

Abstract
Direct evidence for the requirement of transcriptional feedback repression in circadian clock function has been elusive. Here, we developed a molecular genetic screen in mammalian cells to identify mutants of the circadian transcriptional activators CLOCK and BMAL1, which were uncoupled from CRYPTOCHROME (CRY)-mediated transcriptional repression. Notably, mutations in the PER-ARNT-SIM domain of CLOCK and the C terminus of BMAL1 resulted in synergistic insensitivity through reduced physical interactions with CRY. Coexpression of these mutant proteins in cultured fibroblasts caused arrhythmic phenotypes in population and single-cell assays. These data demonstrate that CRY-mediated repression of the CLOCK/BMAL1 complex activity is required for maintenance of circadian rhythmicity and provide formal proof that transcriptional feedback is required for mammalian clock function.
Circadian clocks have been proposed to consist of autoregulatory loops that use transcriptional feedback and regulated protein turnover to maintain 24-h periodicity1–3. However, the universal requirement for transcriptional feedback repression has been questioned recently by two studies showing that it is not required for circadian rhythms in cyanobacteria4,5. Circadian feedback repression in mammals is believed to be mediated by the CRY1 and CRY2 and PERIOD (PER1 and PER2) proteins. CRY and PER are hypothesized to autoregulate their own expression by repressing the heterodimeric complex of the basic helix-loop-helix (bHLH) PER-ARNT-SIM (PAS) domain transcriptional activators CLOCK and BMAL1, which bind to E-box elements in the CRY6 and PER7,8 promoters. Indirect support for this feedback mechanism comes from biochemical, molecular and genetic evidence: (i) CRY and PER physically interact with CLOCK/BMAL1, (ii) CRY and PER repress CLOCK/BMAL1 activity in steady-state transcriptional assays9–12 and (iii) CRYand PER genes are required for maintenance of circadian rhythms in mice13–15. However, direct evidence for the requirement of CRY-mediated repression of CLOCK/BMAL1 transcriptional activity in the maintenance of circadian clock function has been not been shown.
RESULTS
Mutagenesis and functional screening of CLOCK
In order to determine the requirement of feedback repression in circadian clock function, we sought to identify CLOCK alleles that were insensitive to CRY1 repression but that maintained normal transcriptional activity. We generated a random library of ~6,000 mutant human CLOCK alleles. We screened clones individually in cell-based reporter assays with wild-type BMAL1 cDNA and a PER1 promoter-luciferase construct7 in the presence of cotransfected CRY1. Of the approximately 6,000 CLOCK clones screened, three clones (Clock-1, Clock-2 and Clock-3) reproducibly maintained threefold or greater reporter activity in the presence or absence of CRY1 compared with wild-type CLOCK. We verified the reduced CRY1 sensitivity phenotype by 96-well assays of activity of CLOCK mutants with increasing amounts of CRY1 plasmid. When cotransfected with wild-type BMAL1 and the PER1 reporter, the Clock-1, Clock-2 and Clock-3 clones maintained noticeably greater activity than wild-type CLOCK in the presence of 1 or 2.5 ng of CRY1 plasmid (Fig. 1a). However, cotransfection of 5 ng of CRY1 plasmid reduced the transcriptional activity of the mutants to a level similar to that of wild-type CLOCK, indicating that these clones are partially insensitive to CRY1 repression. Notably, these clones demonstrated similar transcriptional activities as wild-type CLOCK in the absence of cotransfected CRY1, suggesting that these mutations do not cause overt alterations in the heterodimerization, nuclear localization, DNA binding and transactivation properties of the mutant CLOCK/BMAL1 complex.
Figure 1.
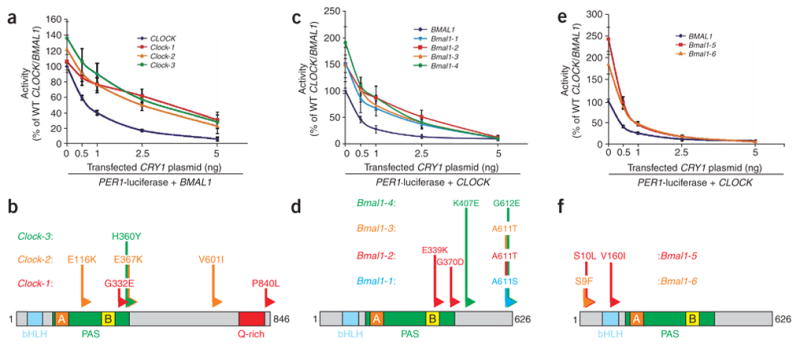
Mutations in CLOCK and BMAL1 confer insensitivity to CRY-mediated transcriptional repression without affecting CLOCK/BMAL1 transcriptional activity. (a,c,e) Results of cell-based transcriptional PER1-luciferase reporter assays with mutant CLOCK and BMAL1 clones in HEK293T cells. Plasmids expressing Flag-tagged wild-type or mutant CLOCK (a) or BMAL1 (c,e) cDNAs were transiently cotransfected with wild-type (WT) BMAL1 or CLOCK, respectively, PER1-luciferase reporter and 0–5 ng of CRY1 plasmid. Activity is expressed as the percentage of normalized PER1-luciferase activity in cells transfected with wild-type CLOCK/BMAL1 alone. Data are mean ± s.e.m. from independent experimental triplicates. (b,d,f) Domain locations of causative mutations. Schematic locations of amino acid changes within the CLOCK PAS-B domain (b) and BMAL1 C terminus (d) that confer CRY1 desensitization are indicated. Locations of protein domains are indicated for CLOCK (bHLH, blue, amino acids 35–85; PAS domain, green, amino acids 113–377; PAS-A repeat, orange, amino acids 128–170; PAS-B repeat, yellow, amino acids 283–329) and BMAL1 (bHLH, blue, amino acids 73–126; PAS domain, green, amino acids 148–439; PAS-A repeat, orange, amino acids 163–206; PAS-B repeat, yellow, amino acids 344–391). (f) Schematic location of amino acid changes within the N terminus of BMAL1 that confer CLOCK/BMAL1 hyperactivity: Bmal1-5, S10L and V160I; Bmal1-6, S9F.
