

Abstract
Light stimuli produce graded hyperpolarizations of the photoreceptor plasma membrane and an associated decrease in a voltagegated calcium channel conductance that mediates release of glutamate neurotransmitter. The Cav1.4 channel is thought to be involved in this process. The CACNA1F gene encodes the poreforming subunit of the Cav1.4 channel and various mutations in CACNA1F cause X-linked incomplete congenital stationary night blindness (CSNB2). The molecular mechanism of the pathology underlying the CSNB2 phenotype remains to be established. Recent clinical investigations of a New Zealand family found a severe visual disorder that has some clinical similarities to, but is clearly distinct from, CSNB2. Here, we report investigations into the molecular mechanism of the pathology of this condition. Molecular genetic analyses identified a previously undescribed nucleotide substitution in CACNA1F that is predicted to encode an isoleucine to threonine substitution at CACNA1F residue 745. The I745T CACNA1F allele produced a remarkable approximately –30-mV shift in the voltage dependence of Cav1.4 channel activation and significantly slower inactivation kinetics in an expression system. These findings imply that substitution of this wild-type residue in transmembrane segment IIS6 may have decreased the energy required to open the channel. Collectively, these findings suggest that a gain-of-function mechanism involving increased Cav1.4 channel activity is likely to cause the unusual phenotype.
Voltage-gated calcium channels are key players in muscle contraction, hormone and neurotransmitter release, and regulation of gene transcription (1). The pore-forming component of voltage-gated calcium channels is provided by the α1 subunit. Each member of the α1 subunit of the voltage-gated calcium channel (CACNA1) family contains four structurally conserved motifs (domains I–IV). Each domain consists of a voltage sensor (formed by the helical segments S1–S4) coupled to a pore region that includes the ion selectivity filter [formed by the S5, P (pore), and S6 helices] (1). The focus of the present study is the CACNA1F gene, encoding the α1 subunit of the Cav1.4 voltage-gated calcium channel, also known as α1F. This gene maps to chromosome Xp11.23 and, when mutated, causes the recessive disorder X-linked incomplete congenital stationary night blindness (CSNB2) (2, 3).
CSNB2 is a nonprogressive retinal disorder with clinical features that include reduced visual acuity, nystagmus, and variable myopia (short-sightedness) or hypermetropia (long-sightedness). Characteristic abnormalities are detected upon electroretinography, which measures the electrical responses of the retina to light stimuli. One such feature is a negative waveform where the ratio of b/a wave amplitudes is <1. Light stimuli activate the phototransduction pathway resulting in hyperpolarization of the photoreceptor plasma membrane, an event thought to contribute to the a wave. Subsequent alterations in the activity of second-order neurons are thought to be similarly reflected in the b wave. Hence, the negative waveform in CSNB2 indicates that outer segment function is preserved in photoreceptors, whereas second-order neuronal activity is impaired (4–6). Additional abnormalities are observed with long-flash electroretinography protocols, implying that the retinal ON and OFF pathways are both affected (7). Collectively, these findings suggested that the vision impairment in CSNB2 patients may arise primarily from a decrease in the effectiveness of synaptic transmission between photoreceptors and second-order neurons. CACNA1F/Cacna1f is expressed in various retinal cell types including photoreceptors (2, 3, 8–10). Accordingly, it has been suggested that the Cav1.4 channel may contribute to a voltage-gated calcium conductance that mediates release of glutamate from photoreceptors (2, 3, 11, 12).
Clinical investigations have recently characterized an X-linked retinal disorder that is similar to, but distinct from, CSNB2 in a New Zealand family (13). Male individuals had congenital nystagmus, severe nonprogressive impairment of visual acuity, frequent hypermetropia, and normal fundi. This phenotype was more severe than that typically observed in CSNB2 (4–6, 13). Female individuals had congenital nystagmus, decreased visual acuity and frequent high myopia. In marked contrast to the present family, in CSNB2, female heterozygotes do not show clinical signs (4–6, 13, 14). Electroretinography in male individuals found several features similar to CSNB2: The ratio of the b/a wave amplitudes was <1, the amplitude for the 30-Hz flicker response was reduced, and both the ON and OFF pathways were severely reduced (4–6, 13). In the present family, all tested female individuals had ERG abnormalities that were similar to, but less severe than, those observed in male individuals. ERG abnormalities of this severity and consistency have not been detected in CSNB2 female heterozygotes (4–6, 13, 14). Finally, some visually impaired male individuals in the present pedigree also had intellectual impairment and were autistic. Nonocular neurological abnormalities have not previously been associated with CSNB2 (4–6, 13).
In the present study, we have identified a previously undescribed CACNA1F mutation in this family. The mutation significantly affected the gating properties of the Cav1.4 channel when exogenously expressed in tsA-201 cells. These findings imply that the molecular mechanism of the pathology is likely to involve a gain, rather than loss, of Cav1.4 channel function and that the substituted residue contributes to gating of the Cav1.4 channel.
