

Abstract
The structural determinants of nicotinic acetylcholine receptor (AChR) trafficking have yet to be fully elucidated. Hydrophobic residues occur within short motifs important for endoplasmic reticulum (ER) export or endocytotic trafficking. Hence, we tested whether highly conserved hydrophobic residues, primarily leucines, in the cytoplasmic domain of the α4β2 AChR subunits were required for cell surface expression of α4β2 AChRs. Mutation of F350, L351, L357, and L358 to alanine in the α4 AChR subunit attenuates cell surface expression of mutant α4β2 AChRs. Mutation of F342, L343, L349, and L350 to alanine at homologous positions in the β2 AChR subunit abolishes cell surface expression of mutant α4β2 AChRs. The hydrophobic nature of the leucine residue is a primary determinant of its function because mutation of L343 to another hydrophobic amino acid, phenylalanine, in the β2 AChR subunit only poorly inhibits trafficking of mutant α4β2 AChR to the cell surface. All mutant α4β2 AChRs exhibit high-affinity binding for [3H]epibatidine. In both tsA201 cells and differentiated SH-SY5Y neural cells, wild-type α4β2 AChRs colocalize with the Golgi marker giantin, whereas mutant α4β2 AChRs fail to do so. The striking difference between mutant α4 versus mutant β2 AChR subunits on cell surface expression of mutant α4β2 AChRs points to a cooperative or regulatory role for the α4 AChR subunit and an obligatory role for the β2 AChR subunit in ER export. Collectively, our results identify, for the first time, residues within AChR subunits that are essential structural determinants of α4β2 AChR ER export.
Keywords: cargo, export, endoplasmic reticulum, Golgi, nicotine, trafficking
Introduction
Neuronal acetylcholine receptors (AChRs) are a family of ACh-gated ion channels assembled from different combinations of α2-α10 and β2-β4 subunits (Millar, 2003). The α4β2 AChRs represent >80% of the [3H]nicotine and [3H]epibatidine binding sites in mammalian brain (Whiting and Lindstrom, 1988). β2 AChR subunit gene knock-out mice show little [3H]nicotine binding in their brains, lose their sensitivity to nicotine in passive avoidance tasks (Picciotto et al., 1995), and show attenuated self-administration of nicotine (Picciotto et al., 1998). α4 AChR subunit gene knock-out mice also lack [3H]nicotine binding sites and exhibit reduced antinociceptive effects of nicotine (Marubio et al., 1999) and loss of tonic control of striatal basal dopamine release (Marubio et al., 2003). Using a knock-in mouse expressing α4β2 AChRs hypersensitive to nicotine, α4β2 AChRs were shown to mediate the essential features of nicotine-addiction, including reward, tolerance, and sensitization (Tapper et al., 2004). α4β2 AChRs have important roles in other neurological diseases because some human epilepsies are caused by mutations within the α4 subunit or the β2 subunit (Raggenbass and Bertrand, 2002) and because significant loss of α4β2 AChRs may contribute to the cognitive deficits in Alzheimer's disease (Nordberg et al., 1992; Court and Clementi, 1995).
The ontogeny of AChRs remains to be fully elucidated. The archetypical muscle-type AChR subunits are translated by endoplasmic reticulum (ER) membrane-associated ribosomal complexes and cotranslationally inserted into the ER membrane via the translocon (Johnson and van Waes, 1999; Alder and Johnson, 2004). The nascent subunit polypeptide subsequently undergoes cleavage of its signal sequence, oxidation of its disulfide bonds (Mishina et al., 1985; Blount and Merlie, 1990), and N-glycosylation of specific residues (Merlie et al., 1982; Smith et al., 1987; Blount and Merlie 1988). Chaperones such as binding protein BiP (Blount and Merlie, 1991; Paulson et al., 1991; Forsayeth et al., 1992) and calnexin (Gelman et al., 1995; Keller et al., 1996, 1998) promote proper folding and maturation of AChR subunits. Unassembled or misfolded AChR subunits are then targeted for degradation by the ER-associated proteasomal degradation machinery (Wanamaker et al., 2003). AChRs are exported in coat protein complex II (COPII) vesicles from the ER to the Golgi presumably after masking of specific ER retention signals (Keller et al., 2001; Wang et al., 2002) and recognition of ER export motifs in properly folded and oligomerized AChR subunits.
Several different trafficking motifs have been identified to date (Nishimura and Balch, 1997; Barlowe, 2003; Robinson, 2004), but little is known about the location or nature of such motifs in AChR subunits. Alignment of AChR subunit sequences shows hydrophobic residues in their large cytoplasmic domain that are highly conserved. Such hydrophobic residues occur within short motifs important for either cargo selection into COPII-coated vesicles for ER export (Duncan and Payne, 2003) or cargo selection into clathrin-coated vesicles for endocytotic trafficking (Aridor and Traub, 2002). We investigated the role of these residues in the α4 and β2 AChR subunits and provide evidence that they are structural determinants of AChR ER export.
Materials and Methods
Generation of constructs and mutants. All constructs were made by PCR using appropriate pairs of forward and reverse synthetic oligonucleotide primers (Life Technologies, Bethesda, MD) and Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA). Rat α3, α4, β2, and β4 AChR subunit cDNAs were cloned into the EcoRI-XbaI sites of the mammalian cell expression vector PEF6/myc-His A. Mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene). For mutagenesis of the α4 AChR subunit, the following primer pairs used were: 5′-GCC TGG GTG CGT AGA GTC GCC CTG GAC ATC GTG CC-3′ and 5′-GG CAC GAT GTC CAG GGC GAC TCT ACG CAC CCA GGC-3′ for F350A; 5′-GTG CGT AGA GTC TTC GCG GAC ATC GTG CCT CGC-3′ and 5′-GCG AGG CAC GAT GTC CGC GAA GAC TCT ACG CAC-3′ for L351A; 5′-GAC ATC GTG CCT CGC GCA GCC TTC ATG AAG CGC CCC-3′ and 5′-GGG GCG CTT CAT GAA GGC TGC GCG AGG CAC GAT GTC-3′ for L357A,L358A. The triple mutant L351A,L357A,L358A in the α4 AChR subunit was generated using primers for the L351A mutation and the plasmid DNA template containing the L357A,L358A mutations. For mutagenesis of the β2 AChR subunit, the following primers pairs were used: 5′-TGG GTC AAG GTG GTC GCC CTG GAG AAG CTG CCC-3′ and 5′-GGG CAG CTT CTC CAG GGC GAC CAC CTT GAC CCA-3′ for F342A; 5′-GTC AAG GTG GTC TTC GCG GAG AAG CTG CCC ACC-3′ and 5′-GGT GGG CAG CTT CTC CGC GAA GAC CAC CTT GAC-3′ for L343A; 5′-GTC AAG GTG GTC TTC TTC GAG AAG CTG CCC ACC-3′ and 5′-GGT GGG CAG CTT CTC GAA GAA GAC CAC CTT GAC-3′ for L343F; 5′-GAG AAG CTG CCC ACC GCG GCC TTC CTG CAG CAG CCA-3′ and 5′-TGG CTG CTG CAG GAA GGC CGC GGT GGG CAG CTT CTC-3′ for L349, 350A; 5′-GAG AAG CTG CCC ACC GCG CTC TTC CTG CAG CAG-3′ and 5′-CTG CTG CAG GAA GAG CGC GGT GGG CAG CTT CTC-3′ for L349A; 5′-G AAG CTG CCC ACC CTG GCC TTC CTG CAG CAG CCA C-3′and 5′-G TGG CTG CTG CAG GAA GGC CAG GGT GGG CAG CTT C-3′ for L350A. The underlined bases indicate mismatches used to generate the appropriate codons for the mutants. The N-terminal FLAG-tagged α4 construct was generated by subcloning the cDNA sequence corresponding to the mature α4 subunit (amino acids 35-630) into an expression vector containing influenza hemagglutinin signal sequence and the FLAG epitope (DYKDDDDA) (Guan et al., 1992).
Antibodies. Monoclonal antibodies (mAbs) against the α4 AChR subunit (mAb 299), the β2 AChR subunit (mAb 295), and the antiserum against the β2 AChR subunit (C-20; Santa Cruz Biotechnology, Santa Cruz, CA) that binds to denatured β2 subunits on immunoblots were used. The goat anti-rat and anti-rabbit horseradish peroxidase-conjugated secondary Abs were obtained from Pierce (Rockford, IL). The Alexa Fluor 488- and Alexa Fluor 647-conjugated goat anti-rat and anti-rabbit secondary Abs were obtained from Molecular Probes (Eugene, OR).