DNA sequencing showed that all three of the CLOCK clones had missense mutations causing changes in amino acids C-terminal to the PAS-B repeat but within the PAS domain16 (also called the PAC domain; Clock-1, G332E; Clock-2, E367K; Clock-3, H360Y), in addition to other mutations (Fig. 1b). Notably, the mutations within the PAS domain were independently determined to be causative in the Clock-1 and Clock-2 mutants (Supplementary Fig. 1 online). In addition, the Clock-E367K mutant also had approximately threefold greater activity than the Clock-2 clone in the absence of CRY1, suggesting that the additional mutations within Clock-2 may cause this difference. We examined the importance of these PAS domain residues by generating the identical mutations in the CLOCK paralogue NPAS2 (also known as MOP4), which regulates circadian gene expression in central and peripheral tissues and is similarly repressed by CRY (refs. 11, 17–19). Corresponding mutations in the NPAS2 PAS domain reduced its sensitivity, compared with wild-type NPAS2, to 1 and 2.5 ng of CRY1 plasmid (Supplementary Fig. 1). Together, these results indicate that these mutations in the PAS domains of CLOCK and NPAS2 can partially relieve CRY1-mediated transcriptional repression.
Recently, the crystal structure of the PAS domain of the Drosophila melanogaster PER protein was solved, and a tryptophan residue was found to be important for homodimerization through intermolecular contacts with the PAS-A repeat20. As this conserved tryptophan (Trp362) in CLOCK is near the cluster of CRY1-desensitizing mutations, a Clock-W362A site-directed mutant was tested in PER1-luciferase reporter assays. The Clock-W362A mutant maintained significantly greater transcriptional activity than wild-type CLOCK in the presence of 1 and 2.5 ng of CRY1 plasmid (Supplementary Fig. 1). Thus, these mutations near the CLOCK PAS-B repeat may affect interactions with PAS-A repeats from other PAS proteins, such as the PER or BMAL1 proteins.
Mutagenesis and functional screening of BMAL1
The results of the CLOCK mutagenesis screen suggested that the corresponding residues near the BMAL1 PAS-B repeat may also be important for CRY-mediated repression. Therefore, we generated BMAL1 clones containing the analogous PAS domain mutations by site-directed mutagenesis and examined their activities in PER1 reporter assays. In contrast to CLOCK, BMAL1 PAS domain mutants had no difference in their sensitivities to CRY1 compared with wild-type BMAL1 (Supplementary Fig. 1). In order to uncover the BMAL1 residues required for sensitivity to CRY, we randomly mutagenized plasmids expressing Flag-tagged BMAL1 and screened them with wild-type CLOCK for reduced sensitivity to CRY1 as above. Of 6,000 clones screened, four BMAL1 clones displayed 2.5-fold or greater desensitization to CRY1. The four BMAL1 clones showed similar desensitized activities to 0.5–2.5 ng of CRY1 plasmid as did the CLOCK PAS domain mutants (Fig. 1c). However, in contrast to the CLOCK mutants, these BMAL1 clones were 1.5- to twofold more active in the absence of transfected CRY1, which may reflect partial insensitivity to endogenous CRY, and were completely repressed by 5 ng of CRY1 plasmid. DNA sequencing showed that the four clones contained mutations causing amino acid changes at position 611 or 612 near the C terminus (Fig. 1d; Bmal1-1, A611S; Bmal1-2, A611T; Bmal1-3, A611T; Bmal1-4, G612E). Notably, a single site-directed Bmal1-G612E mutant phenocopied the CRY1-desensitized activity of Bmal1-4 (Supplementary Fig. 1), which contained an additional mutation outside of the C terminus. These results confirm that, although CLOCK uses the PAS domain for responsiveness to CRY1, amino acid residues within BMAL1 C terminus are important for CRY1-mediated repression.
Identification of hyperactive BMAL1 mutants
In addition to identifying CRY1-desensitized mutants, we isolated two gain-of-function BMAL1 clones that showed 2- to 2.5-fold greater activity in the absence of CRY1 but were normally repressed by CRY1 (Fig. 1e). DNA sequencing detected missense mutations resulting in changes at the N terminus upstream of the bHLH region of BMAL1 (Fig. 1f; Bmal1-5, S10L; Bmal1-6, S9F). BMAL1 is known to be phosphorylated by casein kinase Iε21, which has a prominent role in regulating activities within the core circadian feedback loop22. To address the possibility that these mutations altered the phosphorylation state of these serine residues, we performed site-directed mutagenesis to change Ser9 or Ser10 to alanine, mimicking the unphosphorylated residue, or to glutamate, mimicking the phosphorylated form. Single- or double-alanine and glutamate exchanges in the BMAL1 N terminus as well as deletion of the first 11 N-terminal amino acids (Supplementary Fig. 1) all resulted in enhanced PER1 reporter activity, suggesting that phosphorylation states of Ser9 and Ser10 do not modulate BMAL1 activity.
