

Abstract
Histone acetylation plays an important role in chromatin remodeling and gene expression. The molecular mechanisms involved in cell-specific expression of endothelial nitric-oxide synthase (eNOS) are not fully understood. In this study we investigated whether histone deacetylation was involved in repression of eNOS expression in non-endothelial cells. Induction of eNOS expression by histone deacetylase (HDAC) inhibitors trichostatin A (TSA) and sodium butyrate was observed in all four different types of non-endothelial cells examined. Chromatin immunoprecipitation assays showed that the induction of eNOS expression by TSA was accompanied by a remarkable increase of acetylation of histone H3 associated with the eNOS 5′-flanking region in the non-endothelial cells. Moreover, DNA methylation-mediated repression of eNOS promoter activity was partially reversed by TSA treatment, and combined treatment of TSA and 5-aza-2′-deoxycytidine (AzadC) synergistically induced eNOS expression in non-endothelial cells. The proximal Sp1 site is critical for basal activity of eNOS promoter. The induction of eNOS by inhibition of HDACs in non-endothelial cells, however, appeared not mediated by the changes in Sp1 DNA binding activity. We further showed that Sp1 bound to the endogenous eNOS promoter and associated with HDAC1 in non-endothelial HeLa cells. Combined TSA and AzadC treatment increased Sp1 binding to the endogenous eNOS promoter but decreased the association between HDAC1 and Sp1 in HeLa cells. Our data suggest that HDAC1 plays a critical role in eNOS repression, and the proximal Sp1 site may serve a key target for HDCA1-mediated eNOS repression in non-endothelial cells.
Nitric oxide (NO) is a free radical with diverse functions in many biological systems. In the vasculature NO is mostly generated by endothelial nitric-oxide synthase (eNOS).1 Endothelial NO plays a crucial role in maintaining vascular homeostasis (1). Murine or human eNOS promoter/β-galactosidase (LacZ) transgenic mouse models and human eNOS whole gene-containing introns/green fluorescence protein transgenic mouse model have all demonstrated that the eNOS gene is constitutively expressed in and relatively confined to endothelium (2–4). However, the molecular mechanism involved in endothelium-specific expression of eNOS is not fully understood. A recent study has demonstrated that the human eNOS proximal promoter DNA is heavily methylated in non-endothelial cells, whereas it is hardly methylated in endothelial cells. It is suggested that promoter DNA methylation may play an important role in the cell-specific eNOS expression in the vascular endothelium (5). However, to control cell-specific gene expression, DNA methylation requires cooperation from histone modifications and chromatin remodeling factors (6). It is not clear whether histone deacetylation is involved in the cell-specific eNOS expression, i.e. the repression of eNOS in non-endothelial cells, and whether there is any relationship between DNA methylation and histone deacetylation in cell-specific expression of eNOS.
Modifications of core histones are fundamentally important in alteration of chromatin structure and gene transcription (7). Acetylation of core histone unpacks the condensed chromatin and renders the target DNA accessible to transcriptional machinery, hence contributing to gene expression. In contrast, deacetylation of core histones increases the chromatin condensation and prevents the binding between DNA and transcriptional factors, which lead to transcriptional silence (8, 9). Histone acetyltransferases and histone deacetylases (HDACs) regulate the acetylation of histones and interact with components of the transcription machinery (10). Although histone acetylation is related to gene activation, global inhibition of HDACs does not induce widespread transcription (11, 12). For instance, treatment of human lymphoid cell line with HDACs inhibitor trichostatin A (TSA) revealed a change of expression (up- and down-regulation) in only 8 of 340 genes examined (11). It appears that histone deacetylase inhibitors may only activate some specific genes. Several studies have shown that inhibition of HDACs can selectively induce gene expression in the non-expressing cells (13–16).
In this study we examined the human eNOS mRNA, the eNOS promoter activity, and acetylation of histones associated with the 5′-flanking region of the eNOS in non-endothelial cells treated with HDACs inhibitors. We also investigated the effects of HDACs inhibitor on eNOS promoter DNA methylation status and on the DNA methylation-mediated repression of eNOS promoter activity. Furthermore, we examined whether Sp1 binds to endogenous eNOS promoter and the relationship between Sp1 and HDAC1. We found that eNOS was induced in non-endothelial cell types by HDACs inhibitors. Moreover, Sp1 was found to bind to endogenous eNOS promoter and interacted with HDAC1.
EXPERIMENTAL PROCEDURES
Reagents
TSA, 5-aza-2′-deoxycytidine (AzadC), actinomycin D, and cycloheximide were purchased from Sigma and dissolved in dimethyl sulfoxide (Me2SO) except AzadC, which was dissolved in phosphate-buffered saline. Sodium butyrate (SB) solution was purchased from Upstate Group, Inc.
Cell Culture
HeLa (human cervical epithelial cancer cell line) and 293 (human embryonic renal epithelial cell line) cells were cultured in Dulbecco’s modified Eagle’s medium (Cellgro) with 10% fetal bovine serum at 37 °C with 5% CO2. Human skin fibroblast cells (HSFs) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Human umbilical vein endothelial cells were purchase from Clonetics Corp. (San Diego, CA). Human aortic endothelial cells and human coronary artery smooth muscle cells were purchased from Cell Application, Inc (San Diego, CA). HSFs were cultured in minimum essential medium Eagle’s (ATCC) with 10% fetal bovine serum at 37 °C with 5% CO2. Human coronary artery smooth muscle cells were cultured in smooth muscle cell growth medium (Cell Applications) at 37 °C with 5% CO2. Human umbilical vein endothelial cells and human aortic endothelial cells were cultured at 37 °C with 5% CO2 in EBM-2 endothelial cell basic medium with Bulletkit (Cambrex) containing hydrocortisone, fibroblast growth factor B, vascular endothelial growth factor, R3-insulin-like growth factor-1, ascorbic acid, epidermal growth factor, GA-1000, heparin, and 2% fetal bovine serum. Primary cells were used at passage 4–7 in all experiments. The regents of TSA, SB, actinomycin D, and cycloheximide were added in culture medium for 24 h at concentration of 0.2 μg/ml, 10 mmol/liter, 0.2 μg/ml, and 40 μg/ml, respectively, if not specifically mentioned. For corresponding controls, the same volume of Me2SO or water, whichever was used as solvent, was added in culture medium.
AzadC Treatment
HeLa cells were treated with 7 μmol/liter AzadC for 8 days. Culture medium containing AzadC was replaced every 48 h.