Materials and Methods
Molecular Genetic Analysis. Microsatellite markers (15, 16) were genotyped in DNA samples from 27 family members, and the data were analyzed by two-point linkage analysis.
Intronic primer pairs for exons of CACNA1F, including the known splice variants (17), were used for PCR amplification of a DNA sample from an affected male individual. The PCR products were sequenced in both directions, and two nucleotide substitutions were identified, one in exon 17 and the other in intron 21. The frequencies of the nucleotide substitutions in exon 17 and intron 21 in control samples were determined with, respectively, single-strand conformation polymorphism analysis (180 chromosomes) and a forced restriction length polymorphism assay that created a BsmAI site in the wild-type sequences (101 chromosomes). The latter screen identified four samples that failed to digest, and DNA sequencing revealed two heterozygote females and two hemizygote males with an intron 21 sequence variant identical to that identified in the present family. For more details, see Supporting Methods, which is published as supporting information on the PNAS web site.
Functional Analyses. The CACNA1F mutation identified in the present family was introduced into a human Cav1.4 α1 subunit cDNA (18) cloned into the expression vector pCIneo, which contains an additional poly(A) tail. In particular, the gene splicing by overlap extension technique (19) was used to generate a 2234T → C nucleotide substitution into the Cav1.4 α1 cDNA via an AgeI–Bst1107I cassette (nucleotides 1390–3105) (nucleotide numbering according to GenBank accession no. NM_005183, excluding 62 bp of 5′ untranslated sequence). The corresponding wild-type region of the cDNA was removed from the vector by digestion with AgeI–Bst1107I (nucleotides 1390–3105) and replaced by the mutated cassette. Subsequent DNA sequencing (MWG Biotech, Ebersberg, Germany), in both directions, confirmed introduction of the mutation and integrity of the construct.
TsA-201 cells were cotransfected with wild-type or I745T Cav1.4 α1 subunits together with the α2-δ1 (20) and β2a or β3 subunits (21). Transfection of tsA-201 cells was performed by using the FuGENE 6 transfection reagent, as recommended by the manufacturer (Roche Diagnostics, Mannheim, Germany). Transfection solution was prepared by mixing 1 μg of plasmid DNA and 6 μl of FuGENE 6 transfection reagent in 100 μl of serum-free medium. After incubation at room temperature for 15 min, the mixture was added to tissue culture wells containing 1 × 106 cells in 2 ml of complete culture medium.
Barium (IBa) or calcium (ICa) currents through voltage-gated Ca2+ channels were recorded at 22–25°C by using the patch–clamp technique (22) by means of an Axopatch 200A patch clamp amplifier (Axon Instruments, Foster City, CA). Patch pipettes with resistances of 1–4 MΩ were made from borosilicate glass (Clark Electromedical Instruments, Pangbourne, U.K.) and filled with pipette solution containing 145 mM CsCl, 2 mM MgCl2, 10 mM Hepes, and 10 mM EGTA adjusted to pH 7.25 with CsOH. IBa was measured in a solution containing 20 mM BaCl2, 140 mM choline chloride, 10 mM Hepes, and 1.0 mM MgCl2 (buffered to pH 7.4 with methanesulfonic acid). ICa was measured in a solution containing 5 mM CaCl2, 180 mM choline chloride, 10 mM Hepes, and 1.0 mM MgCl2 (buffered to pH 7.4 with methanesulfonic acid).
The voltage dependence of activation was determined from current–voltage (I–V) curves that were fitted according to the following modified Boltzmann term: I = Gmax(V – Vrev)/(1 + exp[(V0.5,act. – V)/kact]), where Vrev is the extrapolated reversal potential, V is the membrane potential; I is the peak current, Gmax is the maximum membrane conductance, V0.5,act is the voltage for half-maximal activation, and kact is the slope factor. The time course of current activation was fitted to monoexponential {I(t) = A[exp(–t/τ)] + C} or biexponential {I(t) = Afast[exp(–t/τfast)] + Aslow[exp(–t/τslow)] + C} functions, where I(t) is the current at time t; A, Afast, and Aslow are the amplitude coefficients of the time constants τ, τfast, and τslow, respectively; and C is the steady-state current. The slow time constant of current activation was estimated from 3-s current traces.
The voltage dependence of IBa inactivation (inactivation curve) was measured by using a multistep protocol to account for run-down. The pulse sequence was applied every 40 s from a holding potential of –100 mV. Inactivation curves were drawn according to a Boltzmann equation: IBa,inact = Iss + (1 – Iss)/(1 + exp[(V – V0.5,inact)/k)], where V is the membrane potential, V0.5,inact is the midpoint voltage, k is the slope factor, and Iss is the fraction of noninactivating current.