Molecular analyses of CLOCK/BMAL1 double mutants
The identification of CLOCK and BMAL1 mutants partially insensitive to CRY suggested that the double CLOCK/BMAL1 mutant complex may show greater insensitivity. This hypothesis was tested by examining the activities of various combinations of CLOCK and BMAL1 mutants in the presence of 0 to 50 ng of CRY1 plasmid in PER1-luciferase reporter assays. With higher amounts (10–50 ng) of CRY1 plasmid, the activity of the CLOCK and BMAL1 single mutants (Fig. 2a–c) was reduced to a basal level, equivalent to that of the reporter (Fig. 2a–c). In contrast, CLOCK/BMAL1 double mutants showed marked and synergistic desensitization to high levels of CRY1 plasmid (Fig. 2a–c). Even with 25 ng of cotransfected CRY1 plasmid, all CLOCK/BMAL1 double mutants retained approximately 50% of reporter activation compared with wild-type CLOCK/BMAL1 lacking CRY1 plasmid. This synergistic insensitivity to CRY1 repression was also seen with NPAS2/BMAL1 and Clock-W362A/Bmal1-3 double mutants (Supplementary Fig. 2 online) but was not when Bmal1-5 or Bmal1-6 hyperactive alleles were coexpressed with mutant Clock-3 (Supplementary Fig. 2. Thus, BMAL1 hyperactivity alone did not account for synergistic insensitivity to CRY1.
Figure 2.
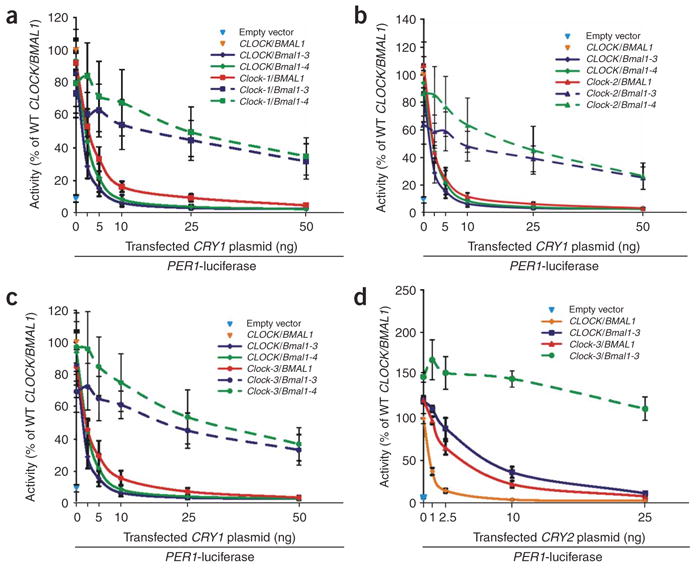
Coexpression of CLOCK and BMAL1 desensitized mutants confers synergistic insensitivity to CRY1 in HEK293T cells. (a–c) Various combinations of single and double CRY-desensitized CLOCK and BMAL1 mutant cDNAs were cotransfected with the PER1 reporter and 0–50 ng of CRY1 plasmid. PER1 reporter activity alone (blue triangle) or with wild-type CLOCK/BMAL1 (orange triangle) are also displayed. Activity is expressed as the percentage of normalized PER1-luciferase activity in cells transfected with wild-type CLOCK/BMAL1 alone. Solid lines: CLOCK and BMAL1 single mutants. Dashed lines: double mutants. (d) Luciferase activities were analyzed from combinations of single and double Bmal1-3 and Clock-3 mutant cDNAs that were cotransfected with the PER1 reporter and 0–25 ng of CRY2 plasmid. PER1 reporter activity alone (blue triangle) or with wild-type CLOCK/BMAL1 (green triangle) are also shown. Activity is expressed as the percentage of normalized PER1-luciferase activity in transfection with wild-type CLOCK/BMAL1 and no CRY1 plasmid. Data are mean ± s.e.m. determined from independent experimental triplicates.
To determine if these mutations also block repression by mammalian Cryptochromes in general, we also examined Clock-3/Bmal1-3 double mutants for sensitivity to CRY2. Notably, single Clock-3 and Bmal1-3 mutants were desensitized to low amounts of CRY2, whereas the double mutants showed synergistic insensitivity to CRY2 (Fig. 2d). In sum, the synergistic, rather than additive, insensitivity of these double mutants suggest that the CLOCK/BMAL1 complex acts as the target of CRY-mediated repression.
Physical interactions between mutant CLOCK/BMAL1 and CRY
Three possible biochemical mechanisms can explain how the mutant CLOCK/BMAL1 heterodimers could overcome CRY repression: enhanced expression or stability of mutant proteins, enhanced formation of mutant complexes, or reduced interactions between mutant CLOCK/BMAL1 complexes and CRY proteins. Protein blotting of epitope-tagged CLOCK and BMAL1 proteins coexpressed in HEK293T cells did not show any significant differences in the mutant protein levels relative to the wild-type forms (Supplementary Fig. 3 online). To determine if the CRY-desensitized mutants show enhanced heterodimer formation in the presence of CRY, we performed native coimmunoprecipitations with antibodies against Flag (anti-Flag) on HEK293T cell extracts containing Flag-tagged wild-type or mutant BMAL1 and untagged wild-type or mutant CLOCK. Protein blotting showed that wild-type CLOCK copurified with wild-type Flag-BMAL1 but did not copurify with the Flag epitope alone (Fig. 3a). In contrast, for single- or double-mutant combinations of CLOCK and BMAL1, there were moderately greater levels of mutant CLOCK/BMAL1 heterodimers than in the wild-type combination (Fig. 3a). This was similar to the increased amounts of wild-type CLOCK that the hyperactive Bmal1-5 or Bmal1-6 proteins bound relative to wild-type BMAL1 in the absence of cotransfected CRY1 (Supplementary Fig. 3>). Notably, the interactions between CLOCK and BMAL1 seemed to be disrupted in the presence of CRY1 (Supplementary Fig. 3) and thus may represent a mechanism contributing to CRY repression.
Figure 3.