Real-time Quantitative Reverse Transcription-PCR
Primers were designed with Beacon Designer 2.0 software. The primers for the human eNOS are: sense, 5′-AGG AAC CTG TGT GAC CCT CA-3′; anti-sense, 5′-CGA GGT GGT CCG GGT ATC C-3′. The primers for housekeeping gene β-actin (human) are: sense, 5′-CTG GAA CGG TGA AGG TGA CA-3′; antisense, 5′-AAG GGA CTT CCT GTA ACA ATG CA-3′. Total RNA was extracted from the cells with Trizol (Invitrogen) according to the manufacturer’s protocol. The RNA solution was treated with 1 unit of RNase-free DNase I (1 units/μl, Promega) in a final volume 20 μl at 37 °C for 15 min to remove the trace amount of genomic DNA contamination. The DNase I was inactivated by incubating at 75 °C for 10 min. One microgram of total RNA was used for reverse transcription. The mRNAs were reverse-transcribed into cDNAs with an iScript cDNA synthesis kit (Bio-Rad) containing mixture of oligo(dT) and random primers. The real-time PCR was performed by using an iCycler iQ real-time PCR detection system (Bio-Rad) with a SYBR Green I Super-mix kit (Bio-Rad) with the program running for 40 cycles at 95 °C for 20 s and 60 °C for 1 min. The PCR efficiency for the primers was examined by serially diluting the cDNA templates. The melting curve analysis was performed over the range 55–95 °C by monitoring SYBR Green fluorescence with increasing temperature (0.5 °C increment changes at 10-s intervals). PCR-specific products were determined as a clear single peak at the melting curves more than 80 °C. The specificity of primers was also confirmed as a single band with the correctly amplified fragment size through an agarose gel electrophoresis of the real-time reverse transcription-PCR products. Real-time PCR was duplicated for each cDNA sample. Each gene mRNA level was acquired from the value of the threshold cycle (Ct) of the real-time PCR as related to that of β-actin through the formula 2ΔCt (ΔCt = β-actin Ct − gene of interest Ct).
Promoter/Reporter Construct and Site-directed Mutagenesis
We amplified the human eNOS proximal promoter (−1140 to +22 bp) with standard PCR techniques from genomic DNA and cloned PCR products into pCR2.1-TOPO cloning vector (Invitrogen) and re-cloned into pGL3-Basic vector at KpnI and HindIII sites. The eNOS promoter in pGL3-Basic plasmid was confirmed by DNA sequencing. Primers with restriction enzyme site (underlined) for cloning eNOS promoter are: sense, 5′-GGT ACC CCT CCA CTG CTT TTC AGA GG-3′; antisense, 5′-AAG CCT CAT GTT ACT GTG CGT CCA CT-3′. Site-directed mutagenesis was performed with a QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacture’s instructions using primers specifically introducing a mutation at the Sp1site of the eNOS promoter-pGL3-Basic vector. The primers used for site-directed mutagenesis are as follows (underlines refers to mutated nucleotides): sense 5′-ATG GGA TAG GGC TCG GTT GAG GGC CAG CAC-3′; antisense 5′-GTG CTG GCC CTC AAC CGA GCC CTA TCC CAT-3′. The Sp1 mutation was confirmed by DNA sequencing.
Luciferase Assay
Plasmid DNA was prepared with by using a plasmid extraction kit (Qiagen). One microgram of DNA was transfected into the cells cultured in a 12-well plate with Lipofectamine 2000 (Invitrogen). TSA reagent was added into medium 4 h after the transfection, and the transfected cells were lysed 28 h after the transfection with cell lysis buffer (Promega). Luciferase activity was measured with a FB12 luminometer (Berthold, Germany) using luciferin (Promega) as the substrate.
Chromatin Immunoprecipitation (ChIP) Assay
The ChIP assay was performed using the histone H3 ChIP assay kit according to the manufacturer’s protocol (Upstate). In brief, ~3 × 107 cells were used per ChIP assay. Cells were cross-linked with 1% formaldehyde at 37 °C for 10 min and rinsed twice with ice-cold phosphate-buffered saline. Cells were harvested by a brief centrifugation. Cell pellets were resuspended in SDS lysis buffer (50 mmol/liter Tris-HCl, pH 8.1, 10 mmol/liter EDTA, 1% SDS, protease inhibitors). Sonication was performed on ice using a Sonifer II 450 (Branson, Danbury, CT) with a 3-mm tip set at duty cycle 20 and an output level 2 to achieve chromatin fragments ranging between 200 and 1000 bp in size followed by centrifugation at 15,000 × g for 10 min at 4 °C. Supernatants were collected and diluted 10-fold in ChIP dilution buffer (a 20-μl aliquot was removed to serve as an input sample) followed with preimmunoprecipitation clearing with 80 μl of a mixture of salmon sperm DNA/protein A/protein G at 4 °C with rotation for 30 min. Immunoprecipitation was carried out with 2 μg of anti-acetylhistone H3 at 4 °C overnight with rotation. After immunoprecipitation, 60 μl of a mixture of salmon sperm DNA/protein A/protein G was added and incubated at 4 °C with rotation for 30 min and followed by brief centrifuge. The precipitates were washed twice (5 min each at 4 °C) with low salt buffer, once with high salt buffer, and once with LiCl buffer. Then the precipitates were washed again with TE buffer twice for 5 min each. The immune complexes were extracted three times with 200 μl of elution buffer. The elutes and the input were heated at 65 °C for at least 4 h to reverse the cross-link by the addition of 20 μl of 5 mol/liter NaCl. After proteinase K treatment, DNA was extracted by phenol/chloroform solution and precipitated with ethanol under the aid of yeast tRNA. The recovered DNA was resuspended in 30 μl of H2O, and 4 μl was used for PCR. The PCR products were analyzed on 1.5% agarose gel. Four pairs of primers, A, B, C, D, were used for covering the 5′-flanking region of the human eNOS gene. The sequence of the primers is the following: A (−238/−13), 5′-AGC CCT GGC CTT TTC CTT AG-3′ and 5′-GCT CCC ACT TAT CAG CCT CA-3′; B (−483/−312), 5′-ACC AGG GGG TCA TAA AGG TC-3′ and 5′-GGG GAG GTG AAG GAG AGA-3′; C (−1304/−1421), 5′-CTG TCT CAG GAG GAG GGA GA-3′ and 5′-GGA TCC AGC CCC TAC TTT TC-3′; D (−4917/−4626), 5′-CTG TGC TGG GAA GGG AAG-3′ and 5′-GCA CTC AGC ATG AGT TAC ATC C-3′. For Sp1 ChIP assay, the procedure is the same as above, except that the Sp1 antibody (Upstate) was used for the immunoprecipitation of cross-linked chromatin and primers of pair A (−238/−13) for amplifying the fragment containing the proximal Sp1 site in the eNOS promoter. Anti-FLAG antibody (Sigma) was served as the negative control in the assay.
