

Abstract
The fidelity of yeast RNA polymerase II (Pol II) was assessed in vivo with an assay in which errors in transcription of can1–100, a nonsense allele of CAN1, result in enhanced sensitivity to the toxic arginine analog canavanine. The Pol II accessory factor TFIIS has been proposed to play a role in transcript editing by stimulating the intrinsic nuclease activity of the RNA polymerase. However, deletion of DST1, the gene encoding the yeast homolog of TFIIS, had only a small effect on transcriptional fidelity, as determined by this assay. In contrast, strains containing a deletion of RPB9, which encodes a small core subunit of Pol II, were found to engage in error-prone transcription. rpb9Δ strains also had increased steady-state levels of can1–100 mRNA, consistent with transcriptional errors that decrease the normal sensitivity of the can1–100 transcript to nonsense-mediated decay, a pathway that degrades mRNAs with premature stop codons. Sequences of cDNAs from rpb9Δ strains confirmed a significantly increased occurrence of transcriptional substitutions and insertions. These results suggest that Rpb9 plays an important role in maintaining transcriptional fidelity, whereas TFIIS may serve a different primary purpose.
Keywords: misincorporation, proofreading, yeast
Many DNA polymerases possess 3′–5′ exonuclease activity with an important role in the fidelity of DNA synthesis. This “proofreading” exonuclease excises nucleotides that have been added to the nascent DNA chain but do not correctly match the template base. Mutations that disable or eliminate the nuclease activities of DNA polymerases have demonstrated the importance of exonucleolytic proofreading to DNA replication fidelity. In cells harboring a nuclease-deficient DNA polymerase, the frequency of both base substitution and frameshift mutations can be substantially increased (ref. 1 and references therein).
Interest in the possibility of an analogous proofreading mechanism during transcription was raised when multisubunit RNA polymerases were shown to have a nuclease activity that cleaves single nucleotides and short oligonucleotides from the 3′ ends of nascent RNAs (reviewed in ref. 2). The nuclease activity is intrinsic to RNA polymerases but in some cases can be stimulated by accessory proteins. The best characterized of such stimulatory proteins are GreA and GreB for Escherichia coli RNA polymerase and TFIIS (or SII) for eukaryotic RNA polymerase (Pol) II, the nuclear enzyme that synthesizes mRNAs. Both the Gre proteins and TFIIS have been proposed to participate directly in the nuclease mechanism by providing two carboxylic acid side chains that might help coordinate an essential Mg2+ ion and position the nucleophilic water molecule (3–6). Consistent with a possible physiological role in transcriptional proofreading, both TFIIS and GreA are able to promote removal of misincorporated nucleotides in vitro (7–9).
Less is known about the RNA polymerase residues that contribute to either the intrinsic or the factor-stimulated nuclease activities. Several lines of investigation have identified an important, although largely uncharacterized, role for a small integral Pol II subunit, Rpb9, in transcript cleavage. Rpb9-deficient Pol II was shown to be less able to respond to stimulation of transcript cleavage by TFIIS in vitro and to generate different cleavage products than wild-type Pol II (10).
The Rpb9 subunit is highly conserved among eukaryotes. Indeed, the human homolog can correct the growth defects due to loss of Rpb9 in yeast (11). Although nonessential in yeast, the subunit is required for viability in Drosophila (12). The other nuclear RNA polymerases, Pol I and Pol III, also have Rpb9 counterparts. Rpc11, the corresponding Pol III subunit, is required for the intrinsic nuclease activity of that enzyme in both Saccharomyces cerevisiae and Schizosaccharomyces pombe (13, 14). In addition, the nuclease activity of archaeal RNA polymerase is mediated by a small polypeptide that is clearly related to the eukaryotic Rpb9 family of subunits, although it is less clear whether the archaeal polypeptide, TFS, should be considered a core polymerase subunit or a dissociable accessory factor (15). Like TFIIS, TFS has been shown to improve transcription fidelity in vitro (16). Intriguingly, all of these polypeptides share a conserved zinc ribbon domain with considerable sequence similarity to the zinc ribbon domain of TFIIS that contains the essential catalytic Asp–Glu dipeptide. The mechanistic role of the zinc domain is not yet clear for the polymerase subunits, although a mutation of one or both of the conserved carboxylic acids of Rpc11 eliminated the nuclease activity of S. pombe Pol III in vitro (14) and inactivated an essential function of the S. cerevisiae subunit in vivo (13).
