

Abstract
Integrin cytoplasmic domain-associated protein 1 (ICAP-1) has been shown to interact specifically with the β1 integrin cytoplasmic domain and to control cell spreading on fibronectin. Interestingly, ICAP-1 also is observed in the nucleus, by immunocytochemical staining, and after biochemical cell fractionation, suggesting that it has additional roles that have yet to be determined. We show that the nucleocytoplasmic shuttling capability of ICAP-1 is dependent on a functional nuclear localization signal. In addition, overexpression of β1 integrin strongly reduced this nuclear localization, suggesting that integrin activity could modulate ICAP-1 shuttling by sequestering it in the cytoplasm. Indeed, the nuclear localization of ICAP-1 is dependent on the stage of cell spreading on fibronectin, and we also show that ICAP-1 expression stimulates cellular proliferation in a fibronectin-dependent manner. This function is dependent on its nuclear localization. Moreover, ICAP-1 is able to activate the c-myc promoter in vitro. Together, these results demonstrate that ICAP-1 shuttles between the nucleus and cytoplasm in a β1 integrin-dependent manner. It could act as a messenger that relays information from sites of integrin-dependent cell adhesion to the nucleus for controlling gene expression and cell proliferation.
INTRODUCTION
Integrin adhesion receptors transduce signals that control complex cell functions such as proliferation, differentiation, and survival, which require the regulation of gene expression. Integrins are major extracellular matrix receptors and connect to the actin cytoskeleton through the assembly of specialized sites known as focal complexes and focal adhesions (Kaverina et al., 2002). Integrin-mediated cell adhesion initiates dramatic cytoskeletal rearrangements and signal transduction processes. The intracellular domain of integrins has no catalytic function, indicating that its interaction with other transducing molecules is crucial for signaling. Recent data have suggested that adhesion receptors and their cytosolic partners can regulate the trafficking of signaling proteins between the cytoplasm and nucleus where they control transcription. One of the best described examples of such a phenomenon is the functional connection between lymphocyte function-associated antigen 1 (LFA-1, a β2 integrin family member), and Jun activation domain binding protein 1 (JAB-1). A two-hybrid screen identified JAB-1 as a protein that binds to the β2 integrin cytoplasmic tail (Bianchi et al., 2000). JAB-1 is also a known coactivator of the c-Jun transcription factor and localizes both in the nucleus and at the plasma membrane. LFA-1 engagement is accompanied by an increase in the nuclear pool of JAB-1 and the subsequent enhancement of activator protein-1–driven promoter activities. Another protein, Hic-5, a paxillin-related focal adhesion protein, can interact with the glucocorticoid receptor transactivation domain and acts as a coactivator of this transcription factor (Thomas et al., 1999; Yang et al., 2000). In a further example involving nonintegrin surface adhesion receptors, CASK/LIN-2, a member of the membrane-associated guanylate kinase family, shuttles between synapses where it binds the transmembrane proteins neurexin and syndecan, and the nucleus interacting with the Tbr-1 transcription factor. The localization of CASK is dependent on the relative levels of these cytoplasmic and nuclear anchors. High levels of Tbr-1 recruit CASK to the nucleus and trigger gene transcription, including that of reelin required for cerebrocortical development (Hsueh et al., 2000). In contrast, a high level of syndecan 3, which acts as the transmembrane anchor, increases the CASK cytoplasmic pool. Similarly, other reports have established direct linkages between transcriptional regulators and cell adhesion molecules at cell-cell junctions. The most compelling evidence for adhesion-associated molecules involved in the regulation of transcription comes from studies on β-catenin. This protein is able to interact with E-cadherin at cell-cell contacts, with the adenomatous polyposis coli and axin complex in the cytoplasm, and with lymphocyte-enhancer binding factor 1/T-cell factor in nucleus (Ben-Ze'ev et al., 2000). The membrane-associated guanylate kinase homologue zonula occludens-1 also moves between cell-cell contacts where it binds α-catenin and the nucleus in a cell density-dependent manner (Ben-Ze'ev et al., 2000). In this way, it might impact on epithelial differentiation and morphogenesis by interacting with Y-box transcription factor and by regulating the expression of erb-2 (Balda and Matter, 2000). Further examples are zyxin, lipoma preferred partner, thyroid receptor interacting protein-6, FHL-2, Ajuba, and p120, all of which are predominantly localized in focal adhesions or cell-cell contacts but additionally shuttle to the nucleus (Wang et al., 1999; Kanungo et al., 2000; Muller et al., 2000; Nix et al., 2001; Morlon and Sassone-Corsi, 2003; Petit et al., 2003; Roczniak-Ferguson and Reynolds, 2003).
Among cytoplasmic proteins interacting with the intracellular tails of integrins, the integrin cytoplasmic domain-associated protein 1 (ICAP-1) has been shown to interact specifically with the β1 integrin chain (Chang et al., 1997). ICAP-1 is a 200-amino acid protein containing a phosphotyrosine binding (PTB) domain that interacts with the cytoplasmic tail of the β1 integrin at a NPXY motif (Chang et al., 2002). Similarly, ICAP-1 recognizes an NPXY amino acid sequence within KRIT-1, a protein involved in the human disease, cerebral cavernous malformation 1 (Zhang et al., 2001; Gunel et al., 2002; Zawistowski et al., 2002). Previous data have suggested that phosphorylation of ICAP-1 on threonine 38 by CaMKII modulates α5β1 integrin function (Bouvard and Block, 1998). A further involvement of ICAP-1 in the regulation of β1 integrin function was suggested by experiments indicating that its overexpression increases β1-dependent cell motility on adhesion substrates such as fibronectin (Zhang and Hemler, 1999). Talin and ICAP-1 compete for binding on the cytosolic domain of the β1 integrin chain and high concentrations of ICAP-1 disrupt focal adhesions (Bouvard et al., 2003). The identification of the Rho family GTPases Cdc42 and Rac1 as binding partners for ICAP-1 (Degani et al., 2002) suggests an implication of ICAP-1 in integrin-dependent signaling pathways. Additionally, we showed recently that ICAP-1 interacts with Nm23-H2 (also called NDPK-B), a protein with nucleoside diphosphate kinase activity (Fournier et al., 2002). Nm23-H2 is linked to a variety of cellular activities, including the suppression of metastasis and activation of c-myc transcription (Postel et al., 1993; Berberich and Postel, 1995). ICAP-1 and Nm23-H2 colocalize in ruffles upon integrin ligation (Fournier et al., 2002). Very little is known, however, about the biological functions of ICAP-1. Interestingly, ICAP-1 was often observed both in the nucleus and cytosol. Because the distribution pattern of a protein within the cell provides important clues to its function, we used a combination of immunofluorescence microscopy and biochemical fractionation to determine the subcellular localization of ICAP-1 in epithelial and fibroblastic cells. Our results show that ICAP-1 has a functional nuclear localization signal (NLS) sequence and belongs to the growing family of nucleocytoplasmic shuttles. We also report a physiological relevance of this shuttling: ICAP-1 nuclear localization is dependent on the stage of cell spreading on fibronectin and is clearly related to an increase in cell proliferation, possibly through the activation of the c-myc promoter.
MATERIALS AND METHODS
Reagents
Fibronectin was extracted from human plasma according to the method described previously (Engvall and Ruoslahti, 1977). Monoclonal antibodies against Nm23-H1 were purchased from Seigakaku America (Rockville, MD), anti-lamin B antibody was from Santa Cruz Biotechnology (Santa Cruz, CA), and anti-actin polyclonal antibodies were from Sigma-Aldrich (St. Louis, MO). Rhodamin-phalloidin was from Sigma-Aldrich. Rabbit anti-ICAP-1 serum was raised in our laboratory by immunizing rabbits with purified recombinant ICAP-1 protein, and mouse monoclonal antibody (mAb) anti-ICAP-1 4D1D6 was prepared using recombinant His-tagged ICAP-1 as antigen, as described previously (Bouvard et al., 2003). The mAb 4B7R, directed against human β1 integrin, was purchased from NeoMarkers (Fremont, CA). Anti-bromodeoxyuridine (BrdU) mAb was from Sigma-Aldrich. Anti-cyclin D1 mAb was from Santa Cruz Biotechnology. Goat anti-mouse IgG and goat anti-rabbit IgG coupled to horseradish peroxidase were from Bio-Rad (Hercules, CA) and Jackson ImmunoResearch Laboratories (West Grove, PA), respectively.