Electrophoresis Mobility Shift Assay (EMSA)
HeLa cells were treated with 0.2 μg/ml TSA or Me2SO for 24 h, and then nuclear proteins were extracted with NE-PER nuclear and cytoplasmic extraction reagents (Pierce) according to the manufacture’s instructions. EMSA was performed with a non-radioisotope method with digoxigenin-11-dUTP (DIG)-labeled probe using DIG gel shift kit (Roche Applied Science) according to the manufacture’s instructions as described before (17). Briefly, the sense strand oligonucleotides of human eNOS proximal Sp1 site (5′-GGA TAG GGG CGG GGC GAG G-3′, −111/−93) were annealed with its antisense strand and labeled at 3′-end with DIG by terminal transferase. The binding reaction was performed with 30 fmol of DIG-labeled probes incubated with 5 μg of nuclear extraction in the binding buffer containing 1 μg of poly(dI-dC) in a final volume of 20 μl at room temperature (RT) for 15 min. The DNA-protein complex was resolved through a 6% native polyacrylamide gel at 80 V (5 V/cm gel) followed by electroblotting onto a positively charged nylon membrane (Roche Applied Science) and UV cross-linking. After blocked in the blocking buffer, the blot was then incubated with anti-DIG antibody conjugated with alkaline phosphatase at RT for 30 min followed by washing and chemiluminescent detecting with CSPD (disodium 3-(4-methoxyspiro{1,2-dioxetane-3,2′-(5′-chloro)tricyclo[3.3.1.13,7]decan}-4-yl)phenyl phosphate) as the substrate and exposed to x-ray film.
Methylation of Promoter/Reporter Constructs
The eNOS promoter/luciferase reporter construct was in vitro methylated by M. Sss I (CpG) methylase (New England Biolabs, Inc.) with buffer (10 mmol/liter Tris-HCl, 50 mmol/liter NaCl, 10 mmol/liter MgCl2, 1 mmol/liter dithiothreitol, pH 7.9) supplemented with 160 μm S-adenosylmethionine at 37 °C for 2 h. Mock methylation reactions did not contain the methylase. The methylation status of the constructs was verified using HpaII (blocked by CpG methylation) and MspI (not blocked by CpG methylation) digestion. Methylated and mock-methylated constructs were purified with Wizard DNA Clean-up Kit (Promega) for transient transfection as described in luciferase assay.
Sodium Bisulfite Genomic Sequencing
To map out the DNA methylation status, bisulfite treatment of DNA was performed as described previously (18) with minor modifications. Briefly, genomic DNA (2 μg) isolated from confluent cells was digested with EcoRI for more than 4 h and denatured with 0.3 m NaOH at 42 °C for 20 min in a volume of 55 μl. The denatured DNA was then treated with 3 m sodium bisulfite and 0.5 mm hydroquinone, overlaid with mineral oil, and incubated at 55 °C for 16 h in the absence of light. DNA was purified with a Wizard DNA clean-up column (Promega). The purified DNA was desulfonated with 0.3 m NaOH for 15 min at 37 °C and then neutralized with ammonium acetate, precipitated by using ethanol and yeast tRNA, dissolved in 20 μl H2O, and stored at −20 °C. The bisulfite-treated DNA (1 μl) was subjected to 35 cycles of PCR amplification with primers covering human eNOS 5′-flanking region (−653 to +3): sense 5′-GGG GAA GGG ATA TTT TAA TGT TAG A-3′, antisense 5′-ACT ACC TAC TCC AAC AAA ACC CTA ACC-3′. The primers were designed with MethPrimer software (19). Nest PCR amplification was performed with 35 cycles with primers covering the human eNOS promoter region (−481 to −41), which showed most CpG dinucleotides methylated in non-endothelial cells (5): sense, 5′-GAG AGA ATT TGT ATG ATT TTA GAG GTT-3′; antisense, 5′-AAC AAA ACC CTA ACC TTT TCC TTA A-3′. The final PCR products were subcloned with pCR2.1-TOPO cloning vector (Invitrogen), and 10–15 individual clones were picked for DNA sequencing.
DNA Affinity Purification Assay (DAPA)
Biotinylated oligonucleotides were custom-synthesized and purified by standard desalting purification (IDT, Coraville, IA). The 5′-end-biotinylated sense strand was annealed with its non-labeled antisense strand. The wild type Sp1 probe contains the sequence corresponding to eNOS promoter high affinity proximal Sp1 site (−111/−93, 5′-biotin/GGA TAG GGG CGG GGC GAG G-3′). The mutated Sp1 probe contains the same mutation as that in the site-directed mutagenesis (−111/−93, 5′-biotin/GGA TAG GGC TCG GTT GAG G-3′; the underlined refers to the mutated nucleotides). The HeLa nuclear extract (200 μg) was incubated with biotinylated DNA probe (1.125 nmol) in a final volume of 500 μl of binding buffer containing 20 mmol/liter HEPES, pH 7.6, 5% glycerol, 50 mmol/liter KCl, 1 mmol/liter MgCl2, 1 mmol/liter dithiothreitol, 10 μmol/liter ZnCl2 for 2 h at RT. One hundred microliters of streptavidin-agarose beads (Sigma) were added to the reaction, and incubation was continued for 1 h at RT. The streptavidin-agarose was in excess to ensure a complete pull-down of DNA-protein complexes. After five washes with 1 ml of ice-cold binding buffer, the bound proteins were released by boiling in SDS loading buffer and then subjected to 10% SDS-PAGE and Western blot analysis.
Co-immunoprecipitation
Co-immunoprecipitation of Sp1 with HDAC1 was performed as described previously (20) with minor modifications. Cells were washed twice with ice-cold phosphate-buffered saline and lysed in mild lysis buffer containing 20 mmol/liter Tris, pH 8.0, 100 mmol/liter NaCl, 1 mmol/liter EDTA, 0.5% Nonidet P-40, 1 mmol/liter phenylmethylsulfonyl fluoride, 1 mmol/liter dithiothreitol, 2 mmol/liter sodium orthovanadate, and complete protease inhibitor mixture (Sigma) on ice for 10 min. The cells were then harvested for centrifugation at 15,000 × g for 15 min at 4 °C, the supernatant was collected as whole cell extracts, and equal amounts (2.5 mg) were incubated in 500 μl of extraction buffer with 4 μg of the respective antibody for 16 h with rotation at 4 °C. After the addition of 40 μl of protein A/G-agarose beads (Santa Cruz Biotechnology), the mixture was further incubated with rotation for 1 h at 4 °C. After five washes with 1 ml of extraction buffer, the bound proteins were released by boiling in 30 μl of SDS loading buffer and then subjected to 10% SDS-PAGE and Western blot analysis. Sp1 was immunoprecipitated and detected with an affinity-purified polyclonal rabbit antibody (Upstate). HDAC1 was immunoprecipitated and detected with an affinity-purified polyclonal rabbit antibody (eBioscience, San Diego, CA). The membranes were then stripped for detection with the respective antibodies used in the immunoprecipitation to confirm the pull-down proteins. Anti-FLAG antibody (Sigma) was used as a non-relevant antibody for negative control for the immunoprecipitation of the endogenous proteins.
Western Blot
Proteins were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes. The membrane was blocked in 5% nonfat milk in TBS-T (50 mmol/liter Tris, pH 7.5, 150 mmol/liter NaCl, 0.05% Tween 20) for 1 h at RT. After incubation with primary antibody in TBS-T containing 1% milk for 2 h at RT or 16 h at 4 °C, the membrane was washed extensively with TBS-T and then incubated with secondary anti-rabbit horseradish peroxidase-conjugated antibody for 1 h at RT. After extensive washes with TBS-T, the membrane was visualized with ECL plus reagents (Amersham Biosciences).