Although TFIIS is an obvious candidate for a role in transcriptional proofreading by Pol II in vivo, other eukaryotic RNA polymerases presumably must rely on their intrinsic nuclease activities if they are to proofread the nascent transcripts. Indeed, it is possible that the intrinsic nuclease activity might suffice for Pol II as well, because we have shown that Pol II can remove a misincorporated nucleotide during rapid chain extension in vitro even in the absence of TFIIS, although the efficiency was higher when TFIIS was present (7).
To determine whether the intrinsic and/or TFIIS-promoted nuclease activity of Pol II contributes to the fidelity of mRNA synthesis in the cell, we developed an assay with which we can detect changes in Pol II transcriptional fidelity in S. cerevisiae. Using this assay, we demonstrated that the loss of the Rpb9 subunit significantly reduced transcriptional accuracy in vivo, whereas deletion of the gene encoding TFIIS had a much more modest effect.
Results and Discussion
Assay for Transcriptional Fidelity in Vivo.
We designed our transcriptional accuracy assay system to have the following essential characteristics: an easily scored phenotype, very high sensitivity, and a very low background from translational errors. Previous assays for transcriptional errors have monitored transcriptional correction of premature stop codons in an upstream region of a reporter gene (17–19). However, a relatively high frequency of translational read-through of nonsense codons can confound such analyses (18). To avoid this problem, we modeled our system after an assay successfully used to isolate decreased fidelity mutants of E. coli RNA polymerase (17). That screen used a highly polar nonsense mutation within the lacZ coding sequence. When this mutant allele was accurately transcribed, the lacZ transcripts were truncated by premature transcription termination. By this means, the background of β-galactosidase production due to translational read-through of the stop codon was depressed sufficiently that the rare transcriptional errors by mutant RNA polymerases could be detected. The decreased accuracy of those polymerase variants was demonstrated in vitro (17), validating the assumptions inherent in the design of the screen.
The nonsense-mediated decay (NMD) pathway may be the eukaryotic counterpart of the prokaryotic phenomenon of transcriptional polarity. The NMD pathway in yeast has been shown to promote rapid degradation of RNAs containing premature stop codons (reviewed in refs. 20 and 21). Not every premature stop codon promotes rapid degradation of the mRNA. Instead, other signals, as yet poorly understood, are required to trigger the response (20). For our fidelity assay, we used a gene with an easily scored phenotype for which a premature nonsense codon had been shown to promote rapid degradation of the mRNA. In this way, we could be confident that RNAs that were faithfully transcribed by Pol II (and therefore contained the stop codon and any other signals required for NMD) would be rapidly degraded. RNAs that had lost the stop codon or other NMD-related sequence information because of a transcriptional error would be stabilized and contribute to production of the protein product. A well characterized allele of the CAN1 gene fulfilled these criteria.
The CAN1 gene encodes a high-affinity permease that transports arginine into cells (22). Canavanine is a toxic arginine analog that is also transported by Can1. can1–100 has an A-to-T mutation at codon 47, changing AAA (Lys) to UAA (Stop) (23). Growth inhibition of CAN1 cells by canavanine can be detected at concentrations as low as 1 μg/ml, whereas can1–100 cells are resistant to high concentrations (at least 300 μg/ml). However, in the absence of a functional NMD pathway, infrequent translational read-through of the nonsense codon produces enough Can1 to render the can1–100 cells sensitive to relatively low concentrations of canavanine (30 μg/ml) (ref. 23; also, see Fig. 2). Together, these observations demonstrated that the can1–100 allele has the essential characteristics for an assay to detect rare transcriptional errors: low background levels due to translation errors and very high sensitivity. Any transcriptional errors that stabilized the can1–100 mRNA against NMD might be expected to contribute to Can1 production, observable as an increase in canavanine sensitivity, by increasing the steady-state level of the transcript. The expectations for our assay are summarized in Fig. 1.
Fig. 2.

Contributions of TFIIS, Rpb9, and the NMD pathway to canavanine sensitivity. (a) Mid-log phase cultures of the various yeast strains were serially diluted to equal concentrations and spotted onto synthetic complete medium lacking arginine and containing the indicated concentrations of canavanine. Growth was evaluated after 2 days. (b) Same as in a for the indicated strains, except that growth was evaluated after 4 days.
Fig. 1.

Design of assay for transcriptional accuracy in vivo. Rare transcriptional errors by an error-prone Pol II correct the premature stop codon and/or stabilize the can1–100 mRNA to NMD, allowing increased production of Can1 protein, which is detected as enhanced sensitivity to canavanine.