Expression Plasmid Construction
Full-length human ICAP-1 was excised from the pBS-ICAP-1 vector as an EcoRI/XbaI fragment and inserted into the pcDNA3.1(+) vector (Invitrogen, Breda, The Netherlands). The 1- to 99-nt cDNA fragment corresponding to the N terminus of ICAP-1 was cut from pet19b/1–99 ICAP-1 by digestion with BglII and BamHI enzymes before cloning into pcDNA3.1 already cut with BamHI. The 100- to 200-nt C-terminal fragment of ICAP-1 was generated by polymerase chain reaction (PCR), allowing to generate a 5′Kpn end and a 3′Xho end to introduce the fragment into the KpnI/XhoI sites of pcDNA3.1. The site-directed mutagenesis for the NLS functional analysis was carried out on pcDNA3.1/ICAP-1 with the QuikChange site-directed mutagenesis kit according to the manufacturer's instructions (Stratagene, La Jolla, CA). The two lysines at position 6–7 were changed to alanines. This vector was called pcDNA3.1/ICAP-1ΔNLS. The human ICAP-1 cDNA fragment was excised from the pcDNA3.1/ICAP-1 vector with BamHI and HincII enzymes and inserted into the pTREhyg vector (BD Biosciences Clontech, Palo Alto, CA) digested with BamHI and EcoRV. The pTREhyg/ICAP-1ΔNLS was generated by mutagenesis by using the same strategy. All sequences were verified by DNA sequencing. The plasmid pcmyc-NHE was kindly provided by E. Postel (Lewis Thomas Laboratory, Princeton, NJ) and the pcDNA3.1/α-actinin vector was a generous gift from A. Duperray (Institut Albert Bonniot, Grenoble, France). The pcDNA3/Nm23-H2 was generously provided by M. L. Lacombe (Faculté de Medecine Saint Antoine, Paris, France). The pECE human β1 integrin was kindly provided by E. Ruoslahti (The Burnham Institute, La Jolla, CA).
Cell Culture and Cell Transfection
Chinese hamster ovary (CHO), HeLa, and GD25 cell lines were cultured in α-minimal essential medium (MEM) supplemented with 10% calf serum and penicillin/streptomycin at 37°C in 5% CO2-humidified chamber. The human lung cell line H358 was grown in RPMI 1640 medium supplemented with 10% calf serum and penicillin/streptomycin. Cells were transiently transfected with the cDNA constructs, by using Exgen 500 (Euromedex, Mundolsheim, France) and the expression of the transgene was assessed between 24 and 48 h after transfection. Transfection of H358/tet-on or Madin-Darby canine kidney (MDCK)/tet-off epithelial cells was carried out with the use of FuGene (Roche Diagnostics, Indianapolis, IN) and Lipofectamine, respectively. To select stable clones after transfection, H358 or MDCK cells were cultured in medium containing either Geneticin (G418) and hygromycin, or puromycin and hygromycin.
A targeted deletion of ICAP-1 was generated by homologous recombination in embryonic stem cells and introduced in mice. The generation and characterization of the ICAP-1 knockout mice will be reported in a separate article. Primary osteoblasts were isolated from wild-type mice and mice that were homozygous null for ICAP-1 by using standard procedure (Globus et al., 1998). Osteoblasts were cultured in α-MEM with 10% calf serum and penicillin/streptomycin. Primary cells were then immortalized using the large T antigen by retroviral infection. Among several clones, SV2.1 and SV6.5 derived from ICAP-1 null mouse and wild-type mouse, respectively, were used in this study. ICAP-1 and EGFP cDNA were inserted as a bicistronic element into pCL-MFG retroviral vector (Naviaux et al., 1996). Retroviral supernatant was produced by transient transfection of phoenix cells and used to infect SV2.1 and SV6.5 cells. Infection was confirmed by fluorescence-activated cell sorting (FACS) analysis for enhanced green fluorescent protein (EGFP) fluorescence, and cell sorting was performed to obtain a homogeneous population.
Protein Immunoblot Analysis
CHO cells were transiently transfected with Exgen 500 (Euromedex). Twentyfour hours after the transfection, the cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing proteases and phosphatases inhibitors for 45 min, and proteins were subjected to electrophoresis and transferred to polyvinylidene diflouride (PVDF) membranes. The samples were analyzed by Western blot with either rabbit polyclonal or mouse monoclonal anti-ICAP-1 antibodies, and with anti-actin, anti-Nm23-H1, and anti-lamin C antibodies. Immunological detection was achieved with horseradish peroxidase-conjugated secondary antibody (Bio-Rad or Jackson ImmunoResearch Laboratories), and the detection was carried out with enhanced chemiluminescence (ECL) according to the manufacturer's instructions (Amersham Biosciences, Piscataway, NJ).
Immunofluorescence Staining of Cells
CHO, GD25, and MDCK cells were cultured as a monolayer in DMEM medium containing 10% fetal calf serum (FCS) and harvested with trypsin/EDTA. The cells were plated on coverslips that were precoated with 10 μg/ml human plasma fibronectin in a 37°C incubator with 5% CO2 to reach 50% confluence the day of transfection. The cells were fixed with 3% paraformaldehyde in phosphate-buffered saline (PBS), permeabilized with 0.2% Triton X-100 in PBS. Nonspecific sites were blocked in 10% goat serum for 1 h at room temperature. Cells were stained for 1 h with either monoclonal or polyclonal antibodies in a moist chamber. Anti ICAP-1 4D1D6 monoclonal antibodies issued from hybridoma supernatant was used at 1:3 and anti-ICAP-1 polyclonal antibodies were used at 1:1000. The 4B7R mAb specific for human β1 integrin was used at 1:200. After rinsing, coverslips were incubated with an appropriate Alexa-conjugated secondary antibody (Molecular Probes, Eugene, OR) for 30 min. The cells were mounted in Mowiol solution and viewed using an Provis AX 70 Olympus microscope.
Fractionation of Cell Lysates
HeLa cells or ICAP-1–expressing CHO cells were washed in PBS and then broken on ice in hypotonic buffer (20 mM Tris, 1 mM EDTA, 1 mM EGTA, and 1 mM phenylmethylsulfonyl fluoride [PMSF]) by using a Dounce homogenizer as described previously (Sadoul et al., 1995). After low-speed centrifugation (2500 rpm, 10 min, 4°C, Heräus Biofuge) to isolate nuclei, the post-nuclear supernatant was centrifuged at 30,000 × g, 30 min, 4°C to obtain a crude membrane pellet and cytosol. The membrane pellet was washed once in high salt buffer (20 mM Tris, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, and 1 M NaCl, pH 7.4) before solubilization with a detergent-containing buffer (20 mM Tris, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 150 mM NaCl, and 1% Triton X-100, pH 7.4). Solubilized membranes were centrifuged at 100,000 × g, 1 h, 4°C to separate Triton-soluble membrane components from insoluble material. The remaining pellet was resuspended directly in SDS-sample buffer for gel electrophoresis. All volumes were kept constant starting with 106 cells/100 μl of hypotonic buffer. Fraction purity was tested by Western blot, by using as a cytoplasmic marker Nm23-H1 and as nuclear marker lamin B.
Cell Proliferation Assay
For proliferation assay, MDCK cells were washed three times in DMEM before replating 2 × 104 cells in 3 ml of DMEM supplemented with 4% FCS. Triplicate wells for each time point were analyzed. The medium was changed every other day. The total number of viable cells per well was determined by counting at days 2, 5, 7, and 9 after plating without reaching confluence.
BrdU Incorporation Assay
Cells (1.6 × 104 cells) were plated on polylysine-, vitronectin-, or fibronectin-coated coverslips, cultured for 24 h in 1% FCS, and then incubated with 10 μM BrdU (Sigma-Aldrich) for 1 h, washed twice with PBS, and fixed in methanol. Cells were then treated with 2 N HCl at 37°C for 60 min, washed twice with 0.1 M NaB4O7, and rinsed with PBS. Cell population that entered the S phase was determined by quantifying the incorporated BrdU by using the mouse anti-BrdU mAb (Sigma-Aldrich). In parallel, ICAP-1 localization was visualized with the polyclonal antibody. After rinsing, cells were incubated with appropriate Alexa-conjugated secondary antibodies (Molecular Probes) for 30 min. The cells were mounted in Mowiol solution and viewed using an Provis AX 70 Olympus microscope for counting or a confocal laser scanning microscope (Zeiss LSM 410).