Expression of recombinant α4β2 AChRs in human embryonic kidney tsA201 and human neuroblastoma SH-SY5Y cells. Human tsA201 cells, a derivative of the human embryonic kidney cell line 293, were cultured at 37°C in a humidified atmosphere of 5% CO2 in either 12- or 24-well plates in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum. Cells at 90% confluency were transfected with various combinations of cDNAs using Lipofectamine 2000 (Invitrogen) per the instructions of the manufacturer and typically used after incubation at 30°C for 48 h or additional incubation at 37°C for another 1-2 h. The levels of wild-type α4β2 AChRs in tsA201 cells were reduced by replating after trypsinization transfected cells with nontransfected cells at ratios of 1:1, 1:2, and 1:5, respectively, or by transfecting lower amounts of total DNA at much reduced levels of cDNA mixes of 1/64 and 1/128 the normal amount. Human neuroblastoma SH-SY5Y cells were cultured at 37°C in a humidified atmosphere of 5% CO2 in Eagle's Minimum Essential Medium (EMEM) (Invitrogen) supplemented with 10% fetal bovine serum. Cells were differentiated in serum-free EMEM media with N2 supplement (Invitrogen) and then transfected with various combinations of cDNAs using Lipofectamine 2000.
Enzyme-linked immunoassay for quantitating cell surface α4β2 AChRs. Cell surface α4β2 AChRs were measured using the methodology described previously (Jeanclos et al., 2001). Transfected tsA201 cells plated in 12-well dishes were washed once in PBS and fixed with 3% paraformaldehyde for 5 min at room temperature. After four washes with PBS, the cells were blocked with 3% BSA in PBS for 30 min and then incubated for 1 h at room temperature with an mAb against the β2 AChR subunit (mAb 295) whose epitope is located in the extracellular domains of the subunit. After six washes with PBS, the cells were incubated with horseradish peroxidase-conjugated goat anti-rat secondary Ab for 1 h, washed eight times with PBS, and finally incubated with the HRP substrate 3,3′,5,5′-tetramethylbenzidine (Sigma, St. Louis, MO) for 30 min. The absorbance of the supernatant was then measured at 655 nm in a Beckman Instruments (Fullerton, CA) spectrophotometer. The nonspecific background binding of Abs to cells was determined using nontransfected cells and was typically <0.5% of the total binding observed for wild-type α4β2 AChRs.
Detergent solubilization of cellular proteins. Transfected tsA201 cells were homogenized by sonication (using five 5 s bursts) in ice-cold lysis buffer (50 mm NaCl, 5 mm Tris, pH 8.0, 5 mm EGTA, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 1 mm benzamidine, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 5 μg/ml pepstatin, 1 mm N-ethylmaleimide, and 2% NP-40), and the proteins were solubilized by gently agitating the cells for 2 h at 4°C. Typically, 1 × 106 cells were lysed in 400 μl of lysis buffer. The clear supernatant of solubilized proteins used in the analysis was obtained after centrifugation at 18,000 × g for 15 min. Proteins were quantitated using the Bio-Rad (Hercules, CA) Dc assay, and protein samples were boiled with sample buffer and separated by SDS-PAGE.
Immunoblot analysis. Proteins were transferred onto polyvinylidene difluoride membrane (IMMUN-BLOT; Bio-Rad) and incubated with diluted rat Ab against the α4 AChR subunit (mAb 299) or the goat polyclonal Ab against the β2 AChR subunit (C-20) in PBS containing 5% nonfat milk powder. Binding of the primary Abs to proteins was detected using Alexa Fluor 680-conjugated secondary Abs (Molecular Probes). Fluorescent signals were detected using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE). Typically, the scan times were adjusted to prevent saturation of fluorescent signals on the detector. The intensities of the fluorescent bands were then quantitated using the Odyssey image analysis software.
Immunostaining of α4β2 AChRs. For immunostaining of cell surface α4β2 AChRs, transfected cells were diluted (1:5), replated on 12 mm circular glass coverslips (Fisher Scientific, Pittsburg, PA), and cultured for an additional 32 h in 24-well dishes. For immunostaining of cell surface α4β2 AChRs in SH-SY5Y cells, cells were cultured in EMEM media containing 10% fetal bovine serum, changed to serum-free EMEM with N2 supplements next day, and then incubated for 24 h. The cells were then transfected with various combinations of plasmids and incubated at 30°C for 48 h or then shifted to 37°C for another 1-2 h. Transfected cells were then fixed with 3% paraformaldehyde in PBS for 5 min at room temperature. After four washes with PBS, cells were blocked with 3% BSA in PBS for 30 min. Cells were then incubated with the Ab against the β2 AChR subunit (mAb 295; 1:1000 dilution) and then, after three washes with PBS, incubated with Alexa Fluor 488-conjugated goat anti-rat secondary Ab (1:1000 dilution). For immunostaining of intracellular α4β2 AChRs, transfected tsA201 as well as SH-SY5Y cells were fixed and then permeabilized with 0.1% Triton X-100 in PBS containing 3% BSA for 30 min at room temperature. The subcellular distribution of α4β2 mutants with respect to the ER network was determined by costaining with a polyclonal rabbit antiserum to calnexin (1:1000 dilution; Stressgen, Victoria, British Columbia, Canada), and its binding was then detected with an Alexa Fluor 647-conjugated goat anti-rabbit secondary Ab (1:1000 dilution). After a temperature shift to 37°C for 1-2 h, the subcellular distribution of α4β2 AChRs in the Golgi complex was detected by costaining with a polyclonal rabbit antiserum to giantin (1:1000 dilution; Covance, Princeton, NJ), and its binding was then visualized with an Alexa Fluor 647-conjugated goat anti-rabbit secondary Ab (1:1000 dilution). As controls, nontransfected cells were subject to the same immunohistochemical process. Additionally, transfected cells were also incubated with secondary antibodies in the absence of the primary antibody. No positive staining was found in these controls (data not shown).
Fluorescence microscopy. Cells were visualized using a deconvolution microscope (Zeiss, Oberkochen, Germany) with ultraviolet illumination from a 50 W xenon arc through a 40× Zeiss fluorescence oil immersion objective (numerical aperture 1.3), and images were captured using the SlideBook software (Intelligent Imaging Innovations, Denver, CO) using 5 μm steps in the z-axis, alternating between FITC and cyanine 5 (Cy5) filters. For image analysis, the captured images were pseudo-colored and processed using Adobe Photoshop software (Adobe Systems, San Jose, CA).
Ligand-binding assays. [3H]Epibatidine binding to α4β2 AChRs was done directly in wells containing cells that were gently fixed with paraformaldehyde. Under these conditions of fixation, the binding affinity of α4β2 AChRs for [3H]epibatidine was not significantly different from that of α4β2 AChRs in normal cells (our unpublished data). Transfected tsA201 cells were fixed with 2% paraformaldehyde diluted with DMEM containing 10% fetal bovine serum for 1 h at room temperature. After four washes with PBS, cells were permeabilized with 0.25% Triton X-100 in PBS for 30 min at room temperature. For saturation binding experiments, permeabilized cells were incubated with 3 nm [3H]epibatidine overnight at 4°C. For inhibition experiments, cells were incubated with 400 pm [3H]epibatidine in the presence or absence of 10, 30, 100, 300, or 1000 nm nicotine. After four washes with PBS containing 0.25% Triton X-100, the bound epibatidine was eluted with a buffer containing 0.5% SDS and 0.5 m NaOH for 1 h. The eluates were then mixed with liquid scintillation fluid, and the amount of radioactivity present was measured with a Winspectral liquid scintillation counter (Wallac, Turku, Finland).
Data analysis. The Hill-type equation was fitted to the nicotine concentration inhibition of [3H]epibatidine binding with a nonlinear least-squares error curve fit method (KaleidaGraph; Abelbeck Software, Reading, PA): I(x) = Imax[xnH/(xnH + IC50nH)], where I(x) is the amount of [3H]epibatidine bound at concentration x of nicotine, Imax is the maximal number of [3H]epibatidine binding sites in the absence of nicotine (set at 100%), IC50 is the nicotine concentration required for half-maximal inhibition, and nH is the Hill coefficient.
Results
Alignment of the first 30 amino acid residues of the long cytosolic loop of AChR subunits reveals highly conserved leucine residues among all AChR subunits (Fig. 1). Such leucine residues occur within short motifs important for cargo selection into COPII-coated vesicles that transport proteins from the ER to the Golgi (for review, see Duncan and Payne, 2003). They also occur in sequences of integral membrane proteins that interact with adaptor proteins (such as AP-1) that select their cargo for endocytosis within clathrin-coated vesicles (for review, see Aridor and Traub, 2002). However, exocytotic or endocytotic motifs that had been described previously were not readily identifiable within this region. Hence, to determine whether these conserved hydrophobic residues in the α4 and β2 AChR subunits were part of a trafficking motif, we mutated them to alanine or other residues and studied the effect of the mutations on the cell surface expression levels of the mutant α4β2 AChRs. Table 1 lists all of the α4 AChR and β2 AChR subunit mutants used in this study and a summary of the experimental results obtained.
