

Abstract
Complete information regarding transcriptional and posttranscriptional gene regulation in stem cells is necessary to understand the regulation of self-renewal and differentiation. Alternative splicing is a prevalent mode of posttranscriptional regulation, and occurs in approximately one half of all mammalian genes. The frequency and functional impact of alternative splicing in stem cells are yet to be determined. In this study we combine computational and experimental methods to identify splice variants in embryonic and hematopoietic stem cells on a genome-wide scale. Using EST collections derived from stem cells, we detect alternative splicing in >1,000 genes. Systematic RT-PCR and sequencing studies show confirmation of computational predictions at a level of 80%. We find that alternative splicing can modify multiple components of signaling pathways important for stem cell function. We also analyze the distribution of splice variants across different classes of genes. We find that tissue-specific genes have a higher tendency to undergo alternative splicing than ubiquitously expressed genes. Furthermore, the patterns of alternative splicing are only weakly conserved between orthologous genes in human and mouse. Our studies reveal extensive modification of the stem cell molecular repertoire by alternative splicing and provide insights into its overall role as a mechanism of generating genomic diversity.
Keywords: genome, exons, introns, transcription
The architecture of most genes in higher eukaryotes consists of interspersed coding exons and noncoding introns (1). The removal of introns and the joining of exons by RNA splicing is an essential step in the assembly of functional mRNAs. Alternative splicing events can assemble different combinations of exons to produce mRNA isoforms with distinct protein coding potentials. Thus, different mRNA isoforms from a single gene can often encode proteins with distinct, sometimes opposite functions (2). Numerous biological processes ranging from sex determination to apoptosis depend on the alternative splicing of specific genes (2, 3). The best studied example is in Drosophila, for which sex determination of the whole organism depends on splicing choices in the sex-lethal and transformer genes (4). The regulatory mechanism of alternative splicing relies on interactions between transacting splicing factors and cis-regulatory elements within the spliceosome, a large macromolecular complex that catalyzes intron excision and exon joining (4).
Sequencing of the human genome and collections of EST have facilitated global detection of alternatively spliced variants (5, 6). Because ESTs are generally derived from mature spliced mRNA populations, they provide a broad sample of mRNA diversity. Computational analyses of cDNA and EST sequences have suggested that alternatively spliced transcripts are produced from >50% of mammalian genes (7-9). Most alternative splicing occurs within protein-coding regions (10, 11). It has been suggested that alternative splicing is a general mechanism to increase the coding capacity and diversity of the genome in metazoans (12).
Recent studies have extensively characterized the global gene expression profiles of various stem cell populations. A large number of genes were found to be preferentially expressed in primitive stem cells (13-16). These studies provided a first estimate of the molecular repertoire available for stem cell regulation. Until now, analyses of alternative splicing in stem cells have been limited to individual genes (17-19). For instance, alternative splicing of the Ikaros gene produces a variety of functionally diverse transcription factors found in hematopoietic stem cells (HSC) and lymphoid progenitors (17). Global analyses of alternative splicing have been limited to heterogeneous cell populations from whole tissues (e.g., brain or bone marrow). Tissue-specific stem cells are very rare (≈1 in 10,000 whole bone-marrow cells). Therefore, it would be difficult to detect splice variants specific to stem cells in whole-tissue analyses. Herein we use a combined computational and experimental approach to analyze alternative splicing in embryonic stem (ES) cells and highly purified HSC on a genome-wide scale. In addition, we investigate genome-wide trends of alternative splicing and establish relationships between levels of transcription, tissue-specific gene expression, and the frequency of alternative splicing.
Materials and Methods
Computational Analysis. Our computational strategy for genome-wide analysis of alternative splicing is outlined in Fig. 1. Human and murine stem-cell-specific EST libraries were extracted from the National Center for Biotechnology Information (NCBI) and Stem Cell Databases (13, 20). The complete list of the queried libraries is provided in Table 2, which is published as supporting information on the PNAS web site. For a reference set, the sequences of all assigned human and mouse genes and their transcripts and corresponding exon-intron structures were obtained from Ensembl (release 32) (21). The blast program (22) was used to align the ESTs with the full-length transcripts. A threshold of 95% identity over 100 nucleotides was used to define sequence identity. Potential sites of alternative splicing were identified as gaps in the alignments within coding regions. Alignment gaps were further analyzed by mapping to exon/intron boundaries.