Reporter Gene Assays
CHO cells were transfected with a pcmyc-driven chloramphenicol acetyltransferase (CAT) reporter gene and with the indicated cDNAs, by using the Exgen 500 reagent (Euromedex). In all cases, the amount of DNA transfected was kept constant by the addition of the pcDNA3 plasmid (the vector backbone for ICAP-1). At the time of harvesting, the cells were placed on ice, washed twice with ice-cold PBS, and lysed directly on wells with lysis buffer provided by the manufacturer. CAT expression was measured in cell lysates 48 h after transfection at 405 nm according instructions from Roche Diagnostics.
RESULTS
Dual Localization of ICAP-1 within Cells
To visualize the subcellular localization of ICAP-1, we performed indirect immunostaining on fibroblast NIH 3T3 or CHO cells overexpressing this protein constitutively (Figure 1A). The use of either monoclonal or polyclonal antibodies revealed that ICAP-1 was localized in both cytoplasmic and nuclear compartments. A similar staining was observed in the lung epithelial cell line H358 (Tet-on) in which ICAP-1 expression was controlled by a tetracycline-dependent promoter. H358 clones with high expression of the rtTA transactivator were transfected with pTREhyg plasmid encoding full-length ICAP-1. On induction with doxycycline, ICAP-1 was observed in the nucleus and the cytoplasm (Figure 1A). Biochemical fractionations were carried out to assay the dual cellular locations of the ICAP-1 protein. In all of the mentioned cells, endogenous ICAP-1 was difficult to detect either by immunofluorescence or Western blot, probably due to low expression levels. Conversely, HeLa cells express detectable amounts of endogenous ICAP-1 with a predominant localization in nucleus as visualized by immunostaining (Figure 1B). Because HeLa cells express physiological levels of endogenous ICAP-1, they were used in a fractionation procedure, separating a cytoplasmic soluble fraction and a nuclear fraction. These fractions were characterized by stripping and reprobing the blot by using antibodies directed against marker proteins of different cellular compartments. As expected, Nm23-H1 was found in the cytoplasmic, actin in nuclear and cytoplasmic fractions, and lamin B was restricted to the nucleus. As shown in Figure 1B, ICAP-1 was detected in both cytoplasmic and nuclear fractions of HeLa cells consistent with the distribution observed in immunofluorescence microscopy experiments.
Figure 1.

Subcellular localization of ICAP-1. (A) Different types of cells were plated on coverslips coated with fibronectin. NIH 3T3 or CHO cells were transiently transfected by pcDNA3/ICAP-1. Twenty-four hours after transfection, the cells were fixed, permeabilized, and stained with anti-ICAP-1 polyclonal antibodies. The H358 cells stably transfected with pTREhyg/ICAP-1 were induced with 1 μg/ml doxycycline during 24 h before fixation, permeabilization, and immunostaining with anti-ICAP-1 polyclonal antibodies. Two photos per cell type are shown to visualize nuclear and cytoplasmic localization of ICAP-1. Bar, 10 μm. Endogenous ICAP-1 was visualized in HeLa cells by immunostaining with anti-ICAP-1 polyclonal antibodies (B) (bar, 10 μm.) and by Western blot (C) by using the fractionation procedure described under Materials and Methods. Fractionation and Western immunoblot analyses of HeLa cells confirmed the subcellular localization of ICAP-1 protein in the cytosol and nucleus. Fractions are labeled as follows: C, cytosol; N, nuclear. The various cell fractions were characterized by reprobing the blots for lamin as a nuclear marker, Nm23-H1 as a cytoplasmic marker and actin as a general marker.
ICAP-1 Has a Functional Nuclear Localization Signal
To identify a putative NLS in ICAP-1, the complete amino acid sequence of ICAP-1 was examined using the PSORT program (Expasy, Swiss Institute of Bioinformatics, Geneva, Switzerland). A “pat 4” residue pattern similar to the monopartite NLS of the SV40 large T antigen was found at position 6–9. This sequence usually encompasses three basic amino acids (K or R) and one histidine. On ICAP-1, this sequence was KKRH. To determine whether this motif was a functional NLS and targets ICAP-1 to the nucleus, mutations were introduced, changing the two lysines at position 6–7 to alanines (Figure 2A). After transfection in CHO cells and immunostaining with anti-ICAP-1 antibodies, exogenous ICAP-1 expression was verified by Western blot (Figure 2B), and a statistical analysis evaluating for the presence of ICAP-1 in nucleus was carried out. Figure 2C shows that ICAP-1 was localized in the nucleus for ∼60% of transfected cells. As expected, the AARH mutant showed a diffuse staining in CHO-transfected cells for >95% of the cells, demonstrating that the K/A substitution within the NLS impaired nuclear localization. This indicated that ICAP-1 has a functional NLS in CHO cells. In addition, ICAP-1 was fused to green fluorescent protein (GFP) to decrease passive diffusion due to the small molecular weight of ICAP-1. Within this construct, an N-terminal ICAP-1 deletion encompassing the KKRH sequence as well as AARH mutation was generated (Figure 2A). The expression of the fusion proteins was verified by Western blot (Figure 2B). Deletion of the NLS sequence resulted in a dramatic decrease in the number of cells displaying nuclear GFP-ICAP-1 (Figure 2C). To confirm the shift of ICAP-1 distribution induced by NLS mutation, CHO cells were transiently transfected with ICAP-1 WT and ICAP-1ΔNLS before performing subcellular fractionation as described under Materials and Methods. ICAP-1ΔNLS was predominantly localized in the cytoplasmic fraction, whereas wild-type ICAP-1 was equally present in both cytoplasmic and nuclear fraction (Figure 2D).
Figure 2.

Mutation in the NLS of full-length ICAP-1 results in the accumulation of the protein in the cytoplasm. (A) Schematic representation of the different mutants of ICAP-1 used for transfection. CHO cells were transfected with pcDNA3.1 plasmids that express different ICAP-1 proteins fused or not with GFP, with or without mutations in NLS sequence (ΔNLS corresponds to the change of the two lysine (KK6–7) of the NLS to alanine and delta 12 corresponds to the deletion of the 12 first amino acids of ICAP-1). (B) Proteins from cells 24 h after transfection were resolved by SDS-PAGE, and the wild-type and mutant ICAP-1 was revealed by Western blot by using ECL substrate. The NT track corresponds to a lysate of CHO cells transfected with the empty pcDNA3.1. (C) At 24 h posttransfection, the cells were prepared for immunofluorescence microscopy by using the anti ICAP-1 polyclonal antibodies, and ICAP-1 distribution was examined. Two patterns were visualized: either nuclear or diffuse. Scoring 100 cells for ICAP-1 protein distribution revealed that ICAP-1 WT resides in nucleus in 60% of cells, whereas mutation of NLS sequence results in the accumulation of ICAP-1 in the cytoplasm for almost 100% of the visualized cells. Experiments were independently reproduced at least three times. (D) Western blot analysis performed after cell fractionation of wild-type CHO and transfected CHO cells overexpressing either wild-type ICAP-1 (WT) or ICAP-1ΔNLS. Note the different distribution of ICAP-1WT and ICAP-1ΔNLS between the nuclear and the cytoplasmic fraction. Fractions are labeled as follows: C, cytoplasmic fraction; L, total lysate; M, membrane fraction; N, nuclear fraction.
β1 Integrin Overexpression Shifts ICAP-1 Localization toward the Cytosol
We then considered whether the nuclear localization of ICAP-1 could be influenced by binding partners of ICAP-1 such as the cytoplasmic domain of the β1 integrin chain. When the human β1 chain was coexpressed with ICAP-1 in CHO cells, ICAP-1 predominantly exhibited a diffuse pattern in 90% of cells, whereas this pattern was only observed in 40% of the control cells transfected with the ICAP-1–encoding plasmid alone (Figure 3). The point mutation Y to S in the NPXY membrane distal (cyto3) domain of the human β1 integrin abolishes the interaction between ICAP-1 and the β1 cytoplasmic tail (Chang et al., 1997, Bouvard et al., 2003). To test whether the modification of ICAP-1 localization by integrin expression was due to a direct interaction with the cytoplasmic tail, this mutant was coexpressed with ICAP-1. Indeed, coexpression of the mutant β1 chain with ICAP-1 no longer shifted ICAP-1 localization toward the cytosol. Approximately one-half of the transfected cells exhibited ICAP-1 in nucleus, a distribution similar to that of the controls (Figure 3). These results suggest that cell adhesion molecules such as β1 integrins may regulate nucleocytoplasmic trafficking of ICAP-1, possibly by sequestering the protein in the cytosol.