Mutations in the CLOCK PAS domain and BMAL1 C terminus abrogate interactions between the CLOCK/BMAL1 complex and transcriptional repressors CRY1 and PER2. (a–c) Native coimmunoprecipitations (co-IPs) were performed on cell extracts from HEK293T cells transiently transfected with plasmids expressing epitope-tagged circadian components. (a) Flag-tagged wild-type or mutant BMAL1 protein was coexpressed with or without CRY1 and various untagged CLOCK alleles and precipitated with anti-Flag. Copurified proteins were visualized by protein blotting with antibodies to Flag or CLOCK. (b) Co-IPs were performed with anti-MYC on extracts from cells coexpressing MYC-tagged CRY1, combinations of various Flag-tagged CLOCK and BMAL1 CRY-desensitized alleles and Flag-tagged PER2. Anti-Flag and CRY1 antibodies were used in protein blotting. (c) Flag-PER2 was coprecipitated (using anti-Flag) from cells coexpressing additional combinations of untagged CLOCK and BMAL1 alleles. Protein blotting with anti-Flag, anti-CLOCK and anti-BMAL1 was used to identify copurified proteins.
Previous studies have shown that CRY can physically interact with the CLOCK/BMAL1 complex10,11,23 and that this interaction is important for CRY-mediated repression6. To examine the interactions with CRY, wild-type and mutant Flag-tagged CLOCK/BMAL1/PER2 complexes were affinity purified with MYC-tagged CRY1 by coimmunoprecipitation. MYC-tagged CRY1 appeared to bind substantially less CLOCK and Bmal1 single-and double-mutant complexes (Fig. 3b) compared with the wild-type complex (Fig. 3b) but maintained direct interactions with PER2, as expected9,11. Notably, coimmunoprecipitations of Flag-PER2 showed that the CRY-desensitizing mutations in CLOCK also blocked interactions with PER2 when coexpressed with either wild-type or mutant BMAL1 (Bmal1-4, Fig. 3c, and Bmal1-3, data not shown). However, interactions with wild-type CLOCK and mutant BMAL1 heterodimers were unaffected (Fig. 3c and data not shown), supporting the earlier notion that the CLOCK PAS domain may mediate interactions with PER. Together, these results suggest that the CRY insensitivity phenotypes resulted from reduced interactions between mutant CLOCK/BMAL1 complexes and the CRY/PER repressor proteins, as well as moderately enhanced CLOCK/BMAL1 heterodimer formation.
Real-time analysis of circadian gene expression
The prevailing transcriptional feedback model predicts that impairment of CRY-mediated repression should have marked effects on circadian expression of the PERIOD genes. This notion is supported by in vivo observations that expression of PER1 and PER2 is constitutively elevated in Cry1/Cry2 double knockout mice14,24. To determine whether these mutations in CLOCK and BMAL1 cause phenotypic changes in circadian gene expression, we performed real-time bioluminescence assays in an oscillating mammalian cell model25,26. This approach allows for a continuous and quantifiable report of circadian promoter activity from the same transfected cells over multiple days and enables statistical evaluation of important parameters of circadian clock function such as period length and amplitude. Mouse NIH3T3 fibroblasts were transfected with plasmids containing destabilized luciferase driven by the PER2 or SV40 promoters along with various combinations of BMAL1 and CLOCK alleles, stimulated with forskolin to synchronize circadian rhythmicity, and then were monitored in real-time for circadian luciferase activity (Fig. 4a,b and Supplementary Fig. 4 online). Cotransfection of wild-type CLOCK and BMAL1 did not substantially alter rhythmicity compared with cells transfected with empty vector, as their period lengths were 21.4 h ± 0.4 h (mean ± s.d., Supplementary Fig. 4). In contrast, transfection of either of the single CLOCK or BMAL1 mutant alleles resulted in substantial impairment of circadian rhythmicity after one or two cycles of oscillations when compared with wild-type CLOCK/BMAL1 (Fig. 4a and Supplementary Fig. 4). Notably, cotransfection of CRY-insensitive mutant CLOCK and BMAL1 together resulted in the loss of circadian PER2 promoter activity (Fig. 4b and Supplementary Fig. 4). Thus, transcriptional repression of CLOCK/BMAL1 by CRY is required for circadian E-box activity.
Figure 4.

Coexpression of CLOCK/BMAL1 mutant heterodimers that are insensitive to CRY repression ablates circadian E-box and RORE activities in NIH3T3 cells. Plasmids expressing Flag-tagged CLOCK and BMAL1 alleles were transiently cotransfected with the PER2-destabilized luciferase (dLuc) reporter plasmid into NIH3T3 cells. (a,b) PER2 promoter activities in NIH3T3 cells transfected with single (a) or double (b) CRY1-insensitive CLOCK and BMAL1 mutants were monitored over 5 d. (c,d) BMAL1 promoter activities in NIH3T3 cells transfected with single (c) or double (d) CRY-insensitive mutants of CLOCK and BMAL1 were monitored over 6 d. All reporter activities were normalized such that the median wild-type luciferase activity over the time-course was 100%.
In addition to PER and CRY, rhythmic expression of BMAL1 mRNA is also under circadian clock regulation12. However, the BMAL1 promoter does not have E-box sites and instead contains retinoic acid–related orphan nuclear receptor binding elements (RORE)25,27, whose activities are reciprocally controlled by the rhythmically expressed transcriptional repressor REV-ERBα27 and activator Rora28,29. As an additional test for circadian clock function, the effects of mutant CLOCK and BMAL1 on rhythmic RORE activity were examined by real-time assays with a BMAL1-destabilized luciferase (BMAL1-dLuc) reporter. Similar to results with the PER2-dLuc reporter, transfection of single CLOCK or BMAL1 mutants resulted in the decreased amplitude of cycling of BMAL1-dLuc activity compared with wild-type CLOCK/BMAL1 (Fig. 4c and Supplementary Fig. 4). Moreover, this decrease in cycling amplitude was further exacerbated upon cotransfection of the double mutant heterodimer (Fig. 4d and Supplementary Fig. 4). These results are probably due to misexpression of endogenous REV-ERBα, which is regulated by CLOCK/BMAL1 and CRY27 through E-box elements in the REV-ERBα promoter. Consistent with this, cotransfection of wild-type CLOCK/BMAL1 inhibited steady-state BMAL1 promoter activity, whereas additional cotransfection of CRY1 caused activation (Supplementary Fig. 5 online). In contrast, CRY-mediated activation of the BMAL1 reporter was abrogated when cotransfected with the CLOCK/BMAL1 double mutants (Supplementary Fig. 5 and data not shown). These results indicate that transcriptional repression of CLOCK/BMAL1 by CRY is also required for circadian BMAL1 expression through ROR elements, which is dependent upon transcriptional, translational and posttranslational actions of endogenous cellular factors.