Fig. 1.

Computational and experimental approach. A reference set of full-length gene transcripts derived from Ensembl was aligned with ESTs from the NCBI and Stem Cell Databases. Potential sites of alternative splicing were identified as gaps in the alignments that correspond to the exon boundaries. For these studies, we focused only on inclusion or exclusion of entire exons within coding sequences. Computationally detected alternative splicing events were confirmed experimentally with RT-PCR, cloning, and sequencing. PECAM, platelet/endothelial cell adhesion molecule.
To measure genome-wide trends of alternative splicing, we performed a separate computational analysis that characterized the distribution of splice variants among different classes of genes. For these analyses, the reference set of 8,100 full-length cDNAs representing known human genes was extracted from SwissProt (release 41), a manually annotated database (23). The EST tissue source information was extracted from the TissueInfo database (24). Detailed descriptions and complete results of the computational analyses are included in Supporting Materials and Methods and Data Sets 1-6, which are published as supporting information on the PNAS web site; additional data also are available from the authors upon request.
Experimental Confirmation of Alternative Splicing. Murine HSCs were purified from bone marrow as described in ref. 13. Panels of total RNA from human and mouse adult tissues were purchased from BD Biosciences Clontech. Specific primers flanking predicted sites of alternative splicing were used for RT-PCR amplifications with TaqGold polymerase (Applied Biosystems). PCR products were separated by agarose gel electrophoresis, and the band intensities were quantified by using the GelDoc imaging system and quantityone software (Bio-Rad). Amplified products were extracted from gels, cloned by using the TOPO cloning kit (Invitrogen), and sequenced to confirm alternative splicing. All primers, splice junction sequences, and detailed descriptions of experimental procedures are included in Supporting Materials and Methods and Data Sets 1-6 (additional data also are available upon request).
Results
Alternative Splicing in Stem Cells. To analyze the effect of alternative splicing in stem cells, we selected human and mouse EST libraries that were generated from ES cells and HSCs (Table 1). Specifically, we used >40,000 sequences produced in our laboratory from murine long- and short-term HSC populations. In addition, we extracted ESTs specific for ES cells and murine HSCs from other libraries available at the NCBI EST database (16). We did not identify an EST data set derived from purified human HSCs in the NCBI EST database. We computationally aligned the stem-cell-specific ESTs to full-length cDNAs from the Ensembl database to identify sites of alternative splicing as gaps in alignments of highly similar sequences (Fig. 1). Comparison of the alignment gaps with exon/intron boundaries led us to discover splice variations within coding regions for >300 and 1,000 genes in HSCs and ES cells, respectively (Table 1). Frequencies of alternatively spliced genes were ≈3% in mouse HSC and ES cells and ≈9% in human ES cells. These values are close to previous estimates, considering the number of available ESTs (7). The observed variations between the stem cell populations may reflect differences in preparation and characterization of corresponding EST libraries. For example, 87% of human ES cell ESTs came from a single study that did not employ normalization or amplification (25). In contrast, the mouse HSC and ES cell libraries were derived from a large number of studies that used normalization and/or amplification (13, 26).