Figure 3.
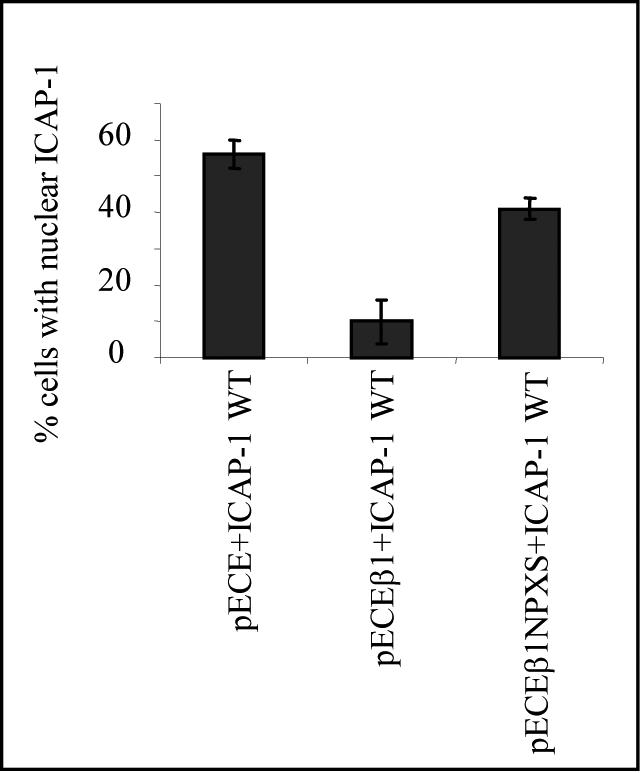
β1 Integrin-dependent cytoplasmic translocation of ICAP-1. CHO cells were cotransfected with two different cDNAs, pcDNA3.1/ICAP and pECE/human β1 or pcDNA3.1/ICAP-1 and pECE or pcDNA3.1/ICAP-1 and pECE/human NPXS β1, as indicated in each histogram. The subcellular distribution of the ectopically expressed proteins was visualized by double immunostaining by using the anti-human β1 mAb 4B7R and polyclonal anti-ICAP antibodies. Two distribution patterns were visualized: either nuclear or diffuse. Scoring 100 cells for ICAP-1 protein distribution revealed that human β1 integrin cotransfection resulted in the sequestration of ICAP-1 in the cytoplasm for 90% of the visualized cells, whereas the cotransfection with NPXS human β1 integrin restores the dual cytoplasmic and nuclear distribution. Experiments were independently reproduced at least three times.
Even more clear-cut results were obtained with GD25 cells (Figure 4). These cells do not express the β1 integrin chain due to a null mutation in both alleles (Fassler et al., 1995). Under our experimental conditions, immunofluorescence microscopy or Western blot analysis did not reveal any detectable staining for endogenous ICAP-1 in GD25 cells but ectopically expressed ICAP-1 localized predominantly in the nucleus. On the other hand, GD25 cells were stably transfected with the human β1A full-length cDNA, called GD25/β1A (Fassler et al., 1995; Retta et al., 1998). In such rescued cells, ICAP-1 localization was mainly cytosolic (Figure 4, A and B). Thus, overexpression of β1 integrins in the cells almost fully abolished the nuclear localization of ICAP-1.
Figure 4.

Subcellular localization of ICAP-1 is modulated by β1 integrin expression GD25 or GD25/β1 cells were transiently transfected with pcDNA3.1/ICAP-1, pcDNA3.1/ICAP-1(1-99), or pcDNA3.1/ICAP-1(100-200). (A) Subcellular distribution of the ectopically expressed proteins was visualized by immunofluorescence by using polyclonal anti-ICAP antibodies. Bar, 10 μm. (B and C) Three distribution patterns were visualized 24 h later as either nuclear, cytoplasmic, or both cytoplasmic and nuclear. Scoring 100 cells for ICAP-1 protein distribution revealed that cells devoid of β1 integrin sequestered ICAP-1 within nucleus (A and B). The 1–99 fragment bearing the NLS sequence was located in the nucleus (A and C) independently of the expression of β1 integrins, whereas the 100–200 fragment was sequestered in the cytoplasm (A and C).
Distinct Contributions of the C- and N-Terminal Halves of ICAP-1 to Its Cellular Localization
As mentioned above, the N- and C-terminal halves of ICAP-1 exhibit unique features: The N-terminal moiety encompasses the NLS sequence, whereas the C-terminal moiety encompasses the binding site of the β1 integrin tail. To address the role of each domain regarding to subcellular localization, we constructed two plasmids expressing either the N-terminal half or the C-terminal half of ICAP-1, respectively. After transfection of these plasmids into GD25 or GD25/β1A cells, the expressed proteins were visualized by immunofluorescence with anti-ICAP-1 polyclonal antibodies. As expected, the N-terminal half (1–99 aa), carrying the NLS sequence, showed a nuclear (∼70% of cells) or diffuse (∼30% of cells) localization in GD25 and GD25/β1A cells (Figure 4, A and C). Conversely, a predominantly cytoplasmic distribution was observed in GD25 and GD25/β1A cells tranfected with the C-terminal half (100–200 aa) bearing the binding domain of integrin and devoid of the NLS sequence (Figure 4, A and C).
ICAP-1 Overexpression Increases Cell Proliferation
In most cases, the end point of the integrin-induced signaling cascade is the regulation of gene expression programs that control cell differentiation or proliferation. To determine whether nuclear ICAP-1 localization affects cell proliferation, we generated stable MDCK clones overexpressing full-length ICAP-1 in an inducible manner (tet-off system) and control cells transfected with the empty vector (MOCK cells). Multiple independent clones of each cell line were isolated. Anti-ICAP-1 immunoblot analysis of cell extracts demonstrated that ICAP-1 protein with the expected size was expressed in all selected clones upon induction (Figure 5 A). Immunofluorescence studies also showed ICAP-1 accumulation in the nucleus (Figure 5B). The proliferation curve of these cell lines, in the presence of 10% serum, was followed for 5 d. The different clones overexpressing full-length ICAP-1 consistently exhibited a proliferation increase of 15% compared with that of control cells (our unpublished data). This small but reproducible result encouraged us to measure cell proliferation in low-serum conditions (4%). Under these conditions and after 7 d of culture, cells had a proliferation rate 2–3 times higher when ICAP-1 was expressed (Figure 5C). The next question to be addressed was to determine whether ICAP-1–induced proliferation was correlated with its nuclear localization. We generated inducible stable MDCK clones expressing ICAP-1 with the KKRH NLS sequence mutated into AARH (Figure 6). The levels of ICAP-1ΔNLS expression were comparable with those achieved with wild-type ICAP-1, as estimated by immunoblot (Figure 6B). According to immunofluorescence microscopy study, it seemed that mutation in the NLS sequence redirected ICAP-1 expression from the nucleus to the cytoplasmic fraction, thereby preventing nuclear accumulation of the mutant protein and contrasting with the location of wild-type ICAP-1 (Figure 6A). Again, we confirmed that the destruction of this NLS sequence abolishes nuclear import of ICAP-1 stably expressed in MDCK cells. Proliferation curves were determined with untransfected MDCK and MOCK-transfected cells, two MDCK clones overexpressing ICAP-1 (clones 4.9 and 8.9), and three MDCK clones overexpressing ICAP-1ΔNLS (clones 3.5, 11.3, and 16.2). During the time course of the experiment, proliferation of MDCK/ICAP-1ΔNLS cells was comparable with that of control MDCK cells (Figure 6C), suggesting that the increase in cell proliferation observed upon ICAP-1 overexpression is dependent on nuclear localization of the ICAP-1 protein.
Figure 5.