Role of circadian feedback repression in single cells
The arrhythmic PER2 expression seen from a population of cells expressing the double mutant CLOCK/BMAL1 complex may be due to the disruption of oscillator function or a lack of synchrony between individual rhythmic cells. In order to address these possibilities, quantitative imaging of PER2-luciferase reporter activity was measured from individual NIH3T3 fibroblasts by an approach similar to that used in analyzing BMAL1 reporter rhythms from single cells30. As with the whole-well assays, the median reporter activity for the population of imaged individual fibroblasts coexpressing wild-type CLOCK/BMAL1 oscillated rhythmically (Fig. 5a). In contrast, the population of individual Clock-1/Bmal1-3 mutant cells (Fig. 5b) was visibly arrhythmic. Individual reporter activities from single wild-type cells were rhythmic (Fig. 5c and Supplementary Fig. 6 online), as expected, whereas individual Clock-1/Bmal1-3 double mutant cells showed arrhythmic reporter activities (Fig. 5d and Supplementary Fig. 7 online). These differences in activity patterns were evaluated by autocorrelation (see Methods) and COSOPT31 analyses, two independent statistical methods that score the circadian rhythmicity of experimental time-course data. Reporter activity traces from each wild-type or double mutant cell were assigned autocorrelation and COSOPT (Multiple Measures Corrected minus β, MMC-β) significance scores (Fig. 5c,d). Statistically significant differences between wild-type (n = 133) and double mutant (n = 133) cells were observed from the individually assigned autocorrelation (Wilcoxon test, P = 6.8 × 10−11) and MMC-β (Wilcoxon test, P = 1.1 × 10−13) scores. Thus, both visual inspection and statistical analyses indicate that the double mutant cells differ from rhythmic wild-type cells. We also observed this arrhythmic behavior in individual Clock-3/Bmal1-4 double mutant cells (Supplementary Fig. 8 online). Therefore, the arrhythmicity seen upon expression of double CLOCK/BMAL1 mutants in population studies was a reflection of the activity of mutant complexes in single cells rather than desynchrony of individual cellular oscillators.
Figure 5.

Coexpression of CLOCK/BMAL1 mutant heterodimers impairs circadian rhythmicity in individual cells. (a–d) PER2-luciferase reporter activities from individual NIH3T3 cells (n = 133) transfected with Flag-tagged wild-type CLOCK/BMAL1 (a,c) or double-mutant Clock-1/Bmal1-3 (b,d) were monitored over 3 d. Reporter activities from each wild-type (a) or double-mutant (b) cell were normalized such that the maximum bioluminescence value was set to 100% for each panel. The mean reporter activity for all analyzed single cells at each time point is indicated by a black line. Normalized bioluminescence activities from each wild-type (c) or double-mutant (d) cell were detrended and ranked according to corresponding significance by autocorrelation (upper panels) or COSOPT (lower panels). Autocorrelation and MMC-β values for each cell are depicted to the right of each heat map from 0 (bottom, dark blue) to 1 (top, white). Red and green denote high and low normalized reporter activities, respectively. Results shown are representative of duplicate experiments.
DISCUSSION
The current molecular model for the mammalian circadian clock includes transcriptional feedback repression by CRY and PER proteins3,32. Evidence for this mechanism is based on observations that CLOCK/BMAL1 activates CRY and PER gene expression, whose protein products, in turn, repress CLOCK/BMAL1 activity in transient transfection assays. Repression is believed to be mediated through physical interactions, as CRY associates with CLOCK/BMAL1 in coimmunoprecipitation and yeast two-hybrid assays. Finally, both Cry1/Cry2 and Per1/Per2 double knockout mice are behaviorally arrhythmic. While this evidence is strongly supportive for such a mechanism, functional requirement for feedback repression in the mammalian circadian clock has not been previously uncovered. Proof of this necessity is of import, as recent studies of the cyanobacterial clock found that circadian oscillations in protein phosphorylation can be maintained in the absence of transcriptional feedback repression in vivo5 and with purified proteins in vitro4. Therefore, we sought to formally test the requirement of CRY-mediated transcriptional feedback repression in the mammalian circadian clock by developing and implementing a new, unbiased cellular genetics approach that uses robust mutagenesis techniques and mammalian cell–based screening.
Using this approach, we identified an allelic series of CLOCK and BMAL1 clones that were uncoupled from CRY-mediated repression. Coexpression of these mutants in fibroblasts disrupted the rhythmic activities of both E-box and ROR elements, the two primary transcription factor binding sites used by the circadian oscillator26. These data provide direct evidence that CRY-mediated feedback repression of the CLOCK/BMAL1 complex is required for mammalian clock function. Notably, some oscillatory properties of the mammalian circadian clock, such as protein phosphorylation, may remain intact upon uncoupling of transcriptional feedback repression. Although it is likely that the mammalian clock is governed by a combination of both transcriptional and nontranscriptional feedback mechanisms, any residual circadian properties that remain upon uncoupling of transcriptional feedback are insufficient to maintain circadian transcriptional output and molecular clock function.
Finally, we predict that application of cellular genetics technology will have a significant impact on mammalian biology as similar approaches have had on prokaryotic and yeast biology.
METHODS
Mutant generation and screening protocols are provided in Supplementary Methods online.