Statistic Analyses
All quantitative data were presented as the mean ± S.D. of three separate experiments and by Student’s t test for between group differences and analysis of variance for comparisons among three or more groups. Changes of CpG methylation status were examined with χ2 analysis. The p < 0.05 was regarded as statistically significant.
RESULTS
Induction of eNOS mRNA Expression by HDACs Inhibitors in Non-endothelial Cells
To investigate whether HDACs are involved in the repression of eNOS expression in non-endothelial cells, we treated HeLa, 293, HSFs, and human coronary artery smooth muscle cells with HDACs inhibitor TSA (0.2 μg/ml) for 24 h. We found that eNOS mRNA level was significantly induced up to 8–14-fold by TSA in all the examined non-endothelial cell types (Fig. 1A). The induction of eNOS mRNA by TSA was dose-dependent in HeLa cells and all other examined cell types (Fig. 1B). SB, a HDAC inhibitor differing from TSA in structure, showed similar results in all the examined cell lines (data not shown). The induction of eNOS mRNA in non-endothelial cells by TSA or SB was not blocked by cycloheximide but was completely blocked by actinomycin D (data not shown). The eNOS mRNA level was quantitated by real-time reverse transcription-PCR and normalized to housekeeping gene β-actin. However, in contrast to its effects on eNOS in non-endothelial cell lines, TSA decreased eNOS mRNA in endothelial cells under a totally different mechanism (21).
Fig. 1. Induction of eNOS mRNA expression in non-endothelial cells by HDACs inhibitor TSA.

A, induction of eNOS mRNA by TSA in four non-endothelial cell types. The cells were treated with 0.2 μg/ml TSA or Me2SO in control cells for 24 h. The eNOS mRNA level was quantitated by real-time reverse transcription-PCR and normalized to the housekeeping gene β-actin. B, TSA dose-dependent induction of the eNOS mRNA in HeLa cells. The eNOS mRNA was quantitated as in Panel A. All data are presented as the mean ± S.D. of three separated experiments. CTR, control. HCSMC, human coronary artery smooth muscle cells.
Basal Activity of eNOS Promoter in Non-endothelial and Endothelial Cells and Activation of eNOS Promoter by HDACs Inhibitors
Previous studies demonstrated that eNOS promoter activity was mainly in the proximal region (−1001 to + 109 bp) (22). In the current experiment, we raised two questions. 1) Do non-endothelial cells possess transcriptional machinery comparable with endothelial cells for eNOS promoter activation? 2) Can TSA directly activate eNOS promoter in non-endothelial cells, and if so, what is the responsive element? The episomal promoter/reporter may help to answer the two questions and is broadly used in TSA activation studies as it can help to identify TSA-responsive elements (13–16), although it may not have the proper methylation status that the corresponding endogenous DNA has. Therefore, we made a luciferase-coupled eNOS proximal promoter (−1140 to +22 bp)/reporter construct. Human umbilical vein endothelial and HeLa cells were transiently transfected with the promoter/reporter constructs together with the pGL3 control plasmid. As shown in Fig. 2, the basal activity of eNOS promoter normalized as percentage of that of pGL3 control vector containing SV40 promoter and enhancer was comparable in both HeLa and human umbilical vein endothelial cells. The results suggest that non-endothelial cells possess endothelial equivalent transcriptional machinery to activate eNOS promoter. Furthermore, the episomal eNOS promoter was activated by TSA in HeLa cells, indicating that TSA could directly activate eNOS promoter in non-endothelial cells. The activation of the episomal eNOS promoter was also observed in TSA-treated 293 and in SB-treated HeLa and 293 cells (data not shown).
Fig. 2. Comparison of basal activity of eNOS proximal promoter in endothelial and non-endothelial cells and activation of the eNOS promoter by the HDACs inhibitor TSA in non-endothelial.

A, basal activity of the eNOS promoter in non-endothelial and endothelial cells. The eNOS promoter/reporter constructs were transfected into the cells, and luciferase activity was measured and normalized as the percentage of the pGL3 control vector. B, eNOS promoter was activated by TSA in HeLa. Luciferase activity was measured 24 h after the cells treated with or without TSA. All data are presented as the mean ± S.D. of three separated experiments. CTR, control.
Increase of Acetylation of Histone H3 at eNOS 5′-Flanking Region in Non-endothelial Cells by HDACs Inhibitors
To examine the effect of HDACs inhibitors on the pattern of acetylation of histones associated with the eNOS gene, we performed the ChIP assay. To examine the chromatin status in a broad 5′-flanking region of the eNOS, we used four pairs of primers complementary to the eNOS 5′-flanking region −4917 to −13 bp before transcription start site in the ChIP assay, including the eNOS enhancer region (−4917 to −4626 bp) (23). Acetylation of histone H3 associated with the eNOS 5′-flanking region (−4917 to −13 bp) in HeLa cells was low or undetectable before TSA treatment. However, a remarkable increase in acetylation of histones H3 in the same region of HeLa cells treated with TSA (0.2 μg/ml, 24 h) was observed (Fig. 3). An increase of acetylation of histone H3 was also observed in TSA-treated 293 and HSF cells and in SB-treated HeLa and 293 cells. Interestingly, the status of histone H3 at all these sites in endothelial cells was highly acetylated as comparing to non-endothelial cells, and TSA treatment did not induce a further increase of acetylation (Fig. 3).
Fig. 3. Effect of TSA on acetylation of histone H3 associated with eNOS 5′-flanking region.

A, schematic graph of the eNOS promoter 5′-flanking region and the location of four pairs of primers designated as A, B, C, and D used for PCR amplification in the ChIP assay. TSS, transcription start site. kb, kilobases. B, accumulation of acetylation of histone H3 associated with eNOS in non-endothelial cells by TSA treatment. ChIP assays were performed with anti-acetylated histone H3 antibody for the cells treated with or without TSA. Primers for the eNOS 5′-flanking region, as indicated in panel A, were used to amplify the DNA isolated from the ChIP assay. The data shown are representative of three separated experiments. IP, immunoprecipitated; HUVEC, human umbilical vein endothelia cells.
Histone Deacetylation Involved in DNA Methylation-mediated Repression of eNOS Promoter
A recent study has demonstrated that DNA methylation strongly repressed the eNOS promoter activity (5). DNA methylation is usually linked with histone deacetylation (6). Methylated DNA can provide a dock for transcription repressors, such as MeCP2, a methylated CpG dinucleotide-binding protein. MeCP2 can recruit HDACs·mSin3A complex to the specific methylated DNA region (24). We wanted to know if this mechanism is also involved in eNOS repression in non-endothelial cells. To investigate whether histone deacetylation is involved in DNA methylation-mediated repression of the eNOS promoter, we methylated the luciferase-coupled eNOS promoter/reporter constructs in vitro using methylase M. Sss I, which specifically methylates cytosine of CpG dinucleotides. The methylated the eNOS promoter/reporter constructs, which were confirmed by MspI and HpaII isoschizomer digestion, were then transiently transfected into HeLa cells. Methylation of the eNOS promoter DNA dramatically decreased its promoter activity to 13% of the mock-methylated promoter activity (Fig. 4). Treatment with HDAC inhibitor TSA partially reversed the repression that was induced by the DNA methylation (Fig. 4), indicating that HDAC is involved in DNA methylation-mediated repression of eNOS promoter.