Figure 1.

Sequence homology among the long cytosolic loop of AChR subunits. Topology of AChR subunits and alignment of a 20 amino acid region of the long cytosolic loop showing location of hydrophobic residues that are conserved in the ER export motif identified in this study. The boxed residues correspond to those that are at homologous positions in all of the AChR subunits and those that were specifically mutated in the α4 (F350, L351, L357, and L358) and β2 (F342, L343, L349, L350) AChR subunits.
Table 1.
Properties of AChR subunit mutants
|
AChR subunit |
% Subunit expression |
% Epibatidine binding |
% Surface expression |
|---|---|---|---|
| α4 + β2 wild type | 100 | 100 | 100 |
| α4F350A | 100 | 112 ± 4** | 70 ± 3.5** |
| α4L351A | 99 | 88 ± 7* | 46 ± 2** |
| α4L357,358A | 60 | 68 ± 4** | 31 ± 7** |
| α4L351,357,358A | 52 | 68 ± 10** | 7 ± 1** |
| β2F342A | 76 | 39 ± 9** | 0.7 ± 0.2** |
| β2L343A | 93 | 54 ± 9** | 0.7 ± 0.4** |
| β2L349,350A | 38 | 38 ± 7** | 0.6 ± 0.1** |
| β2L349A | 66 | 53 ± 5** | 7 ± 0.5** |
| β2L350A
|
67 |
48 ± 8**
|
0.9 ± 0.5**
|
The numbering system used to designate the amino acids is for the full-length AChR subunits, including the leader peptide. *p<0.05; **p<0.001.
Mutations of conserved hydrophobic residues within the α4 AChR subunit attenuate cell surface expression of AChRs
The wild-type α4 AChR subunit, or each of the mutant α4 AChR subunits α4F350A, α4L351A, α4L357,358A, and α4L351,L357,358A, was coexpressed with the wild-type β2 AChR subunit in tsA201 cells. Pairs of samples generated from independently transfected wells were analyzed to account for small variations generated by differences in transfection efficiency. The expression level of the α4 AChR subunits was monitored by immunoblot analysis of NP-40-solubilized protein extracts, separated by SDS-PAGE using an Ab against the α4 AChR subunit. The result of a representative experiment is shown in Figure 2A. Small changes in the overall steady-state level of the mutant α4 AChR subunits were observed (Fig. 2A). The steady-state level of the α4 AChR subunit expressed alone (lanes 11, 12) was comparable with that of the α4 AChR subunit coexpressed with the β2 AChR subunit (lanes 1, 2), suggesting that the stability of α4 AChR subunits was not significantly influenced by subunit assembly. No cross-reactive band was obtained in extracts of tsA201 cells expressing the β2 AChR subunit alone, showing that the binding of the α4Abwas specific (lane 13).
Figure 2.

Subunit expression and assembly of high-affinity [3H]epibatidine binding sites by wild-type and mutant α4 AChR subunits. A, The α4, α4F350A, α4L351A, α4L357, 358A, or α4L351, 357, 358A AChR subunit was coexpressed with the wild-type β2 AChR subunit in tsA201 cells, and 2% NP-40-solubilized membrane proteins were subjected to immunoblot analysis using an mAb against the α4 AChR subunit (mAb 299). Paired lanes represent the expression levels of the AChR subunit from two independently transfected wells from a representative experiment. B, Detergent-permeabilized transfected fixed cells expressing wild-type and mutant α4β2 AChRs were incubated with 3 nm [3H]epibatidine overnight at 4°C. The amount of bound [3H]epibatidine was quantitated by scintillation counting. Data for the mutants were expressed as a percentage of the [3H]epibatidine binding observed for the wild-type α4β2. The data represent the mean ± SE from four experiments.
The ability of the mutated α4 AChR subunit to assemble and form high-affinity ligand-binding sites was monitored using [3H]epibatidine binding assays. Ligand binding was performed as described in Materials and Methods using a saturating concentration of 3 nm [3H]epibatidine. Somewhat lower levels of total number of [3H]epibatidine sites were formed by all of the mutant α4β2 AChRs compared with the wild-type α4β2 AChRs (Fig. 2B; Table 1). The α4 AChR subunit expressed by itself failed to bind [3H]epibatidine.
The relative amount of AChRs expressed in cytosol versus the cell surface membrane was determined qualitatively by detecting the binding of the β2 AChR subunit Ab to AChRs in detergent-permeabilized versus nonpermeabilized cells using immunofluorescence microscopy (Fig. 3A). The relative level of α4β2 AChRs expressed on the cell surface membrane was also quantitated using an enzyme-linked immunoassay. Thus, although all four mutant α4β2 AChRs showed a significant decrease in their cell surface expression levels relative to that of the wild-type α4β2 AChR (Fig. 3B), the simultaneous mutations of all three conserved leucine residues in α4L351,L357,358A AChR was not sufficient to completely abolish cell surface expression. However, the magnitude of the decrease in the cell surface expression levels was significantly greater than the magnitude of the decrease in the total number of [3H]epibatidine binding sites formed by the mutant α4β2 AChRs (Table 1). These results suggest that the mutations primarily affect the transport of α4β2 AChRs to the cell surface membrane.
Figure 3.

Hydrophobic residue mutations in the α4 AChR subunit attenuate surface expression of α4β2 AChRs. A, Transfected tsA201 cells expressing wild-type or mutant α4β2 AChRs, or the α4 AChR subunit alone, were processed for immunohistochemistry and probed with an mAb against the β2 AChR subunit (mAb 295). Ab binding was detected using an Alexa 488-conjugated goat anti-rat secondary Ab and imaged with a deconvolution fluorescence microscope. Ab staining to intracellular α4β2 AChRs was detected in 0.1% Triton X-100-permeabilized cells. Shown are 1 μm optical sections of the cells. B, The cell surface expression of α4β2 AChRs was quantitated using an enzyme-linked colorimetric assay. The absorbance data were expressed as a percentage of the value obtained for the wild-type α4β2 AChRs. The data represents the mean ± SE from four experiments.
Mutation of conserved hydrophobic residues within the β2 AChR subunit abolishes cell surface expression of AChRs
We next determined the effects of mutating the conserved hydrophobic residues in the β2 AChR subunit, at positions that are homologous to the mutated positions in the α4 AChR subunit, on cell surface expression of α4β2 AChRs. The wild-type β2 AChR subunit, or each of the mutant β2 AChR subunits β2F342A, β2L343A, β2L349,350A, β2L349A, and β2L350A, was coexpressed with the wild-type α4 AChR subunit in tsA201 cells. As before, the expression levels of the β2 AChR subunits were monitored by immunoblot analysis using a polyclonal antiserum to the β2 AChR subunit. The result of a representative experiment is shown (Fig. 4A). Quantitation of the band intensities using the Odyssey imaging software suggests that the mutations also decreased the overall steady-state levels of the mutant β2 AChR subunits, particularly for those with multiple mutations (Table 1). The steady-state level of the β2 AChR subunit expressed alone (lanes 15, 16) was comparable with that of the β2 AChR subunit coexpressed with the α4 AChR subunit (lanes 1, 2), and, like the α4 AChR subunit, subunit assembly did not significantly influence its stability. No cross-reactive band was obtained in extracts of tsA201 cells expressing the α4 AChR subunit alone, indicating the relatively high binding specificity of the β2 Ab (lane 17).
Figure 4.

Subunit expression and assembly of high-affinity [3H]epibatidine binding sites by wild-type and mutant β2 AChR subunits. A, The β2, β2F342A, β2L343A, β2L349,350A, β2L349A, β2L350A, β2F342A, or β2L343F AChR subunit was coexpressed with the wild-type α4 AChR subunit in tsA201 cells, and 2% NP-40-solubilized membrane proteins were subjected to immunoblot analysis using a polyclonal antiserum against the β2 AChR subunit. Paired lanes represent the expression levels of the AChR subunit from two independently transfected wells from a representative experiment. B, Detergent-permeabilized transfected fixed cells expressing wild-type and mutant α4β2 AChRs, or the β2 AChR subunit alone, were incubated with 3 nm [3H]epibatidine overnight at 4°C. The amount of bound [3H]epibatidine was quantitated by scintillation counting. Data for the mutants were expressed as a percentage of the [3H]epibatidine binding observed for the wild-type α4β2. The data represent the mean ± SE from four experiments.