Table 1. Computational analysis of alternative splicing in stem cells.
| Organism | Cell type | No. of ESTs (no. of represented genes) | No. of genes with splice variations (%) |
|---|---|---|---|
| Human | ES | 89,047 (12,688) | 1,163 (9.1) |
| Mouse | ES | 169,270 (12,904) | 413 (3.2) |
| Mouse | HSC | 75,085 (13,144) | 336 (2.5) |
Using blast, we compared the identified splice variants with sequences in the NCBI EST database that includes all of the publicly available ESTs. We found that ≈30% of the splice variants identified in the stem cell data sets were not found in ESTs from other tissues. We chose a set of 15 genes encoding diverse proteins, such as transcription factors and transmembrane receptors, for experimental confirmation. Prior knowledge of alternative splicing was not used in the selection of the gene sets. Using cloning and sequencing, we were able to confirm that 12 of 15 (80%) of these genes were expressed with the splice variations (Fig. 2 and Fig. 7, which is published as supporting information on the PNAS web site). Lack of confirmation for 20% of the selected genes was likely due to differences in the stem cell lines, isolation protocols, and cell culture conditions. The RT-PCR profiles across ES, HSC, and eight adult tissues showed that selected genes are expressed in stem cells with various degrees of specificity. Literature searches indicate that some of the confirmed genes play important regulatory roles in stem cell biology. For instance, it was shown that the Polycomb group transcription factor EZH2 is required for maintenance of undifferentiated cells during the blastocyst stage of early mouse development (27). The Bric-a-Brac/Poxvirus and zinc-finger domain protein Zbtb20 is a less characterized transcription factor; however, it has been shown to be involved in developmental neurogenesis (28). In the group of transmembrane genes, we identified splice variants for platelet/endothelial cell adhesion molecule-1, a transmembrane protein involved in migration of hematopoietic stem cells (29). Splice variations were also confirmed for the weakly characterized putative adhesion receptor FAD104 and the apoptotic TNF receptor TNFR-7 (Fig. 7). The majority of our experimentally confirmed variants are not described in the published literature (Table 3, which is published as supporting information on the PNAS web site).
Fig. 2.

Experimental confirmation of computationally identified splice variants in stem cells. A representative set of 15 murine transcription factors and transmembrane receptors was selected for experimental confirmation; 80% (12 of 15) were confirmed. Prior knowledge of alternative splicing was not used in the selection of the gene set. RT-PCR profiles are shown for seven of the confirmed genes. The structure of the splice variants was confirmed by cloning and sequencing as described in Materials and Methods. Additional PCR profiles and sequences are provided in Fig. 7.
To estimate a possible functional impact of splice variations in stem cells, the distribution of the identified variants was analyzed across signaling pathways found in the Proteomic Pathway Project (BioCarta, San Diego). Overall, the identified splice variants showed a statistically significant enrichment for genes encoding components of signaling pathways (P < 0.01), as assessed by the hypergeometric distribution (Supporting Materials and Methods). Because the available EST stem cell data are limited, it is difficult to determine whether some pathways are affected more than others. However, we found that alternative splicing modifies components of several signaling pathways that are known to govern stem cell self-renewal and differentiation (Table 4, which is published as supporting information on the PNAS web site). For instance, splice variations were detected in genes encoding components of the mitogen-activated protein kinase signaling pathway, such as Map4k2 and Mnk1 kinases. We also identified alternative splice variants for multiple genes involved in cell cycle and apoptosis. These included cyclin-dependent kinase 4, cyclin-dependent kinase inhibitor 1, cyclin H, caspase 11, and apoptosis regulator BID. The majority of the identified variants were not described previously. Complete results of the pathway analysis are available from the authors upon request. Collectively, our findings indicate that alternative splicing plays a major role in the diversification of numerous regulatory gene products expressed in stem cells.
Correlations Between Transcription and Alternative Splicing. To further explore the observation that alternative splicing extensively modifies regulatory genes, we analyzed the distribution of splice variants across various expression levels. Because the available stem cell EST collection is limited, we used human EST data from all tissues. More than 4,000,000 human ESTs were aligned to a nonredundant reference set of 8,100 full-length cDNAs derived from SwissProt. This database is manually annotated and therefore reliably defines bona fide gene products. Computational analyses identified 2,471 alternative splice sites within coding regions of human genes. The complete data set is presented in Data Sets 1-3.