All data were digitized by using a Digidata 1200 interface (Axon Instruments) at 5 kHz (300-ms pulses) or 0.2 kHz (3-s pulses) and stored on computer hard disk. The data were filtered at 2 kHz (four-pole Bessel filter). Leak currents were subtracted digitally (using average values of scaled leakage currents elicited by a 10-mV hyperpolarizing pulse) or electronically by means of an Axopatch 200 amplifier. Series resistance and offset voltage were routinely compensated. Data are given as mean ± SEM. Statistical significance was assessed by using Student's unpaired t test.
Expression of CACNA1F/Cacan1f in the Pineal Gland. For RT-PCR analysis, first-strand synthesis was performed on total RNA extracted from human pineal tissue (23) by using oligo(dT) primer and Superscript II reverse transcriptase (Invitrogen) and used for PCR amplification of Cacna1f exons 28–29, 45–46, and 47–48. For in situ hybridization analysis, frozen sections were prepared for pineal glands and retinas obtained from 4-week-old C57BL/6 mice. A 759-bp cDNA fragment from exons 42–48 of mouse Cacna1f (nucleotides 5035–5793, GenBank accession no. AF192497) was cloned into the vector pCR II-TOPO, and antisense and sense cRNA probes were transcribed by using the digoxigenin labeling system (Roche Applied Science). In situ hybridization was performed by following the protocol of Groves and Bronner-Fraser (24) (also see www.rodentia.com/wmc/docs/Big_In_Situ.html). See Supporting Methods for more detailed information.
Results
Identification of a Previously Uncharacterized Mutation in CACNA1F. Segregation analysis of chromosome X microsatellite markers (15, 16) in 27 members of the New Zealand family revealed nine markers (DXS1055-DXS8092) forming a haplotype (2-1-4-1-1-3-2-2-6) that cosegregated with the disorder (Fig. 1a). Recombinant haplotypes in affected male individual VII-48 and unaffected male individual VI-17 defined an ≈5-cM critical region between DXS1126 and DXS991. Two-point analyses between the disease gene and each marker within the critical region gave strong evidence of linkage with logarithm of odds scores at theta = 0.00 ranging from 3.52 to 4.33 (see Table 1, which is published as supporting information on the PNAS web site). The critical region encompassed CACNA1F, the gene mutated in CSNB2 (2, 3), but it was proximal to both NYX and RP2, the genes responsible for CSNB1 (25, 26) and X-linked retinitis pigmentosa 2 (27), respectively.
Fig. 1.

Molecular genetic analysis of the New Zealand family. (a) Segregation of chromosome X markers. Filled symbol, affected male individual; checkered symbol, affected female individual. The recombinant haplotypes in individuals VI-17 and VII-48 and the locations of CSNB2/CACNA1F and RP2 relative to the microsatellite markers are indicated. (b) Identification of a 2234T → C nucleotide substitution in exon 17 of CACNA1F.(c) Alignment of transmembrane segment IIS6 sequences for human CACNA1 family members. Shown in bold are a glycine “hinge” residue that is conserved in the voltage-gated ion channel superfamily, an asparagine residue characteristic of the CACNA1 family, and the residue of interest to the present study.
The CACNA1F gene was subsequently sequenced, and two DNA alterations were identified: a T to C substitution at nucleotide 2234 in exon 17 (Fig. 1b) and a G to A substitution at position 3 of the 5′ splice site of intron 21. The intron 21 nucleotide substitution was present at a low frequency in control samples (4 of 101 X chromosomes screened) and either nucleotide is acceptable in the 5′ splice site consensus sequence (28), i.e., this sequence alteration is a polymorphism rather than the mutation responsible for the disorder in the present family. By contrast, the exon 17 sequence alteration is likely to be pathogenic. First, this change was not detected in any control sample (0 of 180 X chromosomes screened). Second, the 2234T → C nucleotide substitution is predicted to alter the amino acid encoded for residue 745 from isoleucine to threonine (numbering according to the nucleotide and protein sequences NM_005183 and NM_005174, respectively). This residue in transmembrane segment IIS6 is completely conserved in all members of the CACNA1 family (Fig. 1c).
Functional Impact of the I745T CACNA1F Mutation. The effects of the I745T CACNA1F substitution on Cav1.4 channel activity were investigated after expressing wild-type or mutant Cav1.4 α1 subunits together with the α2-δ1 subunit (20) and either β3 or β2a subunits (21) in tsA-201 cells. The outer plexiform layer in retinal sections is labeled by an α1F antibody in wild-type mice, but not in CNS-β2-null mice, implying that expression of the Cav1.4 channel in photoreceptors depends on the presence of the β2 subunit (8). Cav1.4 channels are, however, known to express at relatively low density and better with the β3 than the β2a subunit (18). Most of the functional studies (>15 transfections) were therefore performed on Cav1.4 (β3) channels in high (20 mM) barium solution (Figs. 2 and 4). The current densities in cells transfected with wild-type or I745T Cav1.4 channels were significantly higher than in cells transfected solely with the auxiliary subunits (Fig. 2c, P < 0.05), showing that the Cav1.4 currents were not significantly contaminated by endogenous [e.g., T-type (29)] currents. This was also evident from the equivalent sensitivity of the wild-type and mutant channels to the agonist 1,4-dihydropyridine BAY K 8644 (see Fig. 5, which is published as supporting information on the PNAS web site) (18).