Nuclear localization of ICAP-1 increases cell proliferation. (A) MDCK cells stably transfected with pTREhyg/ICAP-1 were induced for ICAP-1 expression by removing doxycycline during 48 h before lysis of the cells. Total proteins were subjected to gel electrophoresis and then transferred to PVDF membrane before processing of Western blot with the anti-ICAP-1 polyclonal antibodies. The same gel was blotted with anti-actin polyclonal antibodies for normalization. (B) Immunofluorescence of cells after induction shows a nuclear localization of ICAP-1. Bar, 10 μm. (C) After reducing the percentage of serum and maintaining cells in 4% serum, proliferation curves of MDCK cells containing pTREhyg/ICAP-1 (clone 8.9) or pTRE/hyg (ct) alone were registered in the absence (induction) or presence of 2 μg/ml doxycycline (+dox). The clone 8.9 expressing ICAP-1 shows an increase in cell proliferation after 5 d of induction.
Figure 6.

Cellular distribution of ICAP-1 depends on its NLS sequence and exhibits differential effects on cell proliferation. (A) Subcellular localization of ICAP-1 was visualized by indirect immunofluorescence analysis with anti-ICAP-1 polyclonal antibodies in MDCK clones containing pTREhyg (MOCK cells) or pTREhyg/ICAP-1 (ICAP-1 WT) or pTREhyg/ICAP-1ΔNLS (ICAP-1ΔNLS) after induction (-dox) or without induction (+dox) with 2 μg/ml doxycycline. Bar, 10 μm. (B) After 7 d of induction (-dox), different clones expressing ICAP-1 WT (8.9 and 4.9) or ICAP-1ΔNLS (3.5 and 11.3) were lysed and expression of ICAP-1 with expected size was detected by Western blot with anti-ICAP-1 polyclonal antibodies. The same gel was blotted with anti-actin polyclonal antibodies for normalization. (C) The ratio of cell proliferation is shown for untransfected MDCK cells, MOCK cells, ICAP-1–overexpressing MDCK cells (clones 4.9 and 8.9), and ICAP-1ΔNLS–overexpressing MDCK cells (clones 3.5, 11.3, and 16.2) after 7 d of induction.
Next, we address the point whether the loss of ICAP-1 also inhibited cell proliferation. To test this, wild-type (SV6.5) and ICAP-1–deficient (SV2.1) immortalized osteoblasts were used in a BrdU incorporation assay. As expected, ICAP-1 expression was abolished in null cells (SV2.1 cells) and restored in SV2.1/ICAP-1 cells upon retroviral infection (Figure 7A). A homogenous population SV2.1/ICAP-1 cells was sorted by FACS by using the EGFP fluorescence (Figure 7B). Immunofluorescence studies confirmed a homogenous ICAP-1 expression in >95% of the SV2.1/ICAP-1 cell population (Figure 7C). In agreement with our previous observations, we noted a dual localization of ICAP-1 in both cytoplasm and nucleus. Consistent with the positive effect of ICAP-1 on cell proliferation, ICAP-1–deficient cells (SV2.1) had a significant lower proliferation rate assayed by BrdU incorporation compared either with wild-type osteoblasts (SV6.5) or with ICAP-1–rescued SV2.1 cells (SV2.1/ICAP-1) (Figure 7D).
Figure 7.

Loss of ICAP-1 decreases cell proliferation in osteoblasts. (A) ICAP-1–deficient osteoblasts (SV2.1) rescued or not with ICAP-1 were lysed with Laemmli buffer, and total cell lysates were resolved in SDS gel, transferred to a PVDF membrane, and probed with the monoclonal anti-ICAP-1 4D1D6 and anti-actin. (B) ICAP-1–deficient osteoblasts rescued with ICAP-1 (SV2.1/ICAP-1) were sorted by FACS by using EGFP fluorescence. (C) Immunostaining of ICAP-1 with polyclonal anti-ICAP-1 antibodies was performed in SV2.1, SV2.1/ICAP-1, SV6.5, and SV6.5/ICAP-1 cells. (D) BrdU incorporation was carried out in ICAP-1 null osteoblasts (SV2.1), ICAP-1-rescued SV2.1 cells (SV2.1/ICAP-1), wild-type osteoblasts (SV6.5), and SV6.5/ICAP-1. Cells that have incorporated BrdU were counted under the microscope by using anti-BrdU staining, and the percentage of labeled cells was estimated by 4,6-diamidino-2-phenylindole counterstaining. The results are the average of four experiments allowing us to estimate the SD. (D) BrdU incorporation was carried out in ICAP-1 null osteoblasts (SV2.1) or in ICAP-1-rescued SV2.1 cells (SV2.1/ICAP-1) plated either on polylysine (PL) or fibronectin (FN) or vitronectin (VN). Cells that have incorporated BrdU were counted under the microscope by using anti-BrdU staining. The results are the average of three experiments allowing us to estimate the SD. (E) SV2.1/ICAP-1 cells were allowed to spread onto FN or PL during 15 min (early stage of spreading) or 5 h (late stage of spreading), and then they were fixed, permeabilized, and stained with anti-ICAP-1 polyclonal antibodies. Visualization of a single section and image capture were done with a confocal microscope.
We attempted to establish whether the type of matrix could regulate ICAP-1–dependent cell proliferation and the potential shuttling of ICAP-1. BrdU incorporation was carried out in ICAP-1–deficient cells (SV2.1) and ICAP-1–rescued SV2.1 cells (SV2.1/ICAP-1) plated on different substrates. SV2/ICAP-1 cells had a higher proliferation rate when cells were spread on fibronectin compared with polylysine and vitronectin (Figure 7E). In good correlation with these results, a time course of spreading coupled with a confocal analysis showed that the nuclear localization of ICAP-1 is linked to the type of matrix used and is dependent on the stage of spreading of cells: ICAP-1 displayed a cytoplasmic distribution with a nuclear exclusion in case of round cells (corresponding to the initial step of spreading), whereas the distribution of ICAP-1 became nuclear in case of well spread cells (Figure 7F). On the other hand, the distribution of ICAP-1 remained cytoplasmic whatever the stage of cell spreading onto polylysine. This result demonstrated that ICAP-1 shuttling is regulated by cell adhesion on a β1 integrin-dependent substrate. Thus, ICAP-1–dependent signaling and subcellular localization of ICAP-1 seem to be dynamic processes linked to adhesion onto specific matrix proteins.
ICAP-1 Increases Transcriptional Activity from the c-myc P1 and P2 Promoter
c-myc is a proto-oncogene that has been primarily reported to stimulate cell proliferation and cell growth (Grandori et al., 2000; Pelengaris et al., 2000). One of the ICAP-1 partners, Nm23-H2 (Fournier et al., 2002), is a putative suppressor of tumor metastasis in humans. The protein self-assembles into a hexamer and has dual enzymatic functions, acting as an NDP kinase and as a transcriptional activation factor. It binds to a nuclease-hypersensitive element of the c-myc promoter through which it activates c-myc transcription (Postel et al., 1993; Berberich and Postel, 1995). Because ICAP-1 increases cell proliferation and interacts with the transcriptional activator of the c-myc proto-oncogene Nm23-H2, we hypothesized that ICAP-1 may be involved in the regulation of specific genes and more particularly may activate c-myc transcription. To address the potential relationship between c-myc transcription and ICAP-1–induced cell proliferation, we performed reporter gene assays by using a plasmid (pcmycCAT) containing the human c-myc P1 and P2 promoter driving the bacterial CAT expression. This plasmid was used in conjunction with either increased amounts of Nm23-H2 expression vectors, increased amounts of ICAP-1 expression vectors, or an irrelevant protein expressing vector. These plasmids were cotransfected into CHO cells, and the expression of CAT was analyzed 48 h after transfection by using the CAT enzyme-linked immunosorbent assay kit (Roche Diagnostics). Figure 8A shows that cotransfection of pcmycCAT with an empty vector or containing an irrelevant cDNA such as that of α-actinin did not affect the activity of the c-myc promoter and was therefore considered as the basal level of CAT expression. In contrast, a threefold increase in the relative CAT expression was observed from the wild-type myc promoter in the presence of Nm23-H2. This activation level is comparable with the one reported by Postel's laboratory (1993). Similarly, cotransfection with the ICAP-1 plasmid resulted in a concentration-dependent increase in CAT expression, reaching a threefold induction over the basal level (Figure 8A). The expression of cyclin D1 known as a c-myc target involved in cell proliferation also was measured at early and late stage of spreading on fibronectin in ICAP-1–deficient osteoblasts or rescued with ICAP-1. The experiments clearly showed a higher expression of cyclin D1 in the cells rescued with ICAP-1 in the late stage of cell spreading on fibronectin (Figure 8B), which nicely correlated with a nuclear localization of ICAP-1 (Figure 7). These data support a model in which ICAP-1 could increase gene expression from c-myc promoter in a manner comparable with that of Nm23-H2. The following question was to determine whether ICAP-1 could act synergistically with or independently of Nm23-H2 to activate the c-myc promoter. After cotransfection with pcDNA3/ICAP-1 and pcDNA3/Nm23-H2, we were able to detect the threefold induction of CAT expression with amounts of vectors that did not allow such induction when they were used separately (see gray histograms in Figure 8C) that favors the view of a synergistic action.