The ability of mutated β2 AChR subunits to form AChRs that bound cholinergic ligands with high affinity was monitored using [3H]epibatidine binding assays containing a saturating concentration of 3 nm [3H]epibatidine. Substantially lower levels of total numbers of [3H]epibatidine binding sites were formed by all of the mutant α4β2 AChRs compared with the wild-type α4β2 AChRs (Fig. 4B), and the decrease qualitatively matched the lower expression levels detected by immunoblot analysis (Table 1). In addition, we found that coexpression of the mutant α4L351,L357,358A AChR subunit with the mutant β2L349,350A AChR subunit did not further reduce the number of [3H]epibatidine binding sites formed (∼35%; data not shown) by the α4β2L349,350A AChR mutant. Overall, the [3H]epibatidine binding sites formed by β2 AChR subunit mutants were lower than those formed by the α4 AChR subunit mutants (Table 1).
The relative amount of AChRs expressed on the cell surface membrane was determined qualitatively using immunofluorescence microscopy (Fig. 5A). Importantly, very little binding of mAb 295 to cells expressing the β2 AChR subunit alone was observed, despite the fact that the immunoblot showed that its expression level is comparable with that of the assembled β2 AChR subunit. This result shows that high-affinity mAb 295 binding depends on conformational maturation promoted by subunit assembly. Quantitative results from the enzyme-linked immunoassay showed that, in contrast to the α4 AChR mutants, the β2 AChR subunit mutants virtually abolished cell surface expression of mutant α4β2 AChRs (Fig. 5B; Table 1). A very small, but detectable, level of mutant α4β2 AChRs surface expression was observed for the β2L349A AChR. This is consistent with a less conserved role for this leucine, relative to the other leucines, as deduced by the lack of sequence conservation at this position among the other AChR subunits (Fig. 1).
Figure 5.

Hydrophobic residue mutations in the β2 AChR subunit abolish surface expression of α4β2 AChRs. A, Transfected tsA201 cells expressing wild-type, mutant α4β2 AChRs, and the β2 AChR subunit alone were processed for immunohistochemistry and probed with an Ab against the β2 AChR subunit (mAb 295). Ab binding was detected using an Alexa 488-conjugated goat anti-rat secondary Ab and imaged with a deconvolution fluorescence microscope. Ab staining to α4β2 AChRs was detected in 0.1% Triton X-100-permeabilized cells before addition of Abs. Shown are 5 μm optical sections of the cells. B, The cell surface expression of α4β2 AChRs was quantitated using an enzyme-linked colorimetric assay. The data were expressed as a percentage of the value obtained for wild-type a4b2 AChRs. The data represent the mean ± SE from four experiments.
Several lines of evidence (Zwart and Vijverberg, 1998; Marks et al., 1999; Nelson et al., 2003; Zhou et al., 2003; Lopez-Hernandez et al., 2004) support the possibility that α4β2 AChRs, which represent most of the high-affinity nicotine binding sites in the brain, exist in at least two different stoichiometries, (α4)2(β2)3 and (α4)3(β2)2. To determine whether the differences in surface expression of mutant α4 versus the mutant β2 AChR subunits were attributable to differences in the number of subunits present in assembled α4β2 AChRs, we transfected cells with a subunit cDNA ratio of wild-type α4/mutant β2L343A of 3:1 rather than the usual 1:1 ratio and measured surface expression of AChRs. This ratio, which would be predicted to bias the stoichiometry in favor of assembly of (α4)3(β2)2 AChRs, still failed to rescue surface expression of the mutant α4β2 AChR (data not shown). Collectively, these results strongly support the notion that the highly conserved hydrophobic residues in the β2 AChR subunit play an obligatory role in the transport of α4β2 AChRs to the cell surface membrane.
The cell surface expression of the α4β2 AChR measured using the β2 Ab or other Abs correlate well with different expression levels of AChR
The overall levels of [3H]epibatidine binding sites formed by coassembly of mutant β2 AChR subunits with the α4 AChR subunit were significantly (∼35%) below that of the wild-type α4β2 AChR. To rule out the possibility that the failure to detect the mutant α4β2 AChRs formed by the mutant β2 AChR subunits was not a result of a detection threshold for the β2 Ab (mAb 295) used in all of the assays, we tested whether the β2 Ab could detect wild-type α4β2 AChRs at all levels of AChR expression observed in the experiments. Reduction of the α4β2 AChRs per well of cells was achieved by dilution of the total amount of cDNA mixture used to transfect cells. Extreme dilutions of 1/64 and 1/128 of the normal amount of total DNA used (0.8 μg/0.25 × 106 cells/well) were found to be necessary to achieve a significant reduction of ∼33 and ∼12%, respectively (data not shown). To stably and predictably achieve specific levels of reduction in the levels of α4β2 AChR per well, we also replated different fixed ratios (1:1, 1:2, and 1:5) of mixtures of tsA201 cells expressing α4β2 AChRs with those not expressing AChRs in each well and then assayed the surface levels of α4β2 AChRs per well under identical conditions. The Ab binding correlated quite well with the number of [3H]epibatidine binding sites measured in parallel assays under the same conditions. These values included those that were below the ∼35% expression levels of [3H]epibatidine binding sites formed by the α4β2L349,350A AChRs (Fig. 6A).
Figure 6.

Cell surface expression levels of wild-type and mutant α4β2 AChRs measured at different overall expression levels and using different Abs. A, Culture wells containing tsA201 cells expressing different levels of wild-type α4β2 AChRs were fixed and then incubated with 3 nm [3H]epibatidine overnight at 4°C. The amount of bound [3H]epibatidine was quantitated by scintillation counting. The cell surface expression levels of wild-type and mutant α4β2 AChRs was quantitated using an enzyme-linked colorimetric assay. The data represent the mean ± SD from four experiments. B, Transfected tsA201 cells expressing wild-type or mutant α4β2L349,350A AChRs were fixed and probed with mAb against the β2 AChR subunit (mAb 295) or mAb against α4 AChR subunit (mAb 299). The cell surface expression level of α4β2 AChRs was quantitated using an enzyme-linked colorimetric assay. The absorbance data were expressed as a percentage of the value obtained for the wild-type α4β2 AChR. The data represents the mean ± SE from four experiments. C, tsA201 cells were transfected with FLAGα4 and β2 or β2L349,350A subunits. The cell surface expression of α4β2 AChRs was quantitated using an enzyme-linked colorimetric assay with mAb against the β2 AChR subunit (mAb 295) or the M2 Ab against the FLAG epitope in the α4 subunit. The absorbance data were expressed as a percentage of the value obtained for the wild-type FLAGα4β2 AChR. The data represents the mean ± SE from four experiments.
The β2 Ab used to measure α4β2 AChR surface expression is a conformation-dependent Ab that preferably binds mature β2 AChR subunits in assembled α4β2 AChRs. Hence, to rule out the possibility that the mutations in the large cytoplasmic loop of the β2 AChR subunit did not affect the conformation of mutant α4β2 AChR such that the binding affinity of the β2 Ab for them was significantly reduced or abolished, we repeated the cell surface measurements with two other conformation-insensitive Abs. First, we used mAb 299, which binds to the α4 AChR subunit on immunoblots and is not expected to be conformation sensitive. Second, we used the M2 anti-FLAG Ab to measure the cell surface levels of an N-terminal FLAG-tagged α4 AChR subunit (FLAGα4) coassembled with the mutant β2L349,350A AChR subunit. We failed to detect either mutant α4β2L349,350A AChRs (Fig. 6B) or mutant FLAGα4b2 AChRs on the cell surface (Fig. 6C), despite robust expression of the respective wild-type α4β2 AChRs and FLAGα4β2 AChRs on the cell surface.
Mutant a4β2 AChRs do not exhibit significant differences in their ligand affinity
To assess whether the decrease in [3H]epibatidine binding sites formed by the mutant α4β2 AChRs was attributable to a change in their ligand affinity, we compared the relative affinity of mutant versus wild-type α4β2 AChR for nicotine in a competitive inhibition experiment. We used α4L351,L357,358A and β2L349,350A as representative α4 and β2 mutants, respectively, to determine the degree to which these mutations affected ligand affinity. Direct measurement of the Kd for [3H]epibatidine was found to be impractical because of experimental problems with ligand depletion at the extremely low concentrations of epibatidine needed to establish the lower binding values in saturation binding experiments. The data best fit a single-site Hill equation with KI values of 0.65 nm for the α4, 0.77 nm for the α4L351,L357,358A, and 0.74 nm for the β2L349,350A. The results show that the representative α4L351,L357,358A AChR and the β2L349,350A AChR mutants, which formed the lowest levels of total [3H]epibatidine binding sites, show similar affinities for nicotine (Fig. 7), and thus the reduced numbers of binding sites most likely reflect higher degradation rates and/or lower assembly efficiency for the mutant α4β2 AChR.