Fig. 2.

Activation properties of Cav1.4 channels containing β3 subunits with 20 mM Ba2+ as charge carrier. (a and b) Representative barium currents (IBa) through wild-type (a) and I745T (b) channels. Currents were evoked by 300-ms depolarizing pulses from a holding potential of –100 mV to the indicated test potentials. (c) I–V of IBa for wild-type (open circles) and I745T (filled circles) channels and endogenous currents (cells transfected with β3 and α2-δ1 subunits) (filled diamonds). (d) Voltage dependence of IBa activation for wild-type (open circles) and I745T (filled circles) channels. The voltage dependencies of IBa activation kinetics in Cav1.4 channels are shown in e and f. Wild-type channel activation was best described by a monoexponential function (e, open circles). The I745T channels (filled circles) activated with a biexponential time course. The voltage dependencies of the fast and slow time constants are represented in e and f, respectively.
Fig. 4.

Inactivation properties of Cav1.4 channels containing β3 subunits with 20 mM Ba2+ as charge carrier. (a and b) Representative families of IBa for wild-type (a) and I745T (b) channels. (c) Inactivation curve of wild-type (open circles) and I745T (filled circles) channels. IBa was elicited with the following pulse sequence. A control 50-ms pulse to the test potential corresponding to the peak of the I–V curves (–30 mV, I745T; 0 mV, wild type) was followed by a 1-s step to –100 mV, a 3-s conditioning step (–80, –70,..., +20 mV), a 4-ms step to –100 mV, and then a 50-ms test pulse to the test potential (see c Inset; the time axis is not to scale). (d) Voltage dependence of IBa decay (% of peak current) in wild-type (open circles) and I745T (filled circles) channels. IBa was elicited by 3-s depolarizing pulses from –100 mV to the indicated test potentials.
The mutation induced a dramatic leftward shift (–34 mV) in the voltage dependence of channel activation (Fig. 2 a–d). The voltage at half-maximal channel activation (V0.5) was –7.5 ± 0.7 mV (n = 17) in wild-type channels compared with –41.3 ± 0.3 mV (n = 16) in I745T mutant channels (P < 0.01) (Fig. 2d). The activation kinetics of the wild-type Cav1.4 channels near the peak current potential (0 mV) could be well fitted by a monoexponential function with a time constant τ = 1.8 ± 0.3 ms (n = 15) (Fig. 2e). Activation of the I745T Cav1.4 channels near the peak current potential (–30 mV) was best described by a biexponential function with τfast = 3.2 ± 0.5 ms and τslow = 126 ± 25 ms (n = 18) (Fig. 2 e and f).
The voltage dependence of channel activation was also investigated under more physiological conditions. With 5 mM calcium as charge carrier, channel activation occurred at slightly more hyperpolarized potentials (wild type, V0.5 =–14.6 ± 0.4 mV, n = 10; I745T mutant, V0.5 =–46.8 ± 0.4 mV, n = 13) with a similar shift in the voltage dependence of channel activation in the mutant channels (–32 mV; Fig. 3). Coexpression of the β2a subunit induced a similarly large shift (–37 mV) in the voltage dependence of channel activation (wild type, V0.5 =–10.2 ± 0.8 mV, n = 11; mutant channels, V0.5 = –47.2 ± 0.4 mV, n = 9) (Fig. 3).
Fig. 3.

Currents for Cav1.4 channels containing different β subunits with 5 mM Ca2+ as charge carrier. Representative families of ICa through wild-type (a and b) and I745T (c and d) channels coexpressed with β3 (a and c) or β2a (b and d) subunits. Voltage dependence of ICa for wild-type (open circles) and I745T (filled circles) channels, coexpressed with β3 (e) or β2a (f) subunits. ICa was elicited by 300-ms depolarizing pulses from –100 mV to the indicated test potentials.
The amino acid substitution also induced substantial changes in barium current inactivation (Fig. 4). When compared near their respective peak current potentials, the I745T Cav1.4 channels displayed significantly slower inactivation kinetics during a 3-s pulse (–30 mV, 7 ± 1% current decay, n = 13) than the wild-type Cav1.4 channels (0 mV, 48 ± 4% current decay, n = 14) (Fig. 4 a–d).
