacin, levofloxacin, moxifloxacin, and trovafloxacin against Streptococcus pneumoniae in a dynamic in vitro model. Antimicrob. Agents Chemother. 45:673-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacy, M. K., W. Lu, X. Xu, P. R. Tessier, D. P. Nicolau, R. Quintiliani, and C. H. Nightingale. 1999. Pharmacodynamic comparisons of levofloxacin, ciprofloxacin, and ampicillin against Streptococcus pneumoniae in an in vitro model of infection. Antimicrob. Agents Chemother. 43:672-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lister, P. D., and C. C. Sanders. 1999. Pharmacodynamics of trovafloxacin, ofloxacin, and ciprofloxacin against Streptococcus pneumoniae in an in vitro pharmacokinetic model. Antimicrob. Agents Chemother. 43:1118-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lister, P. D., W. E. Sanders, Jr., and C. C. Sanders. 1998. Cefepime-aztreonam: a unique double beta-lactam combination for Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 42:1610-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacGowan, A. P., K. E. Bowker, M. Wootton, and H. A. Holt. 1999. Activity of moxifloxacin, administered once a day, against Streptococcus pneumoniae in an in vitro pharmacodynamic model of infection. Antimicrob. Agents Chemother. 43:1560-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayersohn, M., and M. Gibaldi. 1971. Mathematical methods in pharmacokinetics. II. Solution of the two compartment open model. Am. J. Pharm. Educ. 35:19-28. [Google Scholar]
- 19.Miyazaki, E., M. Miyazaki, J. M. Chen, R. E. Chaisson, and W. R. Bishai. 1999. Moxifloxacin (BAY12-8039), a new 8-methoxyquinolone, is active in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 43:85-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murakawa, T., H. Sakamoto, T. Hirose, and M. Nishida. 1980. New in vitro kinetic model for evaluating bactericidal efficacy of antibiotics. Antimicrob. Agents Chemother. 18:377-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishida, M., T. Murakawa, T. Kamimura, and N. Okada. 1978. Bactericidal activity of cephalosporins in an in vitro model simulating serum levels. Antimicrob. Agents Chemother. 14:6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nuermberger, E. L., T. Yoshimatsu, S. Tyagi, R. J. O'Brien, A. N. Vernon, R. E. Chaisson, W. R. Bishai, and J. H. Grosset. 2004. Moxifloxacin-containing regimen greatly reduces time to culture conversion in murine tuberculosis. Am. J. Respir. Crit. Care Med. 169:421-426. [DOI] [PubMed] [Google Scholar]
- 23.Yoshimatsu, T., E. Nuermberger, S. Tyagi, R. Chaisson, W. Bishai, and J. Grosset. 2002. Bactericidal activity of increasing daily and weekly doses of moxifloxacin in murine tuberculosis. Antimicrob. Agents Chemother. 46:1875-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]