Figure 8.

ICAP-1–mediated activation of the c-myc promoter. (A and C) CHO cells were transiently transfected with the c-myc-CAT P1 and P2 promoter construct (1 μg) along with increasing concentrations of Nm23-H2 expression plasmid (0, 0.5, and 1 μg) or ICAP-1 expression plasmid (0, 0.5, 0.7, and 1 μg). In all cases, the amount of DNA transfected was kept constant by the addition of the pcDNA3 plasmid (the vector backbone for Nm23-H2 or ICAP-1). Extracts were generated 48 h later and assayed for CAT expression. The results are the average of three experiments allowing us to estimate the SD. (B) SV2.1 and SV2.1/ICAP-1 cells were cultured for 24 h in 1% FCS before replating them onto fibronectin. After 15 min or 5 h of spreading, cells were washed with PBS and directly lysed onto petri dishes with RIPA buffer. An amount of 30 μg of proteins per lane was subjected to gel electrophoresis and then transferred to PVDF membrane before processing of Western blot with the anti-cyclin D1 mAb. The same gel was blotted with anti-actin polyclonal antibodies for normalization.
ICAP-1 Control of the c-myc Promoter Requires Translocation into the Nucleus and Is Independent of Its Interaction with β1 Integrin
ICAP-1 contains a canonical PTB, a protein module that is present in a variety of signaling and cytoskeletal proteins. All known PTB domain proteins contain additional protein–protein interaction module(s) and are believed to function as adaptor proteins in cell signaling (Shc, IRS-1), lineage determination (numb), and receptor internalization (X11, Fe65, Disabled). Our previous results have shown that ICAP-1 binds to β1 integrins and is recruited in lamellipodia (Fournier et al., 2002). Via the integrin receptors, ICAP-1 might transmit signals from the cell membrane to the nucleus. On the other hand, some ICAP-1 mutants have been characterized to interact or not with β1 integrins and others to accumulate or not in the nucleus. Therefore, the question was whether the induction of the reporter gene controlled by the c-myc promoter is an indirect consequence of integrin activation or due to the nuclear localization of ICAP-1 and possible association with a transcription complex. As judged by other groups' reports and according to our pull-down assays between glutathione S-transferase-β1 and ICAP-1 mutants (our unpublished data), ICAP-1 WT, the 100–200 fragment and the ICAP-1ΔNLS mutant were all able to interact with the β1 integrin chain, whereas the 1–99 fragment and the ICAP-1 I138A mutant showed a defect in the interaction with the β1 integrin cytoplasmic tail. Among the mutants tested, only the ICAP-1ΔNLS mutant did not accumulate in the nucleus. As shown in Figure 9, neither the 1–99 fragment, nor the 100–200 fragment of ICAP-1 was able to operate as activators of the c-myc promoter. Moreover, the inducibility of the reporter gene was dependent on the nuclear localization of ICAP-1 because ICAP-1ΔNLS, devoid of its NLS sequence, was not able to stimulate the transcriptional activity of the c-myc promoter. On the other hand, the ICAP-1 I138A mutant that is compromised in its interaction with the β1 integrin chain could increase efficiently the transcriptional activity of the c-myc promoter. These observations strongly suggest that ICAP-1 integrin binding and its function of transcriptional activation are two independent events. Together, these data are consistent with a scenario in which ICAP-1 interacts directly or indirectly with the c-myc promoter and thereby increases c-myc promoter activity after its translocation into the nucleus.
Figure 9.

Activation of the c-myc promoter requires a functional NLS of ICAP-1. CHO cells were transiently transfected with the reporter construct pcmycCAT and the different constructs of ICAP-1. After 48 h, after lysis, CAT expression was determined according to the protocol of Roche Diagnostics.
DISCUSSION
ICAP-1 Shuttles between Nucleus and Cytoplasm
Compartmentalization of signaling molecules is an important regulatory mechanism that enables cells to build up an elaborate a network of signaling pathways. We have previously shown a transitory localization of ICAP-1 at the leading edge of spreading cells (Fournier et al., 2002). We now report that endogenous ICAP-1 as well as exogenous ICAP-1 protein can be found both in the nucleus and cytoplasm of a variety of cultured cells. Two monoclonal antibodies directed against the ICAP-1 N terminus and one polyclonal antibody consistently showed such a staining pattern. Cell fractionation experiments provided further evidence for a nuclear, cytosolic, as well as membrane localization of ICAP-1 protein. The relative cellular distribution of ICAP-1 between nucleus and cytoplasmic fractions varied among cell types, but it seems to be regulated by β1 integrin expression. Control of trafficking between the nuclear and cytoplasmic compartments is critical for gene expression in response to a variety of stimuli and therefore provides an important mechanism whereby cell adhesion molecules may regulate biological events. In that sense, the observation that a significant fraction of ICAP-1 protein is localized to the nucleus of epithelial cells prompted us to search for a putative nuclear localization signal within the ICAP-1 amino acid sequence. ICAP-1 protein has a stretch of basic amino acid residues, bordered by glycines, typical of a nuclear localization signal. This NLS sequence was found to be functional because mutation of KKRH motif into AARH or its deletion inhibits nuclear accumulation of the protein. According to our experiments, ICAP-1 nuclear localization seems to be associated with an increase in cell proliferation, possibly through the activation of the c-myc promoter. The subcellular localization of ICAP-1 is probably determined by a balance of nuclear import and export, and the dynamic equilibrium between these two processes may determine the different biological effects of ICAP-1. It is likely that a conformational change in ICAP-1 allows the exposure of either the NLS sequence or the integrin binding domain, although we cannot completely rule out that ICAP-1 partners that localize within the nucleus may direct ICAP-1 nuclear import.
Many transcription factors associated proteins either bind directly to cell adhesion molecules or localize to specialized adhesion sites. The recruitment of ICAP-1 into nucleus follows this general scheme and can be influenced by anchor proteins, such as integrins that could mask the NLS or sequester the protein. Our data clearly indicate that overexpression of β1 integrin strongly reduced nuclear localization of ICAP-1, suggesting that free and/or unpaired integrins could modulate ICAP-1 localization by sequestering it outside the nucleus. ICAP-1 shuttling is regulated by cell adhesion on a β1 integrin-dependent substrate because nuclear localization of ICAP-1 is linked to cell adhesion on fibronectin and is dependent on the stage of spreading of the cells. All these results suggest that integrin-mediated cell adhesion is linked, in some manner, to ICAP-1 nuclear signaling events and relevant to a given physiological activity of β1 integrins.
How Could ICAP-1 Increase Cell Proliferation?