CACNA1F/Cacna1f Are Expressed in the Pineal Gland. Many genes active in photoreceptors are also expressed in pinealocytes (30). Moreover, L-type calcium channel currents have been recorded in pinealocytes (31). We therefore investigated whether CACNA1F/Cacna1f are transcribed in the pineal gland. RT-PCR analysis of human pineal RNA was carried out with three pairs of primers specific to CACNA1F (see Fig. 6a, which is published as supporting information on the PNAS web site). For each primer pair, the pineal gland RNA gave a band of the size expected for spliced CACNA1F mRNA, and DNA sequencing provided confirmation of their identity. The RT-PCR results might possibly have been the result of contamination of pineal tissue with other cells that express Cacna1f, such as immune cells (32). However, subsequent in situ hybridization analysis of the mouse retina and pineal gland detected transcripts for Cacan1f in the outer nuclear, inner nuclear, and ganglion cell layers of the retina, as previously reported (3), and in pinealocytes (see Fig. 6b).
Discussion
In the present study, molecular genetic analyses revealed an I745T CACNA1F allele in a New Zealand family segregating an inherited disorder with clinical features similar to, but distinct from, CSNB2 (Fig. 1, and see Table 1). Our functional studies revealed that I745T causes a dramatic approximately –30-mV shift in the voltage dependence of Cav1.4 channel activation (Figs. 2 and 3) and significantly slower inactivation kinetics (Fig. 4). The shift in the voltage dependence of channel activation was similar in the presence of the β2a subunit, rather than the β3, subunit (Fig. 3). These findings implied that increased Cav1.4 channel activity is responsible for the previously uncharacterized phenotype in this family.
If the mutant channel shows comparable alterations in function within the retina, then there may be major consequences for visual function. In photoreceptors, the membrane potential is approximately –40 mV in darkness with hyperpolarization to approximately –65 mV in response to a strong light stimulus (11, 12). The rate of release of glutamate from photoreceptors in darkness is sufficient to nearly saturate a postsynaptic signaling cascade in bipolar cells (33, 34). This saturation is relieved by light stimuli because closure of L-type calcium channels leads to a decrease in the rate of neurotransmitter release. The approximately –30-mV shift in Cav1.4 channel activation induced by the I745T mutation (Fig. 2 a–c) should dramatically increase the percentage of open Cav1.4 channels at physiological membrane potentials. Reduced inactivation for the I745T mutant Cav1.4 channel would additionally support persistent Ca2+ entry. As a consequence, glutamate release should be higher than normal during darkness and may continue to saturate the postsynaptic bipolar cells following light stimuli.
Mutations in CSNB2 patients that encode early truncations in CACNA1F (35, 36) are likely to represent null alleles. Because CSNB2 patients retain moderate vision despite loss of Cav1.4 channel activity, it has been suggested that other classes of L-type calcium channels may continue to permit some light-dependent modulation of glutamate release (10). Recent functional studies (32, 37) found that a single missense mutation located in transmembrane segment IS6 (G369D) increased, and two other (L1068P and S229P) missense mutations decreased, Cav1.4 channel activity (37). We note that the visual phenotype of affected male individuals in the present New Zealand family had similarities to CSNB2 (13). Detailed examinations of heterozygote females will be required in future genotype:phenotype studies of CACNA1F mutations.
If the CACNA1F gene is subject to X-inactivation and there is no selection bias, female heterozygote individuals should show cellular mosaicism for healthy/mutant Cav1.4 channels (38, 39). Conversely, if CACNA1F is not subject to X-inactivation, both healthy and mutant Cav1.4 channels may be expressed within individual retinal cells. In the present family, all heterozygote female individuals examined had a visual phenotype that was similar to, but less severe than, hemizygote male individuals (13). By comparison, in CSNB2, female heterozygotes do not show clinical signs, whereas rare female homozygotes show a phenotype similar to hemizygote males (6). These clinical findings suggest that the I745T CACNA1F allele acts in a codominant manner, whereas CACNA1F alleles responsible for CSNB2 involve a recessive mechanism.
In the present family, five male individuals with visual impairment had unexplained intellectual impairment; three of these five individuals also were autistic. These five individuals belonged to multiple branches of the pedigree, and there were no individuals with healthy vision who had these signs (13). These observations imply that either the I745T CACNA1F mutation itself, or a damaged gene located nearby on chromosome X, conferred susceptibility to intellectual impairment and autism. The former explanation is supported by the recent finding that Timothy syndrome, a multisystem disorder that includes cognitive abnormalities and autism, is caused by a de novo mutation G406R in transmembrane segment IS6 of CACNA1C. The mutation results in a substantial reduction in voltage-dependent inactivation (40). Thus, abnormal Ca2+ entry via Cav1.4 in the disorder described here and via Cav1.2 in Timothy syndrome may be caused by a similar molecular mechanism (i.e., loss of inactivation induced by a single point mutation in a pore-lining S6 segment). Expression of CACNA1F/Cacna1f in brain was not detected in several studies (2, 3, 9, 32), but it remains possible that expression may occur in a spatially and/or temporally restricted manner. Consistent with the presence of Cacna1f expressed sequence tags in a rat pineal library (www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unigene), this study found CACNA1F/Cacna1f transcripts in the pineal gland (see Fig. 6). A major role of the pineal gland is production of melatonin, a hormone with effects on circadian rhythms (41), sleep (41), and epilepsy (42). A role for Cav1.4 channel activity in pineal gland function remains to be investigated. Nocturnal production of melatonin is, however, reduced in autistic children and adolescents, implying that pineal gland dysfunction may contribute to, or arise from, the disease (43).