To describe the frequency of alternative splicing in a quantitative manner, we defined a new parameter, an exon exclusion fraction, fex (Fig. 3). Computationally, the exon exclusion fraction is defined as the number of ESTs with an excluded exon divided by the total number of ESTs spanning a site of alternative splicing:
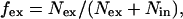 |
where Nex is the number of ESTs with an excluded exon and Nin is the number of ESTs with an included exon. Experimentally, the exon exclusion fraction is defined as the intensity of DNA bands representing the exon exclusion isoform divided by the intensity of all of the bands:
 |
where Iex is the intensity of the exon exclusion isoform (lower band), Iin is the intensity of the exon inclusion isoform (upper band) and L is a fragment length factor. The fragment length factor, defined as the ratio between the long and short isoform lengths, is needed to correct the effect of DNA fragment length on UV absorption. Because tissue-specific EST libraries are not yet sufficiently comprehensive, our calculations were performed across all tissues. For example, 12 ESTs and 14 ESTs represent cases of exon inclusion and exon exclusion in the cAMP-response element-binding protein gene, respectively. In this case, the exon exclusion fraction is 14/(12 + 14) = 0.54.
Fig. 3.

Defining the exon exclusion fraction. To quantitatively characterize frequency of alternative splicing, the exon exclusion fraction is defined as the number of ESTs with an excluded exon divided by the number of ESTs spanning a site of alternative splicing, fex = Nex/(Nex + Nin). This fraction is also determined experimentally as a ratio of DNA band intensities, fex = L[Iex/(Iex + Iin)]. Note the correlation between the computational and experimental values.
To test for correlations between the levels of transcription and alternative splicing, the exon exclusion fractions at each splice site were analyzed as a function of EST numbers covering this site. As shown in Fig. 4A, genes represented by a high number of ESTs show a low frequency of alternative splicing; that is, the majority is represented by a single isoform (i.e., exon exclusion fractions are close to 0 or 1). We chose sets of genes represented with various numbers of ESTs to confirm the computationally identified trends (Fig. 4 B and C). Prior knowledge of alternative splicing was not used in the selection of the gene set. The selected genes with a high number of ESTs encode proteosome components PSB1 and PSB3, the exosomal component RR46, and glucosyl transferase EXT2. According to computational results, the exon exclusion fractions were close to 0.01 for these genes. When tested experimentally, some of these genes do not show detectable splicing variants, whereas others show a very low frequency of alternative splicing (Fig. 4B and Fig. 8, which is published as supporting information on the PNAS web site). In contrast, a higher frequency of alternative splicing was observed computationally and confirmed experimentally for genes represented with low number of ESTs (Fig. 4C and Fig. 9, which is published as supporting information on the PNAS web site). Selected examples include the ion channel P2X5, the hematopoietic transmembrane receptors IL-7R and CD3D, and transcription factor subunit CBFB. For these genes, the exon exclusion fraction was closer to 0.5.
Fig. 4.

Correlation between transcription and frequency of alternative splicing. (A) For each alternative splice site, the exon exclusion fraction fex is plotted versus a number of ESTs covering this site (Nex + Nin). The points corresponding to experimentally tested genes are shown with arrows. Note that experimental data confirm the computational results. (B and C) Representative examples of the ubiquitously expressed (B) and tissue-specific (C) genes. Note that genes with high numbers of ESTs (ubiquitously expressed genes overrepresented in EST libraries) show a low level of exon exclusion and that genes with low numbers of ESTs (tissue-specific genes) show more equal frequencies of exon exclusion and inclusion. Additional PCR profiles and sequences are provided in Figs. 8 and 9.
Because ESTs generally originate from random sequencing efforts, the number of ESTs corresponding to a specific gene is representative of its expression level. Because only 5% of the ESTs in our data set were derived from the most contributing tissue (brain), we assumed that high EST representation characterizes ubiquitously expressed genes. We also assumed that a low number of ESTs characterizes tissue-specific genes. Our RT-PCR data support these assumptions (Figs. 4 B and C, 8, and 9).