Fig. 4. Effects of TSA on DNA methylation mediated-repression of eNOS promoter.

In vitro methylated and mock-methylated eNOS promoter/reporter constructs were transiently transfected into HeLa cells followed by treatment with or without TSA. Luciferase activity was measured 24 h after the treatment. The data shown are the mean ± S.D. of three separated experiments.
Synergistic Effect of TSA and AzadC on the Induction of the eNOS Expression in Non-endothelial Cells
AzadC is an analog of cytosine, which can be incorporated into DNA and irreversibly binds to DNA methyltransferase, thus inhibiting DNA methyltransferase and reversing epigenetic silencing of methylated genes (25). To further investigate the relationship between histone deacetylation and DNA methylation in the repression of the eNOS expression in non-endothelial cells, we treated HeLa cells with DNA methyltransferase inhibitor AzadC (7 μm) alone for 8 days, TAS (1 μg/ml) alone for 24 h, or AzadC for 7 days combined with TSA for another 24 h. TSA and AzadC synergistically induced eNOS mRNA expression in HeLa cells (Fig. 5). However, AzadC alone only mildly induced the eNOS expression compared with the effect of TSA (1 μg/ml) alone (Fig. 5).
Fig. 5. Synergistic induction of eNOS mRNA expression by TSA and AzadC.

HeLa cells were treated with 7 μm AzadC for 8 days, 1 μg/ml TSA for 24 h, or AzadC for 7 days combined with TSA (1 μg/ml) for another 24 h. The eNOS mRNA level was quantitated as in Fig. 1. The data shown are the mean ± S.D. of three separated experiments.
DNA Methylation Status of the eNOS Proximal Promoter of HeLa Cells after Treatment with AzadC or TSA
To investigate the DNA methylation status of the eNOS proximal promoter in HeLa cells after treatment with AzadC or TSA, we performed sodium bisulfite genomic sequencing for this region. The DNA methylation status of eNOS proximal promoter of non-treated and treated HeLa cells was summarized in Fig. 6. There were two CpG dinucleotides within the high affinity proximal Sp1 site (−102 and −97), and they were 36.3 and 54.5% methylated, respectively. However, 40% of the two CpG dinucleotides were unmethylated. Treatment with AzadC resulted in a statistically significant decrease of methylation in 8 of 12 (66.6%) CpG dinucleotides in the eNOS proximal promoter region (Fig. 6A). Moreover, the two CpG dinucleotides (−102 and −97) within the proximal Sp1 site totally lost their methylation after the AzadC treatment (Fig. 6A). Treatment with TSA only resulted in the decrease of methylation in 2 of 12 (16.6%) CpG dinucleotides in the eNOS proximal promoter (Fig. 6B).
Fig. 6. DNA methylation status of the eNOS proximal promoter after treated with AzadC or TSA.

A, bisulfite genomic sequencing results of subcloned PCR products of HeLa cells treated with AzadC (7 μm) for 8 days. B, bisulfite genomic sequencing results of subcloned PCR products of HeLa cells treated with TSA (1 μg/ml) for 24 h. The x axis of each graph indicates the CpG doublet position in the proximal promoter region of eNOS with respect to the start site of transcription. CTR, control. Each column represents the percentage of the methylated CpG of total 10–15 individual clones. p < 0.05 (*) and p < 0.01 (**) are by the χ2 analyses.
Roles of Sp1 Site in Induction of eNOS by TSA in Non-endothelial Cells
The proximal Sp1 site is crucial for eNOS promoter activity (22). Sp1 site is suggested to be a TSA-responsive element in several studies (26–28). It would be necessary to know whether HDAC inhibitors can enhance DNA binding activity of Sp1 transcription factor itself. We performed EMSA with a DIG-labeled probe containing the high affinity proximal Sp1 site of eNOS promoter. Incubation of nuclear extracts from HeLa cells treated with or without TSA with the Sp1 probe resulted in one major retarded band, which was competed by adding a 100-fold molar excess of the unlabeled probe (Fig. 7A). The intensity patterns of the retarded band between the nuclear extracts from TSA-treated and non-treated cells were the same, indicating that DNA binding activity of the Sp1 factor was not activated by TSA. Similar results were also observed in TSA-treated HSF and 293 cells (data not shown). To further examine the role of the proximal Sp1 site in induction of eNOS by TSA in non-endothelial cells, we mutated five nucleotides within the proximal Sp1 site in the luciferase-coupled eNOS promoter/reporter construct and transfected the construct into HeLa cells. A luciferase assay showed that the basal activity of Sp1 site-mutated eNOS promoter was only 4% that of the wild type (Fig. 7B). To examine whether the binding ability of mutant Sp1 site to Sp1 factor was affected, we did a DAPA assay with the biotinylated probes containing the wild type or the mutant Sp1 site as in the promoter/reporter construct. Corresponding to the results of luciferase assay, DAPA assay showed that the mutant Sp1 site significantly decreased its binding ability to only 13% of the wild type according to the density measurement (Fig. 7C). Unexpectedly, the mutation of the proximal Sp1 site did not cause a loss of response to TSA treatment, as compared to the wild type (Fig. 7D).
Fig. 7. The role of the Sp1 in TAS induced eNOS expression in non-endothelial cells.