The molecular basis for the alterations in Cav1.4 channel function associated with replacement of isoleucine, a nonpolar residue, by threonine, a smaller, polar residue, remains to be elucidated. Residue 745 is located toward the intracellular end of transmembrane segment IIS6 (44). Amino acids in the S6 segment have previously been implicated in the gating of voltagegated ion channels (44–49). Activation of voltage-gated ion channels is commonly described by a three-state model
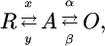 |
where R represents the resting (closed) state, A represents the activated (still closed) state, and O represents the open (conducting) state (see ref. 50 for details). The R-to-A transition describes the movement of the voltage sensing machinery with voltage-dependent rate constants x and y and the A-to-O transition the pore opening with voltage-independent rate constants α and β. The I745T Cav1.4 channel showed a leftward shift in the voltage dependence of activation (Figs. 2 and 3). In the frame of the three-state model, changes in the voltage-independent rate constants (increase in α and decrease in β) will induce leftward shifts in channel activation (48–50). It cannot be decided whether I745T stabilizes the open state (decrease of β), destabilizes the closed activated state (increase of α), or both. The reduction in I745T Cav1.4 channel inactivation (Fig. 4), implying reduced transition from the open to the inactivated state, is consistent with the idea that I745T stabilizes the open channel conformation (49). An activation model with two transitions predicts two time constants of activation. The monoexponential activation observed in wild-type Cav1.4 channels may reflect masking of biexponential activation by inactivation, i.e., state transitions from the open to the inactivated state. The biexponential time course in I745T Cav1.4 channels (Fig. 2 e and f) may be evident because of the substantially reduced inactivation associated with the mutation (compare Fig. 4 a with b). Taken together, I745T appears to reduce the energy required for channel opening. Further studies will clarify the mechanism by which this mutation in the pore region interferes with the voltage-sensing machinery.
Supplementary Material
Acknowledgments
This work was carried out in partnership with the family affected by this disorder. We thank the following individuals for materials and/or advice: Torben Bech-Hansen (CACNA1F primers), Tony Merriman (human control DNA samples), Mark Austin (human pineal gland RNA), Ian McLennan and Ken Turner (tissue preparation), Grant Jacobs (bioinformatics), and Eugen Timin (biophysics). This work was supported by the New Zealand Lottery Grants Board and the Health Research Council of New Zealand, the European Community (HPRN-CT-2000–00082), and Austrian Science Fund (FWF) Grants P15914-B05, P15527, and P17109. A.H.-W. received support from an Otago University Postgraduate Maori Award, the MoRST Mauriora ki te ao Scholarship for Maori in Science, the Te Riutoto Aihe Whanau Trust Board, the Tainui Maori Trust Board Tumate Mahuta Education Grant, the Tanetinorau Opataia Whanau Trust Board, and Retina New Zealand. G.M.C. received support from the Sir William and Lady Stevenson Trust.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Catterall, W. A. (2000) Annu. Rev. Cell Dev. Biol. 16, 521–555. [DOI] [PubMed] [Google Scholar]
- 2.Bech-Hansen, N. T., Naylor, M. J., Maybaum, T. A., Pearce, W. G., Koop, B., Fishman, G. A., Mets, M., Musarella, M. A. & Boycott, K. M. (1998) Nat. Genet. 19, 264–267. [DOI] [PubMed] [Google Scholar]