To further confirm a correlation between a high frequency of alternative splicing and tissue-specific gene expression, we computationally compared distribution of exon exclusion fractions between tissue-specific and ubiquitous genes (Supporting Materials and Methods). Because EST libraries are not comprehensive, we normalized expression levels for genes that are represented with 10 or more ESTs across 102 tissues. Genes were defined tissue-specific if 30% or more of their ESTs originated from a single tissue (tissue specificity P value of <0.001). Genes were defined as ubiquitous if <20% of their ESTs originated from a single tissue (tissue specificity P value of >0.01). We found that the tissue-specific subset included 4-fold more genes with exon exclusion fractions within 0.5 ± 0.3, as compared with the subset of the ubiquitous genes. These experimental and computational data indicated that frequency of alternative splicing is higher for tissue-specific rather than ubiquitous genes. However, as shown in Fig. 4A, there are clearly examples of ubiquitously expressed genes with ratios close to 0.5.
Low Frequency of Exon Exclusion. Upon examination of the genes by RT-PCR, we noticed that they were usually represented by a single isoform. The formation of other isoforms is usually rare and tissue-specific. To explore this trend, we analyzed the distribution of the exon exclusion and exon inclusion events at identified alternative splice sites. If exon exclusion and inclusion were occurring at a similar frequency, we would expect to obtain a Gaussian-like distribution with a peak centered at 0.5. However, as shown in Fig. 5A, the inclusion or exclusion frequencies of individual exons were distributed in a skewed, highly nonrandom fashion. The exon exclusion fraction was <0.2 in nearly 60% of the analyzed alternative splice sites. Thus, at most sites of alternative splicing, exon exclusion was a rare event. Interestingly, very few sites showed an equal representation of both alternatively spliced isoforms. Four examples are presented in Fig. 5B, and the complete data set of 25 genes is included in Fig. 10, which is published as supporting information on the PNAS web site. As expected, the majority of the experimentally tested genes showed predominant formation of the long isoforms, whereas exon exclusion was rare and tissue-specific. The apoptosis regulator MCL1 and transcription factor TF3A gene products shown in Fig. 5B represent examples of predominant exon inclusion. In comparison, the chromodomain-helicase-DNA-binding protein-1 gene CHD1 is an example of predominant exon exclusion. The gene encoding amyloid-like protein APP2 was an example of approximately equal exon inclusion and exclusion frequencies. In general, the computationally determined exon exclusion frequencies were in good agreement with the experimental data (Fig. 5C). These findings suggest that, at the majority of alternative splicing sites, exon inclusion is constitutive and exon exclusion is a rare and tissue-specific event.
Fig. 5.

Low frequency of exon exclusion. (A) Distribution of alternative splice sites as a function of the exon exclusion fraction fex (solid red line). For comparison, a theoretical random distribution profile is shown with the dashed black line. (B) RT-PCR profiles of alternative splicing in representative genes. The gene names follow SwissProt nomenclature, and the exon exclusion values are indicated in parentheses. Here and in Figs. 8-10, note that for most genes, exon inclusion (upper bands) is prevalent in comparison to exon exclusion (lower bands). (C) Correspondence between experimental and computational data. The exon exclusion fractions fex calculated with the EST data Nex/(Nex + Nin) and determined experimentally with RT-PCR L[Iex/(Iex + Iin)] are plotted versus each other. In addition, a linear trend line is added (m = 0.92, r2 = 0.72).
Alternative Splicing Patterns Are Weakly Conserved in Human and Mouse. The genomic structures, specifically, exon and intron boundaries, of orthologous genes are highly conserved between human and mouse species (30). A reasonable assumption is that the patterns of alternative splicing would be similarly conserved. We experimentally analyzed 20 pairs of orthologous genes for conservation of alternative splicing patterns across the same tissues (Figs. 6 and 10). In this group of genes, the analyzed human exons were also present in mouse genes, according to the available transcript data. Surprisingly, no conservation of alternative splicing was detected in 16 of the 20 tested genes. Thirteen of the orthologous genes do not show any evidence for alternative splicing in murine tissues. In the other three cases, the splicing patterns are different. Representative results are shown in Fig. 6. No alternative splicing was detected for the mouse orthologues of the TF3A, CD3D, and KLF6 genes. In addition, different patterns of alternative splicing were detected for the murine orthologue of the CD22 gene. The APP2 gene represents an example of conserved alternative splicing of a single exon. Note that, even in the conserved cases, the ratios of two isoforms in particular tissues were not preserved. Our results suggest that patterns of alternative splicing are not conserved in most of the human and mouse genes.