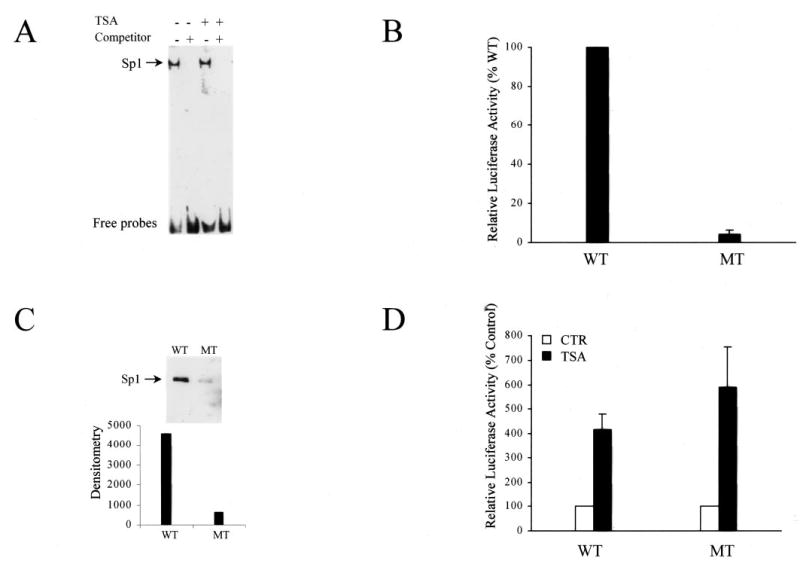
A, DNA binding activity of Sp1 in non-endothelial cells after TSA treatment. EMSA was performed by incubating DIG-labeled Sp1 probe with the nuclear extracts from HeLa cells treated with or without TSA. The competitor was unlabeled probe with a 100-fold molar excess. B, role of the proximal Sp1 site for basal activity of eNOS promoter. The Sp1 site mutant (MT) and wild type (WT) eNOS promoter/reporter constructs were transfected into HeLa cells, and luciferase activity was measured and normalized as the percentage of the wild type. C, Sp1 binding ability between Sp1 site mutant and wild type with DAPA assay. Biotinylated oligonucleotides were incubated with nuclear extracts of HeLa cells and pulled down by streptavidin-agarose and subjected to Western blot. WT, wild type Sp1 probe; MT, mutant Sp1 probe (the mutation is the same as in the mutant Sp1 site eNOS promoter/reporter construct in panel B). D, responsiveness of the proximal Sp1 site to TSA treatment. Wild type and the Sp1 mutated eNOS promoter/reporter constructs were transfected into HeLa cells. Luciferase activity was measured 24 h after the TSA treatment. WT, wild type eNOS promoter/reporter construct; MT, mutant Sp1 site eNOS promoter/reporter construct; CTR, control.
To further investigate the roles of the proximal Sp1 site in TSA-induced eNOS expression in non-endothelial cells, it is important to know whether Sp1 binds to eNOS promoter in vivo in non-endothelial cells. To answer this question, we performed a ChIP assay with Sp1 antibody in HeLa cells. The Sp1 antibody-precipitated chromatin was subjected to DNA extraction and PCR with the primers covering the high affinity proximal Sp1 site (−111/−93). We did four separated experiments and showed that Sp1 bound to the endogenous eNOS promoter in HeLa cells in every assay. As shown in Fig. 8, the occupancy of the endogenous eNOS promoter by Sp1 did not change after treatment with TSA, consistent with our EMSA results. Treatment with AzadC alone also did not alter Sp1 occupancy of the endogenous eNOS promoter (Fig. 8A). However, the combined treatment of AzadC and TSA increased Sp1 occupancy of the endogenous eNOS promoter (Fig. 8A).
Fig. 8. Sp1 binding to endogenous eNOS promoter and associating with HDAC1 in HeLa cells.

A, occupancy of endogenous eNOS promoter by Sp1 after treatment with TSA and AzadC. ChIP assay was performed with anti-Sp1 antibody for non-treated cells or the cells treated with TSA or AzadC or both. The A pair of primers (as shown in Fig. 3), which covers the proximal Sp1 site, was used to amplify the DNA isolated from the ChIP assay. The data are representative of three separated experiments. B, association of HDAC1 with Sp1 in HeLa cells. Whole cell lysates were immunoprecipitated with anti-Sp1 antibody (left panel) or anti-HDAC1 antibody (right panel) and anti-FLAG antibody as negative control in both panels. The immunocomplexes were analyzed by Western blot with antibodies indicated. Input (40 μg of whole cell lysate) was served for position of HDAC1 or Sp1. The membrane was then stripped for detection with the antibodies used in the immunoprecipitation to show the pull-down proteins (lower panel). C, association of HDAC1 with Sp1 after treatment with TSA or TSA and AzadC. Whole cell lysates from HeLa cells treated with TSA (1 μg/ml, 24 h) or AzadC (7 μm) for 7 days combined with TSA (1 μg/ml) for another 24 h were immunoprecipitated by anti-Sp1 antibody. The immunocomplexes were analyzed by Western blot with anti-HDAC1 antibodies. The membrane was stripped for detection with the Sp1 antibody to show the equal pull-down Sp1 protein in the immunocomplexes (lower panel). IP, immunoprecipitation; WB, Western blot.
We further investigated how eNOS was transcriptionally suppressed in non-endothelial cells when Sp1 factor was in binding with the endogenous eNOS promoter or how HDACs were involved in the suppression. The association of Sp1 with HDAC1 was first reported by Doetzlhofer et al. (20), and later the association was shown to be involved in repress genes in several cell lines, including human telomerase reverse transcriptase (29), human luteinizing hormone receptor (30), and transforming growth factor β type II receptor (15). Using a co-immunoprecipitation assay, we tested whether Sp1 was associated with HDAC1 in HeLa cells. As shown in Fig. 8, HDAC1 was detected in anti-Sp1 antibody-precipitated complex, and reversely, Sp1 was also detected in anti-HDAC1 antibody-precipitated complex. But neither Sp1 nor HDAC1 was revealed in negative control anti-FLAG antibody-precipitated complex. Sp1 was shown as a 105-kDa abundant isoform and a 95-kDa less abundant isoform, which may be difficult to be detected by Western blot unless a large amount of proteins are loaded. Notably, anti-HDAC1 antibody pulled down an almost equal amount of the two isoforms, implicating that HDAC1 may preferably interact with the 95-kDa isoform of Sp1. Treatment with TSA or AzadC alone did not alter the association of HDAC1 with Sp1 (Fig. 8C). However, treatment with AzadC and TSA together decreased the association of HDAC1 with Sp1 (Fig. 8).
DISCUSSION
In the present study, we have provided evidence for the first time that HDACs are involved in the repression of the human eNOS expression in non-endothelial cells. The repression of the eNOS could be partially relieved in several non-endothelial cell types by the inhibition of HDACs (Fig. 1). The induction of eNOS in non-endothelial cells by HDACs inhibitors is more likely at the transcriptional level and less likely due to the enhancement of eNOS mRNA stabilization, since actinomycin D (an inhibitor of DNA-dependent RNA synthesis) completely blocked the induction of eNOS by TSA. Moreover, this eNOS induction in non-endothelial cells appears independent of new protein synthesis, since blocking protein synthesis by cycloheximide did not alter the TSA-induced eNOS mRNA expression. Given that the episomal eNOS promoter/reporter was directly activated by TSA, it is implicated that HDACs may be directly involved in the repression of eNOS promoter in non-endothelial cells. Our experiments have further shown that activities of episomal eNOS promoter/reporter were comparable in endothelial and non-endothelial cells (Fig. 2), indicating that endothelial cells and non-endothelial cells (at least among the cell types we have tested) may have equivalent transcriptional machinery necessary for eNOS transcription. Our study also suggests that the induction of eNOS in non-endothelial cells by HDAC inhibitors could be mediated by direct utilization of the existing transcriptional machinery. The reason that non-endothelial cells do not constitutively express eNOS would be more likely due to repressive chromatin structure.