- 3.Strom, T. M., Nyakatura, G., Apfelstedt-Sylla, E., Hellebrand, H., Lorenz, B., Weber, B. H., Wutz, K., Gutwillinger, N., Ruther, K., Drescher, B., et al. (1998) Nat. Genet. 19, 260–263. [DOI] [PubMed] [Google Scholar]
- 4.Miyake, Y., Yagasaki, K., Horiguchi, M., Kawase, Y. & Kanda, T. (1986) Arch. Ophthalmol. 104, 1013–1020. [DOI] [PubMed] [Google Scholar]
- 5.Tremblay, F., Laroche, R. G. & de Becker, I. (1995) Vision Res. 35, 2383–2393. [DOI] [PubMed] [Google Scholar]
- 6.Boycott, K. M., Pearce, W. G. & Bech-Hansen, N. T. (2000) Can. J. Ophthalmol. 35, 204–213. [DOI] [PubMed] [Google Scholar]
- 7.Langrova, H., Gamer, D., Friedburg, C., Besch, D., Zrenner, E. & Apfelstedt-Sylla, E. (2002) Vision Res. 42, 1475–1483. [DOI] [PubMed] [Google Scholar]
- 8.Ball, S. L., Powers, P. A., Shin, H. S., Morgans, C. W., Peachey, N. S. & Gregg, R. G. (2002) Invest. Ophthalmol. Vis. Sci. 43, 1595–1603. [PubMed] [Google Scholar]
- 9.Morgans, C. W., Gaughwin, P. & Maleszka, R. (2001) Mol. Vision 7, 202–209. [PubMed] [Google Scholar]
- 10.Berntson, A., Taylor, W. R. & Morgans, C. W. (2003) J. Neurosci. Res. 71, 146–151. [DOI] [PubMed] [Google Scholar]
- 11.Attwell, D., Borges, S., Wu, S. M. & Wilson, M. (1987) Nature 328, 522–524. [DOI] [PubMed] [Google Scholar]
- 12.Witkovsky, P., Schmitz, Y., Akopian, A., Krizaj, D. & Tranchina, D. (1997) J. Neurosci. 17, 7297–7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hope, C. I., Sharp, D. M., Hemara-Wahanui, A., Sissingh, J. I., Lundon, P., Mitchell, E. A., Maw, M. A. & Clover, G. M. (2005) Clin. Exp. Ophthalmol. 33, 129–136. [DOI] [PubMed] [Google Scholar]
- 14.Rigaudiere, F., Roux, C., Lachapelle, P., Rosolen, S. G., Bitoun, P., Gay-Duval, A. & Le Gargasson, J.-F. (2003) Doc. Ophthalmol. 107, 203–212. [DOI] [PubMed] [Google Scholar]
- 15.Dib, C., Faure, S., Fizames, C., Samson, D., Drouot, N., Vignal, A., Millasseau, P., Marc, S., Hazan, J., Seboun, E., et al. (1996) Nature 380, 152–154. [DOI] [PubMed] [Google Scholar]
- 16.Boycott, K. M., Pearce, W. G., Musarella, M. A., Weleber, R. G., Maybaum, T. A., Birch, D. G., Miyake, Y., Young, R. S. & Bech-Hansen, N. T. (1998) Am. J. Hum. Genet. 62, 865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boycott, K. M., Maybaum, T. A., Naylor, M. J., Weleber, R. G., Robitaille, J., Miyake, Y., Bergen, A. A., Pierpont, M. E., Pearce, W. G. & Bech-Hansen, N. T. (2001) Hum. Genet. 108, 91–97. [DOI] [PubMed] [Google Scholar]
- 18.Koschak, A., Reimer, D., Walter, D., Hoda, J. C., Heinzle, T., Grabner, M. & Striessnig, J. (2003) J. Neurosci. 23, 6041–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horton, R. M., Hunt, H. D., Ho, S. N., Pullen, J. K. & Pease, L. R. (1999) Gene 77, 61–68. [DOI] [PubMed] [Google Scholar]
- 20.Ellis, S. B., Williams, M. E., Ways, N. R., Brenner, R., Sharp, A. H., Leung, A. T., Campbell, K. P., McKenna, E., Koch, W. J., Hui, A., et al. (1988) Science 241, 1661–1664. [DOI] [PubMed] [Google Scholar]
- 21.Castellano, A., Wei, X., Birnbaumer, L. & Perez-Reyes, E. (1993) J. Biol. Chem. 268, 12359–12366. [PubMed] [Google Scholar]
- 22.Hamill, O. P., Marty, A., Neher, E., Sakmann, B. & Sigworth, F. J. (1981) Pflügers Arch. 391, 85–100. [DOI] [PubMed] [Google Scholar]
- 23.Austin, M. C. & O'Donnell, S. M. (1999) J. Neurochem. 72, 2065–2073. [DOI] [PubMed] [Google Scholar]
- 24.Groves, A. K. & Bronner-Fraser, M. (2000) Development (Cambridge, U.K.) 127, 3489–3499. [DOI] [PubMed] [Google Scholar]
- 25.Bech-Hansen, N. T., Naylor, M. J., Maybaum, T. A., Sparkes, R. L., Koop, B., Birch, D. G., Bergen, A. A., Prinsen, C. F., Polomeno, R. C., Gal, A., et al. (2000) Nat. Genet. 26, 319–323. [DOI] [PubMed] [Google Scholar]