Fig. 6.

Comparison of alternative splicing in human and mouse orthologues. Corresponding regions of the mouse and human orthologues were identified using the Ensembl database. All splicing patterns were confirmed by sequencing except those from mouse genes with no detectable alternative splicing. Note the lack of alternative splicing conservation in most of the analyzed genes. Additional PCR profiles and sequences are provided in Fig. 10.
Discussion
The ability to balance self-renewal and differentiation activities is a hallmark property of all stem cell populations. The molecular mechanisms that govern such stem cell fate decisions are largely unknown. Unraveling such mechanisms requires a complete knowledge of the molecular repertoire available for stem cells. A number of microarray studies have revealed multiple genes preferentially expressed in stem cells (14, 15). In comparison, the extent and possible functional consequences of alternative splicing in stem cells are unknown with the exception of a few studies focused on individual genes (17). Therefore, we embarked on a genome-wide identification of splice variants in human and murine ES cells and in murine HSCs. To analyze alternative splicing in an unbiased and global yet grounded manner, we combined computational and experimental approaches. The computational alignment of stem-cell-specific ESTs uncovered hundreds of potential splice variants in ES cells and HSCs. RT-PCR and subsequent sequencing showed an 80% confirmation rate for the computationally predicted splice isoforms. Moreover, alternative splicing was found to extensively affect components of signaling pathways that are functional in stem cells, suggesting an important role of splice variations in self-renewal and differentiation. The frequencies of alternatively spliced genes in various stem cell populations were close to previous estimates, considering the number of available ESTs (7). Further accumulation of EST sequences from stem cells will increase the number of detected splice variants and help to determine their specificity in comparison to other tissues (31, 32). The full significance of the numerous alternatively spliced gene product variants identified in this study awaits comprehensive functional analyses. However, our findings indicate that the repertoire of gene products expressed in stem cells is extensively modified by alternative splicing.
To deepen our understanding of alternative splicing and its regulation and functional consequences, we subsequently analyzed its trends across all tissues. We found that ubiquitously expressed genes show a very low frequency of alternative splicing. It may be that the low-frequency splice variants represent the occasional infidelity of the splicing machinery. Theoretical arguments have estimated a 0.001 frequency of such errors (33). This value is similar to the frequencies at which we detect splicing variants in genes that encode proteosomal components. In contrast, tissue-specific genes appear to show a high frequency of alternative splicing. Previous studies of individual genes have shown that splicing is coupled to transcription by protein-protein interactions between components of the transcription and splicing complexes (34, 35). Taken together, these results suggest that, on the genome-wide level, coupling of transcription and splicing results in diversification of tissue-specific and regulatory gene products, with little effect on ubiquitous “housekeeping” genes. A supporting evolutionary argument is that ubiquitous transcripts responsible for crucial and general cellular processes have evolved not to be modified, whereas diversification is advantageous for tissue-specific gene products. This explanation is further strengthened by our observation of fast evolutionary changes in alternative splicing patterns. We found that these patterns were conserved for only 20% of the examined orthologous genes in the human and mouse species, despite the general conservation of their exon-intron boundaries. These observations are in agreement with results of a recently published study (36) and consistent with previous conclusions regarding the rapid evolution of alternatively spliced exons (37). Lack of conservation of the alternative splicing patterns may contribute to the previously observed differences in functional properties of analogous cell types, such as mouse and human ES cells (38).