Histone acetylation is a critical component of chromatin remodeling and transcriptional regulation (6). The acetylation level of core histone results from the balance between the activities of HDACs and histone acetyltransferases. Inhibition of HDACs by the TSA leads to activation of only specific target genes through increased histone acetylation (11, 12). Our experiments showed that induction of eNOS expression by TSA in non-endothelial cells was accompanied by a remarkable increase in acetylation of histone H3 associated with the eNOS 5′-flanking region (−4917 to −13) (Fig. 3). The increase of the core histone acetylation at the broad 5′-flanking region of the eNOS gene after the TSA treatment indicates that the chromatin structure of eNOS promoter area may become a loose and non-condensed structure, which is usually necessary for the start of transcription (8, 9). In contrast, histone H3 of the proximal promoter and the putative enhancer of eNOS gene in endothelial cells are already in a hyperacetylated status before the TSA treatment (Fig. 3), a typical phenomenon for activated chromatin (8, 9). These data provide further evidence that histone deacetylation plays an important role in the repression of eNOS in non-endothelial cells.
DNA methylation is another important epigenetic process involved in regulating chromatin structure and gene transcription (6). DNA methylation is linked with histone deacetylation through a transcriptional repressor MeCP2 (a methylated CpG-binding protein), which recruits HDACs to specific promoter region, resulting in repressive chromatin structure and suppress gene expression (24, 31). A recent study has demonstrated that eNOS proximal promoter DNA is heavily methylated in non-endothelial cells and that DNA methylation plays an important role in eNOS cell-specific expression (5). Given that our experiment showing histone deacetylation was also involved in the repression of eNOS in non-endothelial cells, it would be reasonable to speculate that DNA methylation and histone deacetylation may be synergistically involved in suppressing eNOS expression in non-endothelial cells. We tested the speculation and observed that the basal activity of the in vitro methylated episomal eNOS promoter was dramatically reduced (Fig. 4) but was partially reversed by TSA treatment (Fig. 4). Considering the cooperation between DNA methylation and HDACs in repression of genes (24, 31), we suggest that HDACs may also be involved in DNA methylation-mediated repression of eNOS promoter activity in non-endothelial cells. Furthermore, even though the bisulfite genomic sequencing has confirmed that DNA methylation of the proximal eNOS promoter was significantly decreased (including complete demethylation of the two CpG dinucleotides within the proximal Sp1 site) by the treatment with AzadC, AzadC alone only mildly induced the eNOS expression in non-endothelial HeLa cells (Figs. 5 and 6A). The results implicate that de-repression by DNA demethylation alone in non-endothelial cells is not sufficient to override the cell-specific suppression of eNOS. On the other hand, inhibition of HDACs by TSA (1 μg/ml) alone resulted in a more dramatic de-repression of the eNOS expression in the non-endothelial cells (Fig. 5). This de-repression by TSA appeared not to be mediated through changing eNOS proximal promoter DNA methylation status (Fig. 6B). In addition, a synergistic effect on the induction of the eNOS expression was observed with a combined treatment of TSA and AzadC (Fig. 5). Taken together, these results suggest that histone deacetylation and DNA methylation may be synergistically involved in repression of eNOS in non-endothelial cells.
The proximal Sp1 site played an important role in HDACs-mediated repression of eNOS in non-endothelial cells. Consensus Sp1 site has been shown to be a responsive element in several HDAC-regulated genes (26–28). In the case of eNOS, the proximal Sp1 site also appeared as a key element in TSA induction of eNOS expression in non-endothelial cells. Consistent with the published data (22), the proximal Sp1 site was responsible for most (96%) of the eNOS proximal promoter basal activity since the mutation within the site resulted in only 13% of Sp1 binding ability and 4% of basal activity (Fig. 7). Furthermore, we demonstrated with the Sp1 ChIP assay that Sp1 still bound to the endogenous eNOS proximal promoter (containing the proximal Sp1 site) in vivo (Fig. 8), although eNOS is repressed in HeLa cells. Treatment with AzadC resulted in unmethylation status of the two CpG dinucleotides within the proximal Sp1 site (Fig. 6A); however, the occupancy of the endogenous eNOS promoter by Sp1 showed no difference between the AzadC-treated and -non-treated HeLa cells (Fig. 8A), indicating that the binding to the endogenous eNOS promoter is not affected by the Sp1 site methylation.
Based on these results it would be logical to ask how eNOS is repressed in non-endothelial cells by HDACs. Our results indicate that the induction of eNOS in non-endothelial cells by HDACs inhibitor is mediated not through increased Sp1 DNA binding activity or its occupancy of endogenous eNOS proximal promoter (Figs. 7A and 8A). Doetzlhofer et al. (20) suggest that Sp1 can serve as a target for HDAC1-mediated transcriptional repression, although Sp1 is usually known as a positive transcription factor. Studies have further shown that the association of HDAC1 or 2 with Sp1 and Sp3 is involved in repressing gene expression in several cell lines (15, 29, 30), which could be a universal mechanism for cell-specific repression of the Sp1 dependent expression of genes. Indeed, our study also has demonstrated the interaction between HDAC1 and Sp1 in HeLa cells (Fig. 8). Given that the proximal Sp1 site is crucial for eNOS promoter activity, Sp1 associates with HDAC1, and Sp1 binds to the endogenous eNOS proximal promoter, it is likely that the proximal Sp1 site may serve as a target for HDAC1-involved eNOS suppression in non-endothelial cells. Based on these findings, we propose that repression of eNOS in non-endothelial cells is mediated by the recruitment of HDACs to the proximal promoter region by the complex of methylated DNA, MeCP2, and other co-repressors and, more importantly, the recruitment of HDAC1 by Sp1 to the proximal Sp1 site. We further speculate that the association of HDAC1 with Sp1 somehow limits the Sp1 transcriptional activating function. The HDAC-specific inhibitor TSA abrogates HDAC1 enzymatic activities of deacetylation and, hence, shifts acetylation status of core histones to hyperacetylation and restores Sp1 transcriptional function. The synergistic effect on eNOS activation in HeLa cells by the combined treatment of AzadC and TSA could be the results of increased Sp1 binding to the endogenous eNOS promoter and decreased association of Sp1 with HDAC1 (Fig. 8). Further studies will be needed to understand how the association of HDAC1 with Sp1 inhibits the Sp1 function and how TSA restores the function of Sp1.
Our experiments have also shown that episomal eNOS promoter was not repressed in non-endothelial cells and was responsive to TSA activation. After the episomal eNOS promoter, which is methylation-free, is transfected into the non-endothelial cells, it will form mini-chromatin but not in a repressive chromatin structure as the endogenous eNOS promoter in these cells. Therefore, the balance between HDAC-related inhibition and histone acetyltransferase-related activation determines the basal activity of the episomal eNOS promoter. Inhibition of HDACs by TSA will shift the balance and lead to the episomal promoter activation.