Figure 7.

Leucine mutations in the α4 and β2 AChR subunits do not affect α4β2 AChR ligand affinity. Transfected cells expressing mutant α4L351,357,358Aβ2 AChR, α4β2L349,350A AChR, or wild-type α4β2 AChR were fixed, permeabilized with 0.5% Triton X-100, and incubated with 400 pm [3H]epibatidine in the presence or absence of 10, 30, 100, 300, and 1000 nm nicotine for 24 h. The amount of bound [3H]epibatidine was determined by liquid scintillation counting. Background binding was determined in nontransfected cells and was subtracted. The data shown was normalized to the amount bound in the absence of nicotine and represent the mean ± SE from four experiments. The data were fitted to the Hill equation, and the KI values obtained are 0.65 ± 0.03 nm (nH = 0.91) for the α4, 0.77 ± 0.04 nm (nH = 0.92) for the α4L351,L357,358A, and 0.74 ± 0.02 nm (nH = 0.92) for the β2L349,350A.
Mutations in the β2 AChR subunit affect ER export
To determine the subcellular location of the mutant α4β2 AChRs that completely fail to traffic to the cell surface membrane, we stained detergent-permeabilized cells simultaneously with Abs against the ER marker calnexin and against the β2 AChR subunit. Staining for both antigens showed complete colocalization (Fig. 8A), as shown for the representative mutant α4L351,L357,358Aβ2 AChRs and α4β2L349,350A AChRs. For muscle AChR, a brief temperature shift from 20°C to 37°C promotes rapid and synchronous exit of preassembled muscle AChR from the ER to the Golgi (Ross et al., 1991). Hence, to unambiguously resolve whether mutant α4β2 AChRs colocalize with the Golgi, we first cultured cells at 30°C and then briefly at 37°C for an additional 1-2 h. When tsA201 cells were temperature shifted, the staining pattern with the β2 Ab colocalized almost completely with the Golgi marker giantin for the wild-type α4β2 AChR but not for the mutant α4β2L349,350A AChR (Fig. 8B). We also investigated whether the Golgi-disruptive agent brefeldin A (BFA) that causes ER-Golgi fusion changed this staining pattern. After BFA treatment (1 μg/ml, 37°C, 1 h), the staining for both giantin and the β2 Ab showed only an ER-like distribution for both the wild-type and the mutant α4β2 AChR expressing cells (Fig. 8C). These results confirm that, under temperature-shift conditions, the difference in staining of the Ab observed between the wild-type α4β2 AChR, which nearly completely overlaps with the giantin stain, and the mutant α4β2L349,350A AChR, which does not do so, corresponds to α4β2 AChRs transiently accumulated in the Golgi and not some other intracellular compartment that fortuitously overlaps with the Golgi. Collectively, these results provide additional evidence that the mutations in the β2 AChR subunit that abolish cell surface expression do so by preventing α4β2 AChR export from the ER.
Figure 8.
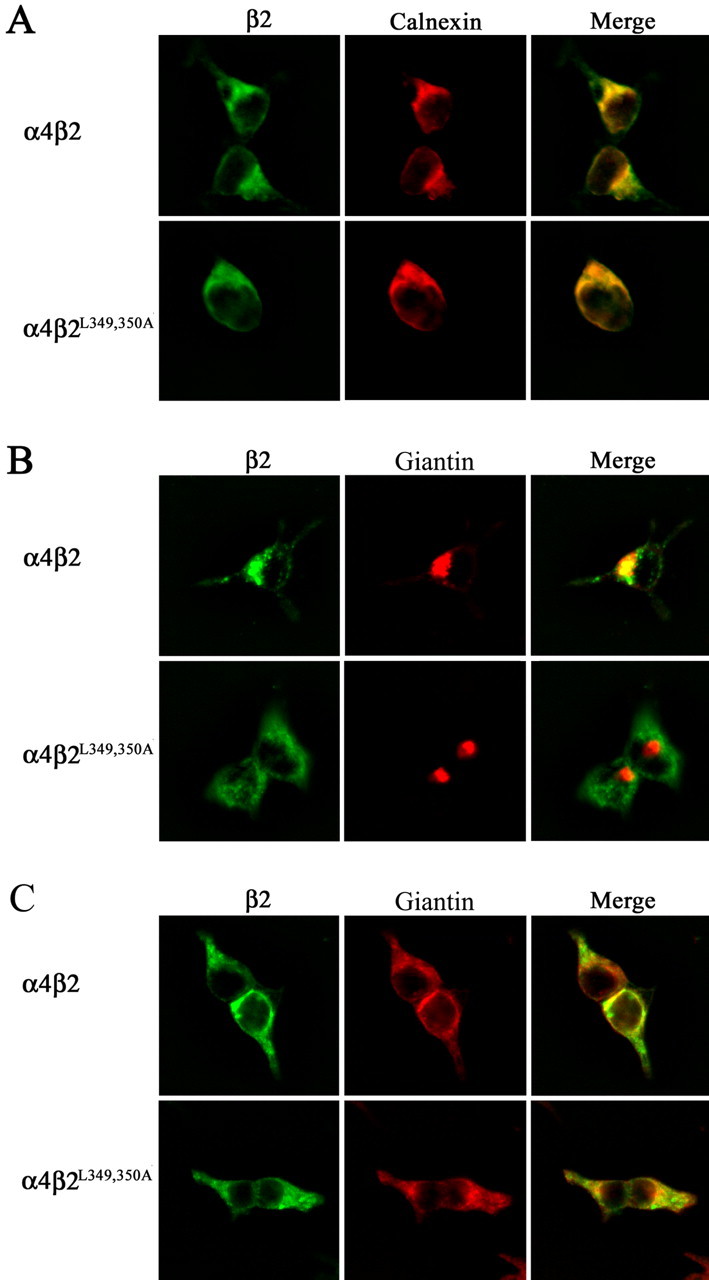
Leucine mutations in β2 AChR subunit block ER export. A, Transfected tsA201 cells expressing wild-type α4β2 AChRs or mutant α4β2L349,350A AChRs were incubated at 30°C, processed for immunohistochemistry, and probed with Abs against the β2 AChR subunit (mAb 295) and against calnexin. Ab binding to the β2 AChR subunit was detected using an Alexa Fluor 488-conjugated goat anti-rat secondary Ab and to calnexin using an Alexa Fluor 647-conjugated goat anti-rabbit secondary Ab. Cells were imaged with a deconvolution fluorescence microscope. Staining of β2 is shown in green, and staining of calnexin is shown in red. Colocalization of the proteins is shown in yellow in the merged image. B, Transfected tsA201 cells expressing wild-type α4β2 AChRs or mutant α4β2L349,350A AChRs were incubated at 30°C and then transferred to 37°C for 2 h. Cells were then processed for immunohistochemistry and probed with Abs against the β2 AChR subunit (mAb 295) and against giantin. Ab binding to the β2 AChR subunit was detected using an Alexa Fluor 488-conjugated goat anti-rat IgG and to giantin using Alexa Fluor 647-conjugated goat anti-rabbit IgG. Cells were imaged with a deconvolution fluorescence microscope. Staining of β2 is shown in green, and staining of giantin is shown in red. Colocalization is shown in yellow in the merged image. C, tsA201 cells were treated under the same procedure as in B except for an additional treatment with brefeldin A (1 μg/ml) for 1 h at 37°C.
Hydrophobicity of conserved leucine residue in the β2 AChR subunit is essential for interaction with the ER export machinery
The residues within the other AChR subunits, which are at homologous positions to the leucines mutated in the α4 and β2 AChR subunits, are hydrophobic in nature (I, F, M, V, and Y). Hence, to test whether retention of the hydrophobic character of leucine alone would be sufficient to retain its function within an ER export motif, we mutated L343 in the β2 AChR subunit (β2L343F) to phenylalanine, a hydrophobic but aromatic residue. As expected, the subunit expression level and number of high-affinity [3H]epibatidine binding sites formed were significantly higher than those of the other β2 AChR mutants. However, in contrast to the effect of mutating this leucine residue to alanine, mutation to phenylalanine does not abolish the cell surface expression of the mutant α4β2 AChR but instead reduces it to ∼57% of that observed for the wild-type β2 AChR subunit (Fig. 5B). This result suggests that the hydrophobic physiochemical property of this leucine residue is a key feature of its function in ER export.