At the molecular level, alternative splicing results from blocking of constitutive splicing sites or activation of weak (cryptic) sites (2, 4). For a large set of alternatively spliced genes, we observed that exon inclusion is predominant, whereas exon exclusion is rare and often tissue-specific. A similar conclusion was obtained in previous computational studies (37). Our experiments confirmed these computationally derived trends, which implies that exon inclusion is a default option in the overall expression process of these genes. Based on these observations, we hypothesize that repression of constitutively used splice sites in primary transcripts is responsible for the formation of most splice variants. Furthermore, such blocking is likely to occur in a tissue-specific manner. However, the observed bias toward exon inclusion may partially reflect an artificial effect from accumulations of ESTs with rare splice errors in ubiquitous genes, as discussed above.
Alternative splicing has been implicated in several cell fate decision systems (2, 4). According to our observations that multiple genes in stem cells undergo alternative splicing and that these genes often encode regulatory proteins, we hypothesize that stem cell molecular networks are more generally dependent on this posttranscriptional mechanism. Thus, understanding stem cell biology will require the complete catalog of splice variations in addition to comprehensive analyses of transcription. Our studies initiate such a catalog.
Supplementary Material
Acknowledgments
This work was supported by funds from the National Institute of Diabetes and Digestive and Kidney Diseases. M.P. was supported by the Burroughs Wellcome Fund Fellowship in Biological Dynamics.
Author contributions: M.P. designed research; M.P. and S.E.W. performed research; M.P., T.T.D., and L.C.K. analyzed data; and M.P. and I.R.L. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HSC, hematopoietic stem cell; NCBI, National Center of Biotechnology Information.
References
- 1.Sharp, P. A. (1994) Cell 77, 805-815. [DOI] [PubMed] [Google Scholar]
- 2.Lopez, A. J. (1998) Annu. Rev. Genet. 32, 279-305. [DOI] [PubMed] [Google Scholar]
- 3.Smith, C. W. & Valcarcel, J. (2000) Trends Biochem. Sci. 25, 381-388. [DOI] [PubMed] [Google Scholar]
- 4.Black, D. L. (2003) Annu. Rev. Biochem. 72, 291-336. [DOI] [PubMed] [Google Scholar]
- 5.Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, et al. (2001) Nature 409, 860-921.11237011 [Google Scholar]
- 6.Modrek, B. & Lee, C. (2002) Nat. Genet. 30, 13-19. [DOI] [PubMed] [Google Scholar]
- 7.Brett, D., Pospisil, H., Valcarcel, J., Reich, J. & Bork, P. (2002) Nat. Genet. 30, 29-30. [DOI] [PubMed] [Google Scholar]
- 8.Modrek, B., Resch, A., Grasso, C. & Lee, C. (2001) Nucleic Acids Res. 29, 2850-2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson, J. M., Castle, J., Garrett-Engele, P., Kan, Z., Loerch, P. M., Armour, C. D., Santos, R., Schadt, E. E., Stoughton, R. & Shoemaker, D. D. (2003) Science 302, 2141-2144. [DOI] [PubMed] [Google Scholar]
- 10.Zavolan, M., van Nimwegen, E. & Gaasterland, T. (2002) Genome Res. 12, 1377-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis, B. P., Green, R. E. & Brenner, S. E. (2003) Proc. Natl. Acad. Sci. USA 100, 189-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maniatis, T. & Tasic, B. (2002) Nature 418, 236-243. [DOI] [PubMed] [Google Scholar]
- 13.Phillips, R. L., Ernst, R. E., Brunk, B., Ivanova, N., Mahan, M. A., Deanehan, J. K., Moore, K. A., Overton, G. C. & Lemischka, I. R. (2000) Science 288, 1635-1640. [DOI] [PubMed] [Google Scholar]