According to our experiments, ICAP-1 may increase cell proliferation by acting directly or indirectly on the c-myc promoter. These data also are supported by studies carried out with ICAP-1–deficient mice that revealed a defect in cell proliferation in vivo (Bouvard, Aszodi, Kostka, Block, Albiges-Rizo, and Fässler, unpublished data). The question remains whether this mechanism belongs to a new signaling pathway or whether is integrated into an already known pathway. Some results suggest that the integrin α5β1 cooperates with receptor tyrosine kinases to promote extracellular signal-regulated kinase (ERK) activation necessary for cell proliferation through two major mechanisms: the Src family/focal adhesion kinase (FAK) pathway, which is activated by most of integrins through the cytoplasmic domain of β subunit, and the Shc/FAK pathway, which is activated by a subset of integrins through the transmembrane segment of α subunit (Wary et al., 1996; Pozzi et al., 1998; Barberis et al., 2000; Hirsch et al., 2002). It seems that the α subunit-dependent pathway increases the level of ERK activation, whereas the β subunit-dependent pathway prolongs this activation and provides crucial signals for ERK nuclear translocation. This latter event could be controlled by Rac because the introduction of a constitutively active Rac rescues nuclear translocation of ERK, which is suppressed in cells carrying a mutation in the β1 cytoplasmic domain (Hirsch et al., 2002). Alternatively, β1 integrins also can activate c-Jun NH2-terminal kinase signaling through the FAK/Cas/Rac pathway (Dolfi et al., 1998; Oktay et al., 1999). Our data indicate that translocation into the nucleus is required for ICAP-1–dependent transactivation of the c-myc promoter and stimulation of cell proliferation, thereby strongly suggesting that the protein may act directly or synergistically with other nuclear factors such as Nm23-H2. ICAP-1–induced transcriptional activation of c-myc and its positive effect on cell proliferation are underlined by higher expression of the cyclin D1, a classical c-myc target, in the well spread cells rescued with ICAP-1. This transcriptional activity correlates with a nuclear localization of ICAP-1 and can explain in part the cell proliferation induced by ICAP-1. However, ICAP-1 also has been shown to interact with Rac and Cdc42 (Degani et al., 2002) with a putative Rho GDP dissociation inhibitor activity, and its association with Nm23-H2 could in turn recruit Nm23-H1 linked to the Rac exchange factor Tiam1. Thus, the interference of ICAP-1 through Rac signaling with other known pathways linking integrins to cell proliferation cannot be excluded.
Regulation of c-myc Expression by β1 Integrins
A recent report has demonstrated the presence of a β1 integrin-signaling pathway that promotes expression of the c-myc protein (Benaud and Dickson, 2001), independently of growth factors, but likely mediated by protein kinase C, Src kinase family, and ERK mitogen-activated protein kinase (MAPK). Indeed, the known pathways leading to c-myc induction, namely, c-Src, ERK1/2 MAPK, and Ras/Akt (Barone and Courtneidge, 1995; Sears et al., 2000; Benaud and Dickson, 2001) are activated by integrin ligation (Frame and Brunton, 2002; Howe et al., 2002). Additionally, the activation of the c-myc promoter via ICAP-1 may correspond to a new pathway linking directly β1 integrin-mediated cell adhesion to cell proliferation. ICAP-1 is transiently colocalized with β1 integrins at the leading edge of spreading cells, and this step also corresponds to c-Src activation. It is tempting to hypothesize that once the interaction between integrin and ICAP-1 is broken during the later stages of adhesion, the protein is sent to the nucleus as a messenger of cell proliferation. This working model could explain how β1 integrins are up-regulating c-myc gene expression.
The profiling of gene expression patterns (>10,000 genes) after c-myc activation in the basal layer of mouse epidermis has recently been carried out (Frye et al., 2003). The major classes of up-regulated genes are involved in synthesis and processing of RNA and proteins that control cell proliferation and differentiation. Conversely, >40% of the downregulated genes code for cell adhesion and cytoskeleton proteins, in correlation with the inhibition in cell adhesion, motility, and wound healing. Impaired cell spreading decreases hemidesmosome formation and reduces the assembly of the actomyosin cytoskeleton in good correlation with the decreased expression of the α6β4 integrin. The authors proposed that c-myc stimulates exit from the stem cell compartment by reducing adhesive interactions with the local microenvironment or niche and that the failure of hair differentiation reflects an inability of keratinocytes to migrate along the outer root sheath to receive hair inductive stimuli (Frye et al., 2003). This general reduction of cell adhesiveness that could result from c-myc activation is consistent with a general role currently attributed to ICAP-1, namely, the inhibition of cell adhesion. Additionally, activation of the c-myc promoter by ICAP-1 may result in a delay or a change in cell differentiation or polarity due to a hyperproliferative effect (Liu et al., 2004). It will be crucial to determine the exact role of ICAP-1 in the nucleus and at the membrane level after integrin binding to extracellular matrix to understand their respective contributions to cell proliferation. In conclusion, our results provide novel insights into the cellular roles of ICAP-1 and open a new avenue for a better understanding of how integrin-mediated cell adhesion is linked to the control of cell proliferation.
Acknowledgments
We thank Jean-Luc Coll for the H358 cells transfected with pTet-on. We also thank Véronique Collin for fluorescence-activated cell sorting analysis. We thank Geneviève Tavernier and Brigitte Peyrusse for technical assistance. We thank Karin Sadoul and Martin Pfaff for critical reading of the manuscript. This work was supported by the Centre National de la Recherche Scientifique, a grant from the Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC), the Association de Recherche pour le Cancer, Alliance des Recherches sur le Cancer, and the Région Rhône-Alpes. H.-N.F. was supported by a fellowship from the Ministère de l'Education Nationale, de la Recherche et de la Technologie. D. B. was supported by a fellowship from ARC.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-08-0744) on February 9, 2005.
References
- Balda, M. S., and Matter, K. (2000). The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J. 19, 2024-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberis, L., Wary, K. K., Fiucci, G., Liu, F., Hirsch, E., Brancaccio, M., Altruda, F., Tarone, G., and Giancotti, F. G. (2000). Distinct roles of the adaptor protein Shc and focal adhesion kinase in integrin signaling to ERK. J. Biol. Chem. 275, 36532-36540. [DOI] [PubMed] [Google Scholar]
- Barone, M. V., and Courtneidge, S. A. (1995). Myc but not Fos rescue of platelet-derived growth factor signalling block caused by kinase-inactive Src. Nature 378, 509-512. [DOI] [PubMed] [Google Scholar]
- Benaud, C. M., and Dickson, R. B. (2001). Regulation of the expression of c-Myc by β1 integrins in epithelial cells. Oncogene 20, 759-768. [DOI] [PubMed] [Google Scholar]
- Ben-Ze'ev, A., Shtutman, M., and Zhurinsky, J. (2000). The integration of cell adhesion with gene expression: the role of β-catenin. Exp. Cell Res. 261, 75-82. [DOI] [PubMed] [Google Scholar]
- Berberich, S. J., and Postel, E. H. (1995). PuF/NM23–H2/NDPK-B transactivates a human c-myc promoter-CAT gene via a functional nuclease hypersensitive element. Oncogene 10, 2343-2347. [PubMed] [Google Scholar]
- Bianchi, E., Denti, S., Granata, A., Bossi, G., Geginat, J., Villa, A., Rogge, L., and Pardi, R. (2000). Integrin LFA-1 interacts with the transcriptional coactivator JAB1 to modulate AP-1 activity. Nature 404, 617-621. [DOI] [PubMed] [Google Scholar]
- Bouvard, D., and Block, M. R. (1998). Calcium/calmodulin-dependent protein kinase II controls integrin α5β1-mediated cell adhesion through the integrin cytoplasmic domain associated protein-1α. Biochem. Biophys. Res. Commun. 252, 46-50. [DOI] [PubMed] [Google Scholar]
- Bouvard, D., Vignoud, L., Dupe-Manet, S., Abed, N., Fournier, H. N., Vincent-Monegat, C., Retta, S. F., Fassler, R., and Block, M. R. (2003). Disruption of focal adhesions by integrin cytoplasmic domain-associated protein-1α. J. Biol. Chem. 278, 6567-6574. [DOI] [PubMed] [Google Scholar]
- Chang, D. D., Hoang, B. Q., Liu, J., and Springer, T. A. (2002). Molecular basis for interaction between ICAP1 α PTB domain and β1 integrin. J. Biol. Chem. 277, 8140-8145. [DOI] [PubMed] [Google Scholar]