In conclusion, our study has demonstrated that HDACs are involved in the eNOS repression in non-endothelial cells. The proximal Sp1 site may be the key target for HDAC1-mediated inhibition of eNOS in non-endothelial cells. The findings that constitutively repressed eNOS gene in non-endothelial cells can be re-activated by the inhibition of HDACs will not only enhance our understanding in tissue-specific eNOS expression but will also have much wider implications in several clinically relevant processes including developmental differentiation, stem cell transformation, and functional restoration after vascular injury. Further investigations in dissecting exact mechanisms about how the association of HDAC1 with Sp1 prevents Sp1 transcriptional activation will be needed to understand tissue-specific expression of genes in endothelium.
Footnotes
This study was supported by National Institutes of Health Grant R01-HL066053.
The abbreviations used are: eNOS, endothelial nitric-oxide synthase; HDAC, histone deacetylase; TSA, trichostatin A; SB, sodium butyrate; ChIP, chromatin immunoprecipitation; AzadC, 5-aza-2′-deoxycytidine; HSF, human skin fibroblast cell; EMSA, electrophoresis mobility shift assay; ChIP, chromatin immunoprecipitation; DAPA, DNA affinity purification assay; DIG, digoxigenin-11-dUTP; RT, room temperature.
References
- 1.Li H, Wallerath T, Munzel T, Forstermann U. Nitric Oxide. 2002;7:149–164. doi: 10.1016/s1089-8603(02)00111-8. [DOI] [PubMed] [Google Scholar]
- 2.Teichert AM, Miller TL, Tai SC, Wang Y, Bei X, Robb GB, Phillips MJ, Marsden PA. Am J Physiol Heart Circ Physiol. 2000;278:1352–1361. doi: 10.1152/ajpheart.2000.278.4.H1352. [DOI] [PubMed] [Google Scholar]
- 3.Guillot PV, Liu L, Kuivenhoven JA, Guan J, Rosenberg RD, Aird WC. Physiol Genomics. 2000;2:77–83. doi: 10.1152/physiolgenomics.2000.2.2.77. [DOI] [PubMed] [Google Scholar]
- 4.van Haperen R, Cheng C, Mees BM, van Deel E, de Waard M, van Damme LC, van Gent T, van Aken T, Krams R, Duncker DJ, de Crom R. Am J Pathol. 2003;163:1677–1686. doi: 10.1016/S0002-9440(10)63524-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan Y, Fish JE, D’Abreo C, Lin S, Robb GB, Teichert AM, Karantzoulis-Fegaras F, Keightley A, Steer BM, Marsden PA. J Biol Chem. 2004;279:35087–35100. doi: 10.1074/jbc.M405063200. [DOI] [PubMed] [Google Scholar]
- 6.Geiman TM, Robertson KD. J Cell Biochem. 2002;87:117–125. doi: 10.1002/jcb.10286. [DOI] [PubMed] [Google Scholar]
- 7.Iizuka M, Smith MM. Curr Opin Genet Dev. 2003;13:154–160. doi: 10.1016/s0959-437x(03)00020-0. [DOI] [PubMed] [Google Scholar]
- 8.Davie JR, Spencer VA. J Cell Biochem Suppl. 1999;32–33:141–148. doi: 10.1002/(sici)1097-4644(1999)75:32+<141::aid-jcb17>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 9.Pazin MJ, Kadonaga JT. Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- 10.Legube G, Trouche D. EMBO Rep. 2003;4:944–947. doi: 10.1038/sj.embor.embor941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Lint C, Emiliani S, Verdin E. Gene Expr. 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- 12.Della Ragione F, Criniti V, Della Pietra V, Borriello A, Oliva A, Indaco S, Yamamoto T, Zappia V. FEBS Lett. 2001;499:199–204. doi: 10.1016/s0014-5793(01)02539-x. [DOI] [PubMed] [Google Scholar]
- 13.Cong YS, Bacchetti S. J Biol Chem. 2000;275:35665–35668. doi: 10.1074/jbc.C000637200. [DOI] [PubMed] [Google Scholar]
- 14.Qiu P, Li L. Circ Res. 2002;90:858–865. doi: 10.1161/01.res.0000016504.08608.b9. [DOI] [PubMed] [Google Scholar]
- 15.Zhao S, Venkatasubbarao K, Li S, Freeman JW. Cancer Res. 2003;63:2624–2630. [PubMed] [Google Scholar]
- 16.Zhang X, Wharton W, Yuan Z, Tsai SC, Olashaw N, Seto E. Mol Cell Biol. 2004;24:5106–5118. doi: 10.1128/MCB.24.12.5106-5118.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Dudley D, Wang XL. Arterioscler Thromb Vasc Biol. 2002;22:e1–e4. doi: 10.1161/01.ATV.0000016248.51577.1F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warnecke PM, Stirzaker C, Song J, Grunau C, Melki JR, Clark SJ. Methods. 2002;27:101–107. doi: 10.1016/s1046-2023(02)00060-9. [DOI] [PubMed] [Google Scholar]
- 19.Li LC, Dahiya R. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 20.Doetzlhofer A, Rotheneder H, Lagger G, Koranda M, Kurtev V, Brosch G, Wintersberger E, Seiser C. Mol Cell Biol. 1999;19:5504–5511. doi: 10.1128/mcb.19.8.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossig L, Li H, Fisslthaler B, Urbich C, Fleming I, Forstermann U, Zeiher AM, Dimmeler S. Circ Res. 2002;91:837–844. doi: 10.1161/01.res.0000037983.07158.b1. [DOI] [PubMed] [Google Scholar]
- 22.Karantzoulis-Fegaras F, Antoniou H, Lai SL, Kulkarni G, D’Abreo C, Wong GK, Miller TL, Chan Y, Atkins J, Wang Y, Marsden PA. J Biol Chem. 1999;274:3076–3093. doi: 10.1074/jbc.274.5.3076. [DOI] [PubMed] [Google Scholar]
- 23.Laumonnier Y, Nadaud S, Agrapart M, Soubrier F. J Biol Chem. 2000;275:40732–40741. doi: 10.1074/jbc.M004696200. [DOI] [PubMed] [Google Scholar]
- 24.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 25.Brown R, Plumb JA. Expert Rev Anticancer Ther. 2004;4:501–510. doi: 10.1586/14737140.4.4.501. [DOI] [PubMed] [Google Scholar]
- 26.Sowa Y, Orita T, Minamikawa S, Nakano K, Mizuno T, Nomura H, Sakai T. Biochem Biophys Res Commun. 1997;241:142–150. doi: 10.1006/bbrc.1997.7786. [DOI] [PubMed] [Google Scholar]
- 27.Kim HS, Park JS, Hong SJ, Woo MS, Kim SY, Kim KS. Biochem Biophys Res Commun. 2003;312:950–957. doi: 10.1016/j.bbrc.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Camarero N, Nadal A, Barrero MJ, Haro D, Marrero PF. Nucleic Acids Res. 2003;31:1693–1703. doi: 10.1093/nar/gkg262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Won J, Yim J, Kim TK. J Biol Chem. 2002;277:38230–38238. doi: 10.1074/jbc.M206064200. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Dufau ML. J Biol Chem. 2002;277:33431–33438. doi: 10.1074/jbc.M204417200. [DOI] [PubMed] [Google Scholar]
- 31.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]