- 14.Ivanova, N. B., Dimos, J. T., Schaniel, C., Hackney, J. A., Moore, K. A. & Lemischka, I. R. (2002) Science 298, 601-604. [DOI] [PubMed] [Google Scholar]
- 15.Ramalho-Santos, M., Yoon, S., Matsuzaki, Y., Mulligan, R. C. & Melton, D. A. (2002) Science 298, 597-600. [DOI] [PubMed] [Google Scholar]
- 16.Sharov, A. A., Piao, Y., Matoba, R., Dudekula, D. B., Qian, Y., VanBuren, V., Falco, G., Martin, P. R., Stagg, C. A., Bassey, et al. (2003) PLoS Biol. 1, 410-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molnar, A. & Georgopoulos, K. (1994) Mol. Cell. Biol. 14, 8292-8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klug, C. A., Morrison, S. J., Masek, M., Hahm, K., Smale, S. T. & Weissman, I. L. (1998) Proc. Natl. Acad. Sci. USA 95, 657-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salesse, S., Dylla, S. J. & Verfaillie, C. M. (2004) Leukemia 18, 727-733. [DOI] [PubMed] [Google Scholar]
- 20.Boguski, M. S., Lowe, T. M. & Tolstoshev, C. M. (1993) Nat. Genet. 4, 332-333. [DOI] [PubMed] [Google Scholar]
- 21.Hubbard, T., Barker, D., Birney, E., Cameron, G., Chen, Y., Clark, L., Cox, T., Cuff, J., Curwen, V., Down, T., et al. (2002) Nucleic Acids Res. 30, 38-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990) J. Mol. Biol. 215, 403-410. [DOI] [PubMed] [Google Scholar]
- 23.Bairoch, A. & Apweiler, R. (2000) Nucleic Acids Res. 28, 45-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skrabanek, L. & Campagne, F. (2001) Nucleic Acids Res. 29, E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brandenberger, R., Wei, H., Zhang, S., Lei, S., Murage, J., Fisk, G. J., Li, Y., Xu, C., Fang, R., Guegler, K., et al. (2004) Nat. Biotechnol. 22, 707-716. [DOI] [PubMed] [Google Scholar]
- 26.Okazaki, Y., Furuno, M., Kasukawa, T., Adachi, J., Bono, H., Kondo, S., Nikaido, I., Osato, N., Saito, R., Suzuki, H., et al. (2002) Nature 420, 563-573.12466851 [Google Scholar]
- 27.O'Carroll, D., Erhardt, S., Pagani, M., Barton, S. C., Surani, M. A. & Jenuwein, T. (2001) Mol. Cell. Biol. 21, 4330-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitchelmore, C., Kjaerulff, K. M., Pedersen, H. C., Nielsen, J. V., Rasmussen, T. E., Fisker, M. F., Finsen, B., Pedersen, K. M. & Jensen, N. A. (2002) J. Biol. Chem. 277, 7598-7609. [DOI] [PubMed] [Google Scholar]
- 29.Yong, K. L., Watts, M., Shaun Thomas, N., Sullivan, A., Ings, S. & Linch, D. C. (1998) Blood 91, 1196-1205. [PubMed] [Google Scholar]
- 30.Batzoglou, S., Pachter, L., Mesirov, J. P., Berger, B. & Lander, E. S. (2000) Genome Res. 10, 950-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu, Q., Modrek, B. & Lee, C. (2002) Nucleic Acids Res. 30, 3754-3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeo, G., Holste, D., Kreiman, G. & Burge, C. B. (2004) Genome Biol. 5, R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graveley, B. R. (2001) Trends Genet. 17, 100-107. [DOI] [PubMed] [Google Scholar]
- 34.Auboeuf, D., Honig, A., Berget, S. M. & O'Malley, B. W. (2002) Science 298, 416-419. [DOI] [PubMed] [Google Scholar]
- 35.Bentley, D. (2002) Curr. Opin. Cell Biol. 14, 336-342. [DOI] [PubMed] [Google Scholar]
- 36.Yeo, G. W., Van Nostrand, E., Holste, D., Poggio, T. & Burge, C. B. (2005) Proc. Natl. Acad. Sci. USA 102, 2850-2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Modrek, B. & Lee, C. J. (2003) Nat. Genet. 34, 177-180. [DOI] [PubMed] [Google Scholar]
- 38.Daheron, L., Opitz, S. L., Zaehres, H., Lensch, W. M., Andrews, P. W., Itskovitz-Eldor, J. & Daley, G. Q. (2004) Stem Cells 22, 770-778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.