- Chang, D. D., Wong, C., Smith, H., and Liu, J. (1997). ICAP-1, a novel β1 integrin cytoplasmic domain-associated protein, binds to a conserved and functionally important NPXY sequence motif of β1 integrin. J. Cell Biol. 138, 1149-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degani, S., Balzac, F., Brancaccio, M., Guazzone, S., Retta, S. F., Silengo, L., Eva, A., and Tarone, G. (2002). The integrin cytoplasmic domain-associated protein ICAP-1 binds and regulates Rho family GTPases during cell spreading. J. Cell Biol. 156, 377-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolfi, F., Garcia-Guzman, M., Ojaniemi, M., Nakamura, H., Matsuda, M., and Vuori, K. (1998). The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc. Natl. Acad. Sci. USA 95, 15394-15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engvall, E., and Ruoslahti, E. (1977). Binding of soluble form of fibroblast surface protein, fibronectin, to collagen. Int. J. Cancer 20, 1-5. [DOI] [PubMed] [Google Scholar]
- Fassler, R., Pfaff, M., Murphy, J., Noegel, A. A., Johansson, S., Timpl, R., and Albrecht, R. (1995). Lack of β1 integrin gene in embryonic stem cells affects morphology, adhesion, and migration but not integration into the inner cell mass of blastocysts. J. Cell Biol. 128, 979-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier, H. N., Dupe-Manet, S., Bouvard, D., Lacombe, M. L., Marie, C., Block, M. R., and Albiges-Rizo, C. (2002). Integrin cytoplasmic domain-associated protein 1α (ICAP-1α) interacts directly with the metastasis suppressor Nm23–H2, and both proteins are targeted to newly formed cell adhesion sites upon integrin engagement. J. Biol. Chem. 277, 20895-20902. [DOI] [PubMed] [Google Scholar]
- Frame, M. C., and Brunton, V. G. (2002). Advances in Rho-dependent actin regulation and oncogenic transformation. Curr. Opin. Genet. Dev. 12, 36-43. [DOI] [PubMed] [Google Scholar]
- Frye, M., Gardner, C., Li, E. R., Arnold, I., and Watt, F. M. (2003). Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development 130, 2793-2808. [DOI] [PubMed] [Google Scholar]
- Globus, R. K., Doty, S. B., Lull, J. C., Holmuhamedov, E., Humphries, M. J., and Damsky, C. H. (1998). Fibronectin is a survival factor for differentiated osteoblasts. J. Cell Sci. 111, 1385-1393. [DOI] [PubMed] [Google Scholar]
- Grandori, C., Cowley, S. M., James, L. P., and Eisenman, R. N. (2000). The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 16, 653-699. [DOI] [PubMed] [Google Scholar]
- Gunel, M., Laurans, M. S., Shin, D., DiLuna, M. L., Voorhees, J., Choate, K., Nelson-Williams, C., and Lifton, R. P. (2002). KRIT1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc. Natl. Acad. Sci. USA 99, 10677-10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, E., et al. (2002). Defective Rac-mediated proliferation and survival after targeted mutation of the β1 integrin cytodomain. J. Cell Biol. 157, 481-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe, A. K., Aplin, A. E., and Juliano, R. L. (2002). Anchorage-dependent ERK signaling–mechanisms and consequences. Curr. Opin. Genet. Dev. 12, 30-35. [DOI] [PubMed] [Google Scholar]
- Hsueh, Y. P., Wang, T. F., Yang, F. C., and Sheng, M. (2000). Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 404, 298-302. [DOI] [PubMed] [Google Scholar]
- Kanungo, J., Pratt, S. J., Marie, H., and Longmore, G. D. (2000). Ajuba, a cytosolic LIM protein, shuttles into the nucleus and affects embryonal cell proliferation and fate decisions. Mol. Biol. Cell 11, 3299-3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaverina, I., Krylyshkina, O., and Small, J. V. (2002). Regulation of substrate adhesion dynamics during cell motility. Int. J. Biochem. Cell Biol. 34, 746-761. [DOI] [PubMed] [Google Scholar]
- Liu, H., Radisky, D. C., Wang, F., and Bissell, M. J. (2004). Polarity and proliferation are controlled by distinct signaling pathways downstream of PI3-kinase in breast epithelial tumor cells. J. Cell Biol. 164, 603-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlon, A., and Sassone-Corsi, P. (2003). The LIM-only protein FHL2 is a serum-inducible transcriptional coactivator of AP-1. Proc. Natl. Acad. Sci. USA 100, 3977-3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, J. M., Isele, U., Metzger, E., Rempel, A., Moser, M., Pscherer, A., Breyer, T., Holubarsch, C., Buettner, R., and Schule, R. (2000). FHL2, a novel tissue-specific coactivator of the androgen receptor. EMBO J. 19, 359-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux, R. K., Costanzi, E., Haas, M., and Verma, I. M. (1996). The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70, 5701-5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix, D. A., Fradelizi, J., Bockholt, S., Menichi, B., Louvard, D., Friederich, E., and Beckerle, M. C. (2001). Targeting of zyxin to sites of actin membrane interaction and to the nucleus. J. Biol. Chem. 276, 34759-34767. [DOI] [PubMed] [Google Scholar]
- Oktay, M., Wary, K. K., Dans, M., Birge, R. B., and Giancotti, F. G. (1999). Integrin-mediated activation of focal adhesion kinase is required for signaling to Jun NH2-terminal kinase and progression through the G1 phase of the cell cycle. J. Cell Biol. 145, 1461-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelengaris, S., Rudolph, B., and Littlewood, T. (2000). Action of Myc in vivo - proliferation and apoptosis. Curr. Opin. Genet. Dev. 10, 100-105. [DOI] [PubMed] [Google Scholar]
- Petit, M. M., Meulemans, S. M., and Van de Ven, W. J. (2003). The focal adhesion and nuclear targeting capacity of the LIM-containing lipoma-preferred partner (LPP) protein. J. Biol. Chem. 278, 2157-2168. [DOI] [PubMed] [Google Scholar]
- Postel, E. H., Berberich, S. J., Flint, S. J., and Ferrone, C. A. (1993). Human c-myc transcription factor PuF identified as Nm23–H2 nucleoside diphosphate kinase, a candidate suppressor of tumor metastasis. Science 261, 478-480. [DOI] [PubMed] [Google Scholar]
- Pozzi, A., Wary, K. K., Giancotti, F. G., and Gardner, H. A. (1998). Integrin α1β1 mediates a unique collagen-dependent proliferation pathway in vivo. J. Cell Biol. 142, 587-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retta, S. F., Balzac, F., Ferraris, P., Belkin, A. M., Fassler, R., Humphries, M. J., De Leo, G., Silengo, L., and Tarone, G. (1998). β1-Integrin cytoplasmic subdomains involved in dominant negative function. Mol. Biol. Cell 9, 715-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak-Ferguson, A., and Reynolds, A. B. (2003). Regulation of p120-catenin nucleocytoplasmic shuttling activity. J. Cell Sci. 116, 4201-4212. [DOI] [PubMed] [Google Scholar]
- Sadoul, K., Lang, J., Montecucco, C., Weller, U., Regazzi, R., Catsicas, S., Wollheim, C. B., and Halban, P. A. (1995). SNAP-25 is expressed in islets of Langerhans and is involved in insulin release. J. Cell Biol. 128, 1019-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears, R., Nuckolls, F., Haura, E., Taya, Y., Tamai, K., and Nevins, J. R. (2000). Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 14, 2501-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, S. M., Hagel, M., and Turner, C. E. (1999). Characterization of a focal adhesion protein, Hic-5, that shares extensive homology with paxillin. J. Cell Sci. 112, 181-190. [DOI] [PubMed] [Google Scholar]
- Wang, Y., Dooher, J. E., Koedood Zhao, M., and Gilmore, T. D. (1999). Characterization of mouse Trip 6, a putative intracellular signaling protein. Gene 234, 403-409. [DOI] [PubMed] [Google Scholar]
- Wary, K. K., Mainiero, F., Isakoff, S. J., Marcantonio, E. E., and Giancotti, F. G. (1996). The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 87, 733-743. [DOI] [PubMed] [Google Scholar]
- Yang, L., Guerrero, J., Hong, H., DeFranco, D. B., and Stallcup, M. R. (2000). Interaction of the tau2 transcriptional activation domain of glucocorticoid receptor with a novel steroid receptor coactivator, Hic-5, which localizes to both focal adhesions and the nuclear matrix. Mol. Biol. Cell 11, 2007-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawistowski, J. S., Serebriiskii, I. G., Lee, M. F., Golemis, E. A., and Marchuk, D. A. (2002). KRIT1 association with the integrin-binding protein ICAP-1, a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum. Mol. Genet. 11, 389-396. [DOI] [PubMed] [Google Scholar]
- Zhang, J., Clatterbuck, R. E., Rigamonti, D., Chang, D. D., and Dietz, H. C. (2001). Interaction between KRIT1 and ICAP1α infers perturbation of integrin β1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum. Mol. Genet. 10, 2953-2960. [DOI] [PubMed] [Google Scholar]
- Zhang, X. A., and Hemler, M. E. (1999). Interaction of the integrin β1 cytoplasmic domain with ICAP-1 protein. J. Biol. Chem. 274, 11-19. [DOI] [PubMed] [Google Scholar]