Cell-based reporter assays
Luciferase reporter assays were performed in triplicate wells of 96-well white Co-Star bioassay plates (Corning). Total plasmid DNA (750 ng) was incubated with 150 μl of serum-free Dulbecco’s modified Eagle’s minimal essential medium (DMEM; GIBCO) and 2.25 μl FuGENE6 for 30 min in microcentrifuge tubes. For the PER1 reporter assays, we used the following amounts of plasmid DNA per well: 25 ng pGL3-mPER1, 50 ng pCMV-mBMAL1, 120 ng pCMV-hCLOCK or hNPAS2 and 5 ng pCMV-β-GALACTOSIDASE. In assays with CRY1 or CRY2, indicated amounts of pCMV-mCRY plasmid were compensated with empty pCMV-Sport6 vector to total 50 ng. HEK293T cells were cultured to 80% confluence at 37 °C with 5% CO2 and were harvested in DMEM/20% fetal bovine serum (FBS; GIBCO)/0.2 mM nonessential amino acids (NEAA; GIBCO)/2× penicillin-streptomycin-glutamine (PSG; GIBCO) at 8 × 105 cells ml−1. We dispensed 50 μl of harvested cells into individual wells in triplicate of the 96-well bioassay plate. After 30 min, 50 μl of plasmid DNA/DMEM/FuGENE6 mix was dispensed into the appropriate well with cells and incubated at 37 °C (5% CO2) for 24 h. Luciferase and β-galactosidase activities were analyzed with Dual Light Kit (Tropix) and Acquest reader (LJL Biosystems) according the manufacturers’ specifications. Ratios of luciferase:β-galactosidase activities from technical triplicates were averaged and normalized as the percent of luciferase:β-galactosidase activity ratios with wild-type CLOCK and BMAL1 plasmids in the absence of CRY. Final percentage activities were calculated from three independent experiments.
Native coimmunoprecipitations and protein blotting
We seeded 6 × 106 HEK293T cells on 10-cm Petri dishes with 7 ml DMEM/10% FBS/0.1 mM NEAA/1× PSG and incubated them at 37 °C/5% CO2 for 20 h. Cells were then transfected with 6 μg of total plasmid DNA in 100 μl of serum-free DMEM and 18 μl of FuGENE6. Twenty-four hours posttransfection, cells were washed and harvested with Dulbecco’s phosphate buffered saline (DPBS, GIBCO) and pelleted by centrifugation. Cell pellets were solubilized in 1 ml TGED buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 5% (vol/vol) glycerol, 0.5 mM DTT), complete protease inhibitors (Roche Applied Sciences) and 0.5% Triton X-100. Soluble lysate was cleared by centrifugation for 15 min at 13,000g at 4 °C. Six microliters of anti-Flag (Sigma-Aldrich) or anti-MYC (Pierce Biotechnology) conjugated to Sepharose beads were added to 1 ml of cleared lysate and incubated for 2 h at 4 °C with agitation. Bound proteins were then washed twice with TGED + 0.5% Triton, twice with TGED + 1% Triton, three times with TGED at 250 mM NaCl + 1% Triton and twice with TGED. Bound proteins were eluted with 50 μl of Laemmeli’s Reducing Sample Buffer (Biorad Laboratories), heated to 42 °C for 5 min and resolved by SDS-PAGE electrophoresis (Biorad Laboratories). Copurified proteins were identified by protein blotting with rabbit polyclonal antibodies to Flag (Sigma-Aldrich), CLOCK (Affinity BioReagents), BMAL1 (Affinity BioReagents) or mCRY1 (a gift from S. Panda, The Salk Institute, La Jolla, California). For comparison of protein expression from cotransfected CLOCK and BMAL1 cDNAs, equal amounts of cleared cell lysates were processed with 1× sample buffer and resolved by protein blotting with anti-Flag or appropriate antibodies. Results of co-IP and protein blotting experiments were independently repeated three times.
Additional plasmid construction
All site-directed mutations were made with the QuikChange Mutagenesis kit (Stratagene). Wild-type and mutant untagged pCMV-BMAL1, CLOCK, NPAS2, CRY1 and CRY2 constructs were generated by cloning the coding sequences for each gene into the pCMV-Sport6 vector (Invitrogen) by PCR and standard recombinant DNA techniques. The Flag-mPER2 plasmid was constructed by PCR amplification and cloning into the SpeI and NotI restriction sites of pFlag. MYC-tagged CRY1 and BMAL1 constructs were generated by PCR amplification with primers containing in-frame EcoRI and XhoI restriction sites and cloning into the identically digested pTag3C vector (Stratagene). The PER2-dLuc and PER2-Luc reporters were generated by cloning the mouse PER2 promoter (−219 to +76 nucleotides from transcriptional start site) into pGL3-dLuc25 and pGL3-Basic (Promega), respectively. The BMAL1-dLuc reporter was generated by cloning the mouse BMAL1 promoter from pGL3-BMAL1 (ref. 28) into the dLuc plasmid. The DNA sequences of all constructs generated in this study were verified. The pGL3P-mPER1 reporter7 and Rora28 plasmids are described elsewhere.
Real-time whole-well circadian reporter assays
Whole-well, real-time circadian assays were performed as previously described26 with the following modifications. NIH3T3 cells (American Type Culture Collection) were grown in DMEM supplemented with 10% FBS (JRH Biosciences) and antibiotics (25 units ml−1 penicillin, 25 μg ml−1 streptomycin; GIBCO). Cells were plated at 5 × 104 cells per well in 24-well plates 24 h before transfection. Cells were transfected with FuGENE6 according to the manufacturer’s instructions. Cells in each well were transfected with 0.32 μg (total) of plasmid (0.08 μg of PER2, BMAL1 or SV40-promoter reporter plasmids and 0.24 μg of pEGFP-N3 or pFlag-BMAL1/CLOCK variants). After 72 h, medium in each well was replaced with 500 μl of culture medium (DMEM/10% FBS) supplemented with 10 mM HEPES (pH 7.2), 0.1 mM luciferin (Promega), antibiotics and 0.01 μM forskolin (Nacalai Tesque). Bioluminescence was measured with photomultiplier tube (PMT) detector assemblies (Hamamatsu Photonics). The modules and cultures were maintained in a darkroom at 30 °C and interfaced with computers for continuous data acquisition. Photons were counted for 2 min at 24-min intervals.