- 26.Pusch, C. M., Zeitz, C., Brandau, O., Pesch, K., Achatz, H., Feil, S., Scharfe, C., Maurer, J., Jacobi, F. K., Pinckers, A., et al. (2000) Nat. Genet. 26, 324–327. [DOI] [PubMed] [Google Scholar]
- 27.Schwahn, U., Lenzner, S., Dong, J., Feil, S., Hinzmann, B., van Duijnhoven, G., Kirschner, R., Hemberger, M., Bergen, A. A., Rosenberg, T., et al. (1998) Nat. Genet. 19, 327–332. [DOI] [PubMed] [Google Scholar]
- 28.Shapiro, M. B. & Senapathy, P. (1987) Nucleic Acids Res. 15, 7155–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berjukow, S., Doring, F., Froschmayr, M., Grabner, M., Glossmann, H. & Hering, S. (1996) Br. J. Pharmacol. 118, 748–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furukawa, T., Morrow, E. M., Li, T., Davis, F. C. & Cepko, C. L. (1999) Nat. Genet. 23, 466–470. [DOI] [PubMed] [Google Scholar]
- 31.Letz, B., Schomerus, C., Maronde, E., Korf, H.-W. & Korbmacher, C. (1997) J. Physiol. 499, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McRory, J., Hamid, J., Doering, C. J., Garcia, E., Parker, R., Hamming, K., Chen, L., Hildebrand, M., Beedle, A. M., Feldcamp, L., et al. (2004) J. Neurosci. 24, 1707–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sampath, A. P. & Rieke, F. (2004) Neuron 41, 431–443. [DOI] [PubMed] [Google Scholar]
- 34.Berntson, A. & Taylor, W. R. (2003) Vis. Neurosci. 20, 621–626. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura, M., Ito, S., Terasaki, H. & Miyake, Y. (2001) Invest. Ophthalmol. Vis. Sci. 42, 1610–1616. [PubMed] [Google Scholar]
- 36.Zito, I., Allen, L. E., Patel, R. J., Meindl, A., Bradshaw, K., Yates, J. R., Bird, A. C., Ersine, L., Cheetham, M. E., Webster, A. R., et al. (2003) Hum. Mutat. 21, 169. [DOI] [PubMed] [Google Scholar]
- 37.Hoda, J.-C., Zaghetto, F., Koschak, A. & Striessnig, J. (2005) J. Neurosci. 25, 252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reese, B. E., Necessary, B. D., Tam, P. P. L., Faulkner-Jones, B. & Tan, S.-S. (1999) Eur. J. Neurosci. 11, 2965–2978. [DOI] [PubMed] [Google Scholar]
- 39.Smallwood, P. M., Olveczky, B. P., Williams, G. L., Jacobs, G. H., Reese, B. E., Meister, M. & Nathans, J. (2003) Proc. Natl. Acad. Sci. USA 100, 11706–11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Splawski, I., Timothy, K. W., Sharpe, L. M., Decher, N., Kumar, P., Bloise, R., Napolitano, C., Schwartz, P. J., Joseph, R. M., Condouris, K., et al. (2004) Cell 119, 19–31. [DOI] [PubMed] [Google Scholar]
- 41.Arendt, J. & Skene, J. D. (2005) Sleep Med. Rev. 9, 25–39. [DOI] [PubMed] [Google Scholar]
- 42.Musshoff, U. & Speckmann, E.-J. (2003) Life Sci. 73, 2603–2610. [DOI] [PubMed] [Google Scholar]
- 43.Tordjman S., Anderson, G. M., Pichard, N., Charbuy, H. & Touitou, Y. (2005) Biol. Psychiatry 57, 134–138. [DOI] [PubMed] [Google Scholar]
- 44.Lipkind, G. M. & Fozzard, H. A. (2001) Biochemistry 40, 6786–6794. [DOI] [PubMed] [Google Scholar]
- 45.Kraus, R. L., Sinnegger, M. J., Koschak, A., Glossmann, H., Stenirri, S., Carrera, P. & Striessnig, J. (2000) J. Biol. Chem. 275, 9239–9243. [DOI] [PubMed] [Google Scholar]
- 46.Stotz, S. C. & Zamponi, G. W. (2001) J. Biol. Chem. 276, 33001–33010. [DOI] [PubMed] [Google Scholar]
- 47.Shi, C. & Soldatov, N. M. (2002) J. Biol. Chem. 277, 6813–6821. [DOI] [PubMed] [Google Scholar]
- 48.Yifrach, O. & MacKinnon, R. (2002) Cell 111, 231–239. [DOI] [PubMed] [Google Scholar]
- 49.Zhao, Y., Yarov-Yarovoy, V., Scheuer, T. & Catterall, W. A. (2004) Neuron 41, 859–865. [DOI] [PubMed] [Google Scholar]
- 50.Zagotta, W. N., Hoshi, T., Dittman, J. & Aldrich, R. W. (1994) J. Gen. Physiol. 103, 279–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}