Conservation of mutant AChR trafficking defects in differentiated SH-SY5Y neural cells
To further examine whether the trafficking deficits observed for mutant α4β2 AChRs in tsA201 cells were conserved in polarized cells of neural origin, we examined whether the mutant α4β2L349,350A AChR behaved similarly in differentiated human neuroblastoma SH-SY5Y cells. These cells were chosen because they do not express detectable levels of wild-type α4β2 AChRs that could interfere with our analysis and because they exhibit reasonable transfection efficiencies. SH-SY5Y cells were first differentiated in serum-free media and then transfected with appropriate cDNAs to express wild-type α4β2 AChRs or mutant α4β2L349,350A AChRs in them. At 30°C, wild-type α4β2 AChRs were strongly expressed on the surface of the soma and processes, but mutant α4β2L349,350A AChRs failed to show surface expression despite high levels of expression in the cytoplasm (Fig. 9A). When SH-SY5Y cells expressing α4β2 AChRs were shifted from 30°C to 37°C for ∼2 h, internal pools of wild-type α4β2 AChRs showed the ability to synchronously move from the ER to almost completely colocalize with the Golgi marker giantin (red), whereas the mutant α4β2L349,350A AChRs failed to do so (Fig. 9B). These results provide additional evidence that the conserved hydrophobic residues are essential determinants of an ER export motif.
Figure 9.
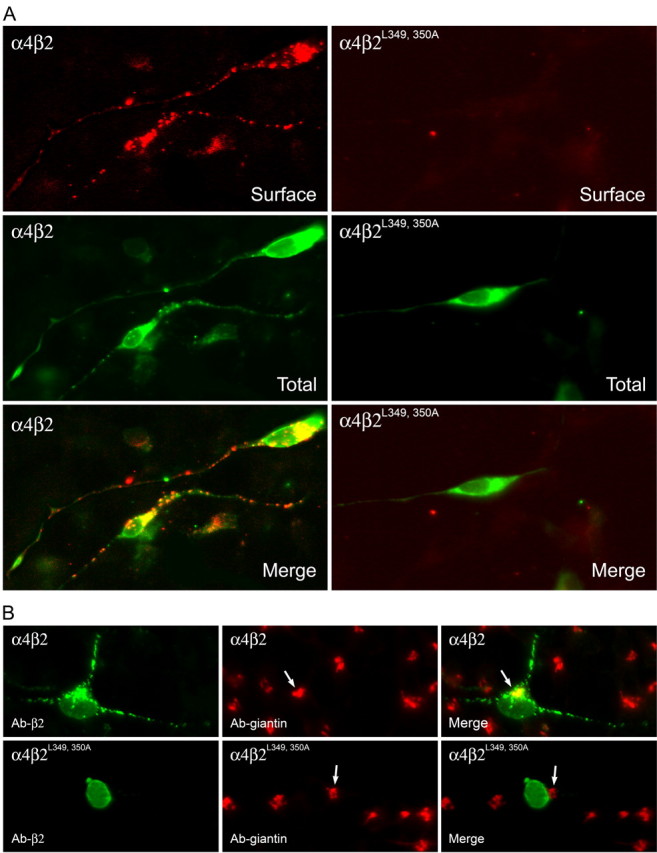
Subcellular distribution of wild-type α4β2 AChRs and mutant α4β2L349,350A AChRs in differentiated SH-SY5Y neural cells. A, Wild-type α4β2 AChRs are expressed on the surface membrane of the soma and processes, but mutant α4β2L349, 350A AChRs fail to do so even in cells that have high expression levels of the proteins in the cytoplasm. Transfected SH-SY5Y cells expressing wild-type α4β2 and mutant α4β2L349, 350A AChRs were incubated at 30°C for 2 d and then fixed for immunohistochemical processing. The cells were probed with Ab against the β2 AChR subunit (mAb 295) before permeabilization, and the Ab binding was detected with the Alexa Fluor 647-conjugated goat anti-rat Abs. These same cells were then permeabilized and probed with the β2 AChR subunit Ab (mAb 295) again, and the Ab binding was detected using the Alexa Fluor 488-conjugated goat anti-rat Abs. B, Wild-type α4β2 AChRs (green) colocalize with giantin (red), whereas mutant α4β2L349, 350A AChRs (green) do not. Transfected SH-SY5Y cells expressing wild-type α4β2or mutant α4β2L349,350A AChRs were incubated at 30°C and then shifted to 37°C for 2 h. The cells were fixed, permeabilized, and probed with the β2 AChR subunit Ab (mAb 295) and the giantin Ab. Ab binding to the β2 AChR subunit was visualized using Alexa Fluor 488-conjugated goat anti-rat Abs and to giantin using Alexa Fluor 647-conjugated goat anti-rabbit Abs. Colocalization of the proteins is shown in yellow in the merged image.
Other heteromeric AChR combinations assembled from mutant α4 and β2 AChR subunits also show trafficking deficits
To determine whether the cell surface trafficking of other heteromeric AChR combinations containing the mutant α4L351,357,358A or the mutant β2L349,350A AChR subunit would be affected, we expressed wild-type α3β2 AChRs and mutant α3β2L349,350A AChRs in tsA201 cells and measured their surface expression using the β2 Ab (mAb 295). Similarly, we expressed wild-type α4β4 AChRs and mutant α4L351,357,358Aβ4 AChRs in tsA201 cells and measured their surface expression levels using the α4 Ab (mAb 299). The results (Fig. 10) show that the mutant α4L351,357,358A AChR subunit attenuated and the mutant β2L349,350A AChR subunit abolished cell surface expression of the corresponding heteromeric AChRs containing these mutant AChR subunits.
Figure 10.

Mutant α4 and β2 AChR subunits also attenuate or abolish cell surface expression of other heteromeric AChRs containing them. Transfected tsA201 cells expressing wild-type α4β4 or mutant α4L351,357,358A β4 AChRs were fixed and probed with an Ab against the α4 AChR subunit (mAb 299), and those expressing wild-type α3β2 AChRs or mutant α3β2L349,350A AChRs were probed with an Ab against the β2 AChR subunit (mAb 295). The cell surface expression of AChRs was quantitated using an enzyme-linked colorimetric assay. The data were expressed as a percentage of the value obtained for wild-type α4β4 AChRs or the wild-type α3β2 AChRs. The data represent the mean ± SE from four experiments.
Discussion
The goal of this study was to elucidate the structural determinants for trafficking of AChRs. The principal finding of this study demonstrates that highly conserved hydrophobic residues within the long cytoplasmic loop of AChR subunits are critical structural determinants for ER export of fully assembled AChRs. We show that cell surface expression of α4β2 AChRs is attenuated when F350, L351, L357, and L358 in the α4 AChR subunit are mutated to alanine and abolished when F342, L343, L349, and L350 in the β2 AChR subunit are mutated to alanine. The mutant AChR subunits assemble to form mutant α4β2 AChRs that colocalize with the endoplasmic marker calnexin and exhibit high-affinity for [3H]epibatidine. Furthermore, in temperature-shift experiments designed to promote rapid ER exit of assembled α4β2 AChRs, internal pools of wild-type α4β2 AChRs colocalize almost completely with the Golgi marker giantin, whereas mutant α4β2 AChRs fail to do so. Collectively, these results suggest that the hydrophobic conserved residues are essential structural determinants of α4β2 AChR ER export.
Mutations of the conserved hydrophobic residues to alanine in both the α4 AChR and the β2 AChR subunits, with the exception of the α4F350A mutant, also decreased the subunit expression levels and the total number of high-affinity ligand-binding sites formed. Thus, it is likely that the decreased subunit stability also contributed to the decrease observed in cellular levels of α4β2 AChRs. Nonetheless, because all of the mutant AChR subunits studied assemble to form >35% of the high-affinity [3H]epibatidine binding sites observed with the wild-type AChR subunits, it is likely that the mutation of these conserved residues have their most significant functional effect on the trafficking of α4β2 AChRs. In this regard, the α4F350A mutant notably showed virtually no decrease in its subunit stability or in the number of high-affinity ligand-binding sites formed but yet exhibited a significant (∼30%) decrease in cell surface expression of mutant α4β2 AChRs. This result, and those obtained for the mutant β2 AChR subunits that abolish α4β2 AChR cell surface expression, in aggregate, strongly support the conclusion that the mutations primarily affect α4β2 AChR trafficking.