Real-time single-cell bioluminescence imaging
We plated 1 × 105 NIH3T3 fibroblasts on 35-mm dishes 24 h before transfection. Plated fibroblasts were then transfected for 72 h with 0.6 μg pGL3-PER2 and 1 μg each of CLOCK or BMAL1 alleles and 3 μl FuGENE6 and were stimulated for 2 h with 0.01 μM forskolin. For Flag-tagged wild-type CLOCK/BMAL1 and Clock-1/Bmal1-3 transfections, medium was replaced with 2 ml fresh medium containing luciferin as described for whole-well assays. We sealed the 35-mm culture dishes with cover slips, placed them on the stage of a luminescence microscope (Olympus) and incubated them at 30 °C (Olympus). Bioluminescence was imaged by an Olympus 10× objective and transmitted to a cooled CCD camera (ORCA-AG C4742-80-12AG, Hamamatsu Photonics) mounted on the bottom port of the microscope. We used 4 × 4 binning of the 336 × 256 pixel array for single-cell measurement. Time-lapse images were collected at 30-min intervals with 25-min exposures by a computer using an image analysis program (AquaCosmos, Hamamatsu Photonics). The protocol for analysis of Clock-3/Bmal1-4 single cells is provided in Supplementary Methods.
Image and data processing for single-cell imaging
Images were analyzed using MetaMorph (Universal Imaging) and corrected for bias and dark current by subtracting a background image. Luminescence intensity was measured within a region of interest defined manually for each cell. The position of the region was adjusted, if necessary, to accommodate cell movement. Cosmic ray artifacts were removed by comparing the bioluminescence intensity with the corresponding value calculated from temporally adjacent images. When bioluminescence intensity of a cell at a certain time point was 50% greater than the average of those of the temporally adjacent data, the value was replaced with the average of the adjacent data. In some experiments, cosmic ray artifacts were removed by pixel-wise minimization of temporally adjacent images. Remaining data processing of extracted single-cell bioluminescence intensity data was conducted with Mathematica (Wolfram Research).
Rhythmicity, period length and amplitude analysis of real-time bioluminescence data
Bioluminescence time-series data beginning 21 h after forskolin stimulation were used for analysis in order to distinguish endogenous circadian oscillation from acute effects of stimulation. Bioluminescence data were detrended by using the trend curve calculated by the smoothing spline method33. The smoothing parameter for this calculation was set such that its frequency-response was 50% at a frequency of about two cycles (42 h) of the typical circadian period (~21 h) observed in fibroblasts transfected with empty vector at 30 °C. Then, autocorrelation of the detrended bioluminescence time-series data was calculated within the range of 16–28 h to determine the circadian period of oscillation. Statistical significance (with 0 as most significant and 1 as least significant) of circadian oscillation was evaluated by comparing the strongest autocorrelation of the detrended data within the range of 16–28 h against that of white noise (theoretical value for the autocorrelation of white noise is , where N is the number of time points). COSOPT calculations were performed as previously described31 with period length parameters of 16–28 h.
The effects on amplitude of cycling by coexpression of CRY-insensitive mutants on circadian oscillation in whole-well assays were determined by measuring the amplitude difference (percentage variation) between maximum and minimum bioluminescence relative to wild-type CLOCK/BMAL1 within 24–48 h from forskolin stimulation. In some circumstances, real-time recordings were omitted when peak and trough points from the luminescence traces could not be clearly identified.
Supplementary Material
Acknowledgments
This research was supported by the Novartis Research Foundation (L.J.M. and J.B.H.), a Rena and Victor Damone Postdoctoral Fellowship from the American Cancer Society (T.K.S.), US National Institute of Health (NIH) grants (D.K.W. and S.A.K.), Scripps Florida (T.K.S., J.E.B., and J.B.H.), RIKEN Center for Developmental Biology (H.R.U.), NIH/Silvio O. Conti Center for Neuroscience grant P50 MH074924-01 (J.E.B. and J.H.), RIKEN Strategic Programs (H.U. and H.R.U.), New Energy and Industrial Technology Organization (NEDO) Scientific Research grant (H.R.U.) and Scientific Research grant and Genome Network Project grant from the Japanese Ministry of Education, Culture, Sports, Science and Technology (H.R.U.). This is manuscript number 17558-CB of The Scripps Research Institute. We thank N. Gekakis, S. Reppert, C. Joazeiro, A. Curtis and G. FitzGerald for plasmids; S. Panda for anti-mCRY1; J. Zhang and T. Orth for robotics support; T. Kondo for high-throughput monitoring systems; M. Ukai-Tadenuma, J. Cartzendafner and J. Geskes for technical support and S. Panda, R. Van Gelder, T. Reyes, M. Pletcher, K. Hayes, B. Miller, M. Conkright and M. Givens for critical reading of the manuscript.
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
AUTHOR CONTRIBUTION STATEMENT
T.K.S. and J.B.H. developed the mutagenesis screening concept. R.G.Y., H.U., and H.R.U. developed the high through-put real-time assay concept. T.K.S. developed, constructed and screened the mutant libraries, performed the steady-state reporter and biochemical analyses, and created the related figures. R.G.Y. developed the statistical method specific for whole-well and single cell real-time bioluminescence experiments, analyzed the data and created the related figures. H.U. developed the high throughput real-time assays and performed the related experiments. J.E.B. generated data for Figure 2a, the Clock-W362A mutant, Supplementary Figure 6 and performed additional steady-state reporter and biochemical analyses. L.J.M. developed the screening protocol and constructed and screened the mutant libraries. T.J.K. and H.U. performed single-cell imaging experiments, analyzed the data and generated related figures. D.K.W. and S.A.K. performed single-cell imaging experiments and analysis. H.R.U. developed and analyzed all real-time assays. R.G.Y., H.U., T.J.K. and H.R.U. wrote the sections relevant to real-time assays. T.K.S. and J.B.H. wrote the remaining sections. All authors discussed the results and commented on the manuscript text.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96:271–290. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- 2.Young MW, Kay SA. Time zones: a comparative genetics of circadian clocks. Nat Rev Genet. 2001;2:702–715. doi: 10.1038/35088576. [DOI] [PubMed] [Google Scholar]
- 3.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 4.Nakajima M, et al. Reconstitution of circadian oscillation of cyanobacterial KaiC phosphorylation in vitro. Science. 2005;308:414–415. doi: 10.1126/science.1108451. [DOI] [PubMed] [Google Scholar]
- 5.Tomita J, Nakajima M, Kondo T, Iwasaki H. No transcription-translation feedback in circadian rhythm of KaiC phosphorylation. Science. 2005;307:251–254. doi: 10.1126/science.1102540. [DOI] [PubMed] [Google Scholar]
- 6.Etchegaray JP, Lee C, Wade PA, Reppert SM. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature. 2003;421:177–182. doi: 10.1038/nature01314. [DOI] [PubMed] [Google Scholar]
- 7.Gekakis N, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 8.Yoo SH, et al. A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Proc Natl Acad Sci USA. 2005;102:2608–2613. doi: 10.1073/pnas.0409763102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffin EA, Jr, Staknis D, Weitz CJ. Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science. 1999;286:768–771. doi: 10.1126/science.286.5440.768. [DOI] [PubMed] [Google Scholar]
- 10.Sangoram AM, et al. Mammalian circadian autoregulatory loop: a timeless ortholog and mPer1 interact and negatively regulate CLOCK-BMAL1-induced transcription. Neuron. 1998;21:1101–1113. doi: 10.1016/s0896-6273(00)80627-3. [DOI] [PubMed] [Google Scholar]
- 11.Kume K, et al. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98:193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- 12.Shearman LP, et al. Interacting molecular loops in the mammalian circadian clock. Science. 2000;288:1013–1019. doi: 10.1126/science.288.5468.1013. [DOI] [PubMed] [Google Scholar]
- 13.van der Horst GT, et al. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature. 1999;398:627–630. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- 14.Vitaterna MH, et al. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc Natl Acad Sci USA. 1999;96:12114–12119. doi: 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng B, et al. Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell. 2001;105:683–694. doi: 10.1016/s0092-8674(01)00380-4. [DOI] [PubMed] [Google Scholar]
- 16.Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- 17.Hogenesch JB, Gu YZ, Jain S, Bradfield CA. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci USA. 1998;95:5474–5479. doi: 10.1073/pnas.95.10.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McNamara P, et al. Regulation of CLOCK and MOP4 by nuclear hormone receptors in the vasculature: a humoral mechanism to reset a peripheral clock. Cell. 2001;105:877–889. doi: 10.1016/s0092-8674(01)00401-9. [DOI] [PubMed] [Google Scholar]
- 19.Reick M, Garcia JA, Dudley C, McKnight SL. NPAS2: an analog of clock operative in the mammalian forebrain. Science. 2001;293:506–509. doi: 10.1126/science.1060699. [DOI] [PubMed] [Google Scholar]
- 20.Yildiz O, et al. Crystal structure and interactions of the PAS repeat region of the Drosophila clock protein PERIOD. Mol Cell. 2005;17:69–82. doi: 10.1016/j.molcel.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 21.Eide EJ, Vielhaber EL, Hinz WA, Virshup DM. The circadian regulatory proteins BMAL1 and cryptochromes are substrates of casein kinase Iepsilon. J Biol Chem. 2002;277:17248–17254. doi: 10.1074/jbc.M111466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowrey PL, et al. Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science. 2000;288:483–491. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee C, Etchegaray J, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 24.Okamura H, et al. Photic induction of mPer1 and mPer2 in cry-deficient mice lacking a biological clock. Science. 1999;286:2531–2534. doi: 10.1126/science.286.5449.2531. [DOI] [PubMed] [Google Scholar]
- 25.Ueda HR, et al. A transcription factor response element for gene expression during circadian night. Nature. 2002;418:534–539. doi: 10.1038/nature00906. [DOI] [PubMed] [Google Scholar]
- 26.Ueda HR, et al. System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat Genet. 2005;37:187–192. doi: 10.1038/ng1504. [DOI] [PubMed] [Google Scholar]
- 27.Preitner N, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–260. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 28.Sato TK, et al. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43:527–537. doi: 10.1016/j.neuron.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 29.Akashi M, Takumi T. The orphan nuclear receptor RORalpha regulates circadian transcription of the mammalian core-clock Bmal1. Nat Struct Mol Biol. 2005;12:441–448. doi: 10.1038/nsmb925. [DOI] [PubMed] [Google Scholar]
- 30.Welsh DK, Yoo SH, Liu AC, Takahashi JS, Kay SA. Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr Biol. 2004;14:2289–2295. doi: 10.1016/j.cub.2004.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panda S, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- 32.Lowrey PL, Takahashi JS. Mammalian circadian biology: elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet. 2004;5:407–441. doi: 10.1146/annurev.genom.5.061903.175925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cook E, Peters K. The smoothing spline: A new approach to standardizing forest interior tree-ring width series for dendroclimatic studies. Tree-Ring Bull. 1981;41:45–53. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.