High-resolution fluorescence imaging experiments show that the wild-type but not the mutant α4β2 AChRs can traffic from the ER to the Golgi. The temperature-shift experiment, as described previously (Ross et al., 1991), was crucial in helping unambiguously resolve whether the mutant α4β2 AChRs traffic to the Golgi for two reasons. First, standard fluorescence image deconvolution protocols showed significant overlap between staining for the Golgi marker giantin and the ER marker calnexin, which was not attributable to “bleed through” between the filters used to visualize the fluorophores Alexa 488 (FITC) and Alexa 647 (Cy5). Second, under steady-state conditions, it is likely that only a small fraction of the α4β2 AChRs is localized in the Golgi, making its visualization rather difficult over the signals from the larger pool of α4β2 AChRs localized in the ER. The brief temperature shift helped mobilize this larger pool of assembled α4β2 AChRs to synchronously exit the ER and traffic to the Golgi, effectively eliminating ER staining while transiently enriching the Golgi pool, which then enhanced our ability to visualize α4β2 AChRs in the Golgi. We obtained similar results using both tsA201 cells and differentiated SH-SY5Y neural cells. Indeed, in SH-SY5Y cells, wild-type α4β2 AChRs are not only expressed on the cell surface of the soma but are also transported down the surface membrane of the processes, whereas the mutant α4β2 AChRs, in contrast, fail to express on the cell surface and remain localized within the soma. These results further support our hypothesis that these conserved hydrophobic residues are determinants of an ER export motif for α4β2 AChRs.
The residues at positions that are homologous to those of the conserved leucines in the α4 AChR and the β2 AChR subunit are not fully conserved, but their hydrophobic nature is highly conserved in all AChR subunits (Fig. 1). For example, the first residue of the first pair of hydrophobic residues is a phenylalanine, isoleucine, or leucine, and the second residue is a leucine or an isoleucine. Similarly, the first residue of the second pair of hydrophobic residues shown is a isoleucine, tryptophan, valine, phenylalanine, or a tyrosine at this position, and a leucine or a methionine at the second position. Such hydrophobic residues, particularly phenylalanine or leucine, occur within short motifs important for cargo selection into COPII-coated vesicles that transport proteins from the ER to the Golgi. To test the possibility that conservation of the hydrophobic character of these leucine residues is a key feature of their function within the ER export motif, we mutated L343 to phenylalanine in the β2 AChR subunit. Because a very significant level (∼60%) of cell surface expression for the mutant α4β2 AChR was observed, we speculate that the hydrophobic residues found at homologous positions in the other AChR subunits may also have conserved functions.
The leucine mutations in the two AChR subunits affected the cell surface expression of mutant α4β2 AChRs to strikingly different degrees. The three leucine mutations in the α4 AChR subunit, even when present together (as in α4L351,357,358A) only attenuated AChR cell surface expression, whereas the individual mutations in the β2 AChR subunits were sufficient to virtually abolish AChR cell surface expression. These results suggest that the α4 AChR subunit has a cooperative or regulatory role and the β2 AChR subunit an obligatory role in their functional interaction with the ER export machinery. Hence, it is possible that other regulatory proteins, yet to be identified, could interact with these residues in the α4 AChR subunit and influence the export rate of α4β2 AChRs from the ER. Coassembly of the mutant α4L351,357,358A or the mutant β2L349,350A AChR subunit with other complementary subunits in the AChR family to form the α3β2 AChR and α4β4 AChR revealed that the functional effects of these mutations with respect to trafficking are conserved even in other heteromeric combinations of AChRs.
Hydrophobic residues occur in trafficking motifs of a number of integral membrane proteins. For example, mutations of leucine residues in the intracellular domains of the kainate receptor KA2 (Ren et al., 2003), the β2 subunit of the GABAA receptor (Herring et al., 2003), and the δ-opioid receptor (Wang et al., 2003) inhibit their endocytotic trafficking. Interestingly, the motif F(X)6(I/L)(I/L), which has significant similarities to the ER export motif FL(X)5LL identified in this study in the α4 and β2 AChR subunits, was found to be present in the C-terminal tail of a very large number, but not all, G-protein-coupled receptors (Duvernay et al., 2004). Mutation of the hydrophobic residues F or L was shown to inhibit ER export of the α2B-adrenergic receptor and thus to represent structural determinants of G-protein-coupled receptor ER export. This motif is spaced approximately three residues away from the seventh transmembrane domain of G-protein-coupled receptors, but its functional homolog in AChR subunits is spaced ∼17 residues away from its third transmembrane domain. Thus, these motifs may function in an autonomous manner because their interaction with the ER export machinery does not appear to be critically dependent on their distance from the ER cytosolic membrane surface.
Other signals that participate in the quality control of AChR biogenesis have been described. The ER retention dibasic signal R313K314 in the α1 AChR subunit, when masked by subunit assembly with the β, γ, and δ AChR subunits, permits trafficking of muscle AChRs from the ER to the Golgi (Keller et al., 2001). Interestingly, ubiquitination of K313 was found to function as a post-Golgi modulator of trafficking. A second ER retention signal, PLYXXN, in the first transmembrane domain of the α1 AChR subunit serves as an additional signal in the quality control of AChR subunit oligomerization (Wang et al., 2002).
The amino acid sequence of the long cytosolic loop is highly divergent among the AChR subunits, and recent work indicates that native AChRs assemble with a dizzying array of subunit complexity (Champtiaux et al., 2003; Moretti et al., 2004). The differential effect of mutating the conserved leucines in the α4 AChR subunit versus the β2 AChR subunit on ER export reveals another level of complexity in the functional roles of well conserved cytoplasmic motifs. Motifs within different AChR subunits, when present in AChRs of different subunit compositions, may show heterogeneity in their functional contributions. Complementary progress has been made in identifying at least two proteins, 14-3-3η (Jeanclos et al., 2001) and ric-3 (Halevi et al., 2002, 2003), which influence the earliest steps in the biogenesis of AChRs. Furthermore, it is of interest to note that VILIP-1, a myristoylated calcium sensor protein whose interaction with the α4 AChR subunit upregulates α4β2 AChR surface expression (Lin et al., 2002), binds in protein-protein interaction screens to the ∼30 amino acid region in the loop encompassing the ER export motif identified in this study. Future experiments need to be done to determine the extent to which this ER export site overlaps, if at all, with that of VILIP-1.
Footnotes
This work was supported in part by grants from the National Science Foundation (R.A.), the National Institutes of Health (R.A., J.M.L.), the Millennium Trust Health Excellence Fund (R.A., J.R.), and Philip Morris USA and Philip Morris International (R.A., J.M.L.). M.W.T. is supported in part with funds from the Grace Benson Fund for Parkinson's Research. A Centers of Biomedical Research Excellence Grant from the National Institutes of Health (National Center for Research Resources) supports the Imaging and Molecular Core Facilities of the Neuroscience Center used in this study. We thank Dr. Gregg Wells for critical comments on this manuscript.
Correspondence should be addressed to Dr. Rene Anand, Neuroscience Center of Excellence and Department of Neurology, 2020 Gravier Street, Suite D, Louisiana State University Health Sciences Center, New Orleans, LA 70112. E-mail: ranand@lsuhsc.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256676-11$15.00/0
References
- Alder NN, Johnson AE (2004) Cotranslational membrane protein biogenesis at the endoplasmic reticulum. J Biol Chem 279: 22787-22790. [DOI] [PubMed] [Google Scholar]
- Aridor M, Traub LM (2002) Cargo selection in vesicular transport: the making and breaking of a coat. Traffic 3: 537-546. [DOI] [PubMed] [Google Scholar]
- Barlowe C (2003) Signals for COPII-dependent export from the ER: what's the ticket out? Trends Cell Biol 13: 295-300. [DOI] [PubMed] [Google Scholar]
- Blount P, Merlie JP (1988) Native folding of an acetylcholine receptor alpha subunit expressed in the absence of other receptor subunits. J Biol Chem 263: 1072-1080. [PubMed] [Google Scholar]
- Blount P, Merlie JP (1990) Mutational analysis of muscle nicotinic acetylcholine receptor subunit assembly. J Cell Biol 111: 2613-2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount P, Merlie JP (1991) BIP associates with newly synthesized subunits of the mouse muscle nicotinic receptor. J Cell Biol 113: 1125-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Lena C, Clementi F, Moretti M, Rossi FM, Le Novere N, McIntosh JM, Gardier AM, Changeux JP (2003) Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci 23: 7820-7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court J, Clementi F (1995) Distribution of nicotinic subtypes in human brain. Alzheimer Dis Assoc Disord 9: 6-14. [DOI] [PubMed] [Google Scholar]
- Duncan MC, Payne GS (2003) ENTH/ANTH domains expand to the Golgi. Trends Cell Biol 13: 211-225. [DOI] [PubMed] [Google Scholar]
- Duvernay MT, Zhou F, Wu G (2004) A conserved motif for the transport of G protein-coupled receptors from the endoplasmic reticulum to the cell surface. J Biol Chem 279: 30741-30750. [DOI] [PubMed] [Google Scholar]
- Forsayeth JR, Gu Y, Hall ZW (1992) BiP forms stable complexes with unassembled subunits of the acetylcholine receptor in transfected COS cells and in C2 muscle cells. J Cell Biol 117: 841-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman MS, Chang W, Thomas DY, Bergeron JJ, Prives JM (1995) Role of the endoplasmic reticulum chaperone calnexin in subunit folding and assembly of nicotinic acetylcholine receptors. J Biol Chem 270: 15085-15092. [DOI] [PubMed] [Google Scholar]
- Guan XM, Kobilka TS, Kobilka BK (1992) Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. J Biol Chem 267: 21995-21998. [PubMed] [Google Scholar]
- Halevi S, McKay J, Palfreyman M, Yassin L, Eshel M, Jorgensen E, Treinin M (2002) The C. elegans ric-3 gene is required for maturation of nicotinic acetylcholine receptors. EMBO J 21: 1012-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevi S, Yassin L, Eshel M, Sala F, Sala S, Criado M, Treinin M (2003) Conservation within the RIC-3 gene family. Effectors of mammalian nicotinic acetylcholine receptor expression. J Biol Chem 278: 34411-34417. [DOI] [PubMed] [Google Scholar]
- Herring D, Huang R, Singh M, Robinson LC, Dillon GH, Leidenheimer NJ (2003) Constitutive GABAA receptor endocytosis is dynamin-mediated and dependent on a dileucine AP2 adaptin-binding motif within the beta 2 subunit of the receptor. J Biol Chem 278: 24046-24052. [DOI] [PubMed] [Google Scholar]
- Jeanclos EM, Lin L, Treuil MW, Rao J, DeCoster MA, Anand R (2001) The chaperone protein 14-3-3eta interacts with the nicotinic acetylcholine receptor alpha 4 subunit. Evidence for a dynamic role in subunit stabilization. J Biol Chem 276: 28281-28290. [DOI] [PubMed] [Google Scholar]
- Johnson AE, van Waes MA (1999) The translocon: a dynamic gateway at the ER membrane. Annu Rev Cell Dev Biol 15: 799-842. [DOI] [PubMed] [Google Scholar]
- Keller SH, Lindstrom J, Taylor P (1996) Involvement of the chaperone protein calnexin and the acetylcholine receptor beta-subunit in the assembly and cell surface expression of the receptor. J Biol Chem 271: 22871-22877. [DOI] [PubMed] [Google Scholar]
- Keller SH, Lindstrom J, Taylor P (1998) Inhibition of glucose trimming with castanospermine reduces calnexin association and promotes proteasome degradation of the alpha-subunit of the nicotinic acetylcholine receptor. J Biol Chem 273: 17064-17072. [DOI] [PubMed] [Google Scholar]
- Keller SH, Lindstrom J, Ellisman M, Taylor P (2001) Adjacent basic amino acid residues recognized by the COP I complex and ubiquitination govern endoplasmic reticulum to cell surface trafficking of the nicotinic acetylcholine receptor alpha-Subunit. J Biol Chem 276: 18384-18391. [DOI] [PubMed] [Google Scholar]
- Lin L, Jeanclos EM, Treuil M, Braunewell KH, Gundelfinger ED, Anand R (2002) The calcium sensor protein visinin-like protein-1 modulates the surface expression and agonist sensitivity of the alpha 4beta 2 nicotinic acetylcholine receptor. J Biol Chem 277: 41872-41878. [DOI] [PubMed] [Google Scholar]
- Lopez-Hernandez GY, Sanchez-Padilla J, Ortiz-Acevedo A, Lizardi-Ortiz J, Salas-Vincenty J, Rojas LV, Lasalde-Dominicci JA (2004) Nicotine-induced up-regulation and desensitization of alpha4beta2 neuronal nicotinic receptors depend on subunit ratio. J Biol Chem 279: 38007-38015. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Whiteaker P, Calcaterra J, Stitzel JA, Bullock AE, Grady SR, Picciotto MR, Changeux JP, Collins AC (1999) Two pharmacologically distinct components of nicotinic receptor-mediated rubidium efflux in mouse brain require the beta2 subunit. J Pharmacol Exp Ther 289: 1090-1103. [PubMed] [Google Scholar]
- Marubio LM, Arroyo-Jimenez MM, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d'Exaerde, A, Huchet M, Damaj MI, Changeux JP (1999) Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature 398: 805-810. [DOI] [PubMed] [Google Scholar]
- Marubio LM, Gardier AM, Durier S, David D, Klink R, Arroyo-Jimenez MM, McIntosh JM, Rossi F, Champtiaux N, Zoli M, Changeux JP (2003) Effects of nicotine in the dopaminergic system of mice lacking the alpha4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci 17: 1329-1337. [DOI] [PubMed] [Google Scholar]
- Merlie JP, Sebbane R, Tzartos S, Lindstrom J (1982) Inhibition of glycosylation with tunicamycin blocks assembly of newly synthesized acetylcholine receptor subunits in muscle cells. J Biol Chem 257: 2694-2701. [PubMed] [Google Scholar]
- Millar NS (2003) Assembly and subunit diversity of nicotinic acetylcholine receptors. Biochem Soc Trans 31: 869-874. [DOI] [PubMed] [Google Scholar]
- Mishina M, Tobimatsu T, Imoto K, Tanaka K, Fujita Y, Fukuda K, Kurasaki M, Takahashi H, Morimoto Y, Hirose T (1985) Location of functional regions of acetylcholine receptor alpha-subunit by site-directed mutagenesis. Nature 313: 364-369. [DOI] [PubMed] [Google Scholar]
- Moretti M, Vailati S, Zoli M, Lippi G, Riganti L, Longhi R, Viegi A, Clementi F, Gotti C (2004) Nicotinic acetylcholine receptor subtypes expression during rat retina development and their regulation by visual experience. Mol Pharmacol 66: 85-96. [DOI] [PubMed] [Google Scholar]
- Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J (2003) Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol 63: 332-341. [DOI] [PubMed] [Google Scholar]
- Nishimura N, Balch WE (1997) A di-acidic signal required for selective export from the endoplasmic reticulum. Science 277: 556-558. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Alafuzoff I, Winblad B (1992) Nicotinic and muscarinic subtypes in the human brain: changes with aging and dementia. J Neurosci Res 31: 103-111. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Ross AF, Green WN, Claudio T (1991) Analysis of early events in acetylcholine receptor assembly. J Cell Biol 113: 1371-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y, LeNovere N, Vincent P, Pich EM, Brulet P, Changeux JP (1995) Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature 374: 65-67. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, Fuxe K, Changeux JP (1998) Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 391: 173-177. [DOI] [PubMed] [Google Scholar]
- Raggenbass M, Bertrand D (2002) Links Nicotinic receptors in circuit excitability and epilepsy. J Neurobiol 53: 580-589. [DOI] [PubMed] [Google Scholar]
- Ren Z, Riley NJ, Garcia EP, Sanders JM, Swanson GT, Marshall J (2003) Multiple trafficking signals regulate kainate receptor KA2 subunit surface expression. J Neurosci 23: 6608-6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS (2004) Adaptable adaptors for coated vesicles. Trends Cell Biol 14: 167-174. [DOI] [PubMed] [Google Scholar]
- Ross AF, Green WN, Hartman DS, Claudio T (1991) Efficiency of acetylcholine receptor subunit assembly and its regulation by cAMP. J Cell Biol 113: 623-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MM, Lindstrom J, Merlie JP (1987) Formation of the alpha-bungarotoxin binding site and assembly of the nicotinic acetylcholine receptor subunits occur in the endoplasmic reticulum. J Biol Chem 262: 4367-4376. [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA (2004) Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science 306: 1029-1032. [DOI] [PubMed] [Google Scholar]
- Wanamaker CP, Christianson JC, Green WN (2003) Regulation of nicotinic acetylcholine receptor assembly. Ann NY Acad Sci 998: 66-80. [DOI] [PubMed] [Google Scholar]
- Wang JM, Zhang L, Yao Y, Viroonchatapan N, Rothe E, Wang ZZ (2002) A transmembrane motif governs the surface trafficking of nicotinic acetylcholine receptors. Nat Neurosci 5: 963-970. [DOI] [PubMed] [Google Scholar]
- Wang W, Loh HH, Law PY (2003) The intracellular trafficking of opioid receptors directed by carboxyl tail and a di-leucine motif in Neuro2A cells. J Biol Chem 278: 36848-36858. [DOI] [PubMed] [Google Scholar]
- Whiting PJ, Lindstrom JM (1988) Characterization of bovine and human neuronal nicotinic acetylcholine receptors using monoclonal antibodies. J Neurosci 8: 3395-3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Nelson ME, Kuryatov A, Choi C, Cooper J, Lindstrom J (2003) Human α4β2 acetylcholine receptors formed from linked subunits. J Neurosci 23: 9004-9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, Vijverberg HP (1998) Four pharmacologically distinct subtypes of alpha4beta2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol Pharmacol 54: 1124-1131. [PubMed] [Google Scholar]