We constructed a set of yeast strains with the can1–100 allele at the chromosomal CAN1 locus and verified that the canavanine-resistant phenotype of these strains was suppressible by a suppressor tRNA (data not shown). To test the importance of TFIIS to transcriptional fidelity, we deleted the DST1 gene, which encodes the yeast TFIIS homolog. We also deleted the RPB9 gene, which encodes a nonessential Pol II subunit. In vitro, Rpb9 contributes to the nuclease activity of Pol II and is required for efficient recognition of pause and arrest sites at which cleavage takes place (10).
Rpb9 Increases Transcriptional Fidelity Independent of TFIIS.
Fig. 2a shows an experiment comparing the canavanine sensitivities of the wild-type, dst1Δ, and rpb9Δ can1–100 strains. Although loss of the DST1 gene caused a small increase in sensitivity (observable at 300 μg/ml canavanine), deletion of the RPB9 gene had a very large effect, conferring sensitivity to as little as 50 μg/ml canavanine. The observed canavanine sensitivity was entirely dependent on expression of the can1–100 allele, because can1Δ cells were completely resistant to 300 μg/ml canavanine, independent of the presence of TFIIS or Rpb9 (data not shown). These results are consistent with an important role for RPB9, and perhaps to a lesser extent DST1, in Pol II transcription fidelity.
We considered the possibility that increased sensitivity to canavanine in the experiment of Fig. 2a resulted indirectly from decreased levels of intracellular arginine rather than directly from increased levels of the Can1 transporter. Published microarray data showed that the deletion of RPB9 has little effect on global gene expression; however, the level of ARG1 transcripts in an rpb9Δ strain was reduced to half of that observed for wild-type cells (24). We supplied an additional copy of ARG1 on a centromere-based plasmid in the rpb9Δ can1–100 cells and observed no effect on canavanine sensitivity (data not shown). This result provides further support that increased canavanine sensitivity in our assay reflects an increased frequency of transcriptional errors in the can1–100 gene, resulting in increased Can1 production.
We designed our assay based on the assumption that NMD-mediated degradation of can1–100 mRNAs would be required for detection of rare transcriptional errors above the background of translational errors. To test the validity of this assumption, we inactivated the NMD pathway by deleting NMD2 in the can1–100 strains (Fig. 2b). Cells lacking NMD2 failed to grow on 40 μg/ml canavanine, even in the presence of the wild-type RPB9 allele. Thus, potential effects of error-prone transcription resulting from deletion of RPB9 were impossible to detect in the absence of a functional NMD pathway. Maderazo et al. (23) showed that deletion of NMD2 increased arginine import activity ≈2.5-fold compared with that measured for an otherwise isogenic can1–100 strain. The loss of canavanine resistance in the nmd2Δ strain, proposed to result primarily from translational read-through of the stop codon (23), illustrates the high level of sensitivity of this assay. Transcriptional errors either eliminating the stop codon or stabilizing the mRNA to NMD would reasonably be expected to occur at even lower frequency than translational read-through. Consistent with that expectation, the rpb9Δ strain with a wild-type NMD pathway was only partially sensitive to 40 μg/ml canavanine (Fig. 2b).
If, as hypothesized, sensitivity to canavanine in the rpb9Δ strain were due to transcriptional errors that stabilized the RNA against NMD, we would expect to observe a higher steady-state level of can1–100 transcripts in rpb9Δ cells than in the other strains. To test this prediction, we isolated RNA from CAN1 and can1–100 strains and used both Northern analysis (Fig. 3) and real-time PCR to measure transcript levels. The results of the two assays were very similar (Table 1). In the CAN1 strains, deletion of either RPB9 or DST1 did not affect the transcript levels, which were the same within experimental error for the three strains. As expected, each strain showed a reduction in transcript levels when the CAN1 allele was replaced with can1–100. The magnitude of reduction due to the can1–100 mutation in the DST1RPB9 strain background is consistent with the reported magnitude of destabilization of the can1–100 transcript by the NMD pathway (23).
Fig. 3.

Northern blot of CAN1 transcripts. Total RNA was isolated from the indicated strains and processed as described in Materials and Methods.
Table 1.
Quantification of CAN1 transcript levels in can1–100 and CAN1 strains
| Strains being compared | Ratio of CAN1 transcript levels |
|
|---|---|---|
| Northern blots | Real-time PCR | |
| dst1Δcan1–100/DST1can1–100 | 1.2 ± 0.2 | 1.1 ± 0.2 |
| rpb9Δcan1–100/RPB9can1–100 | 1.7 ± 0.1 | 1.6 ± 0.1 |
| CAN1/can1–100 | 2.9 ± 0.8 | 3.5 ± 0.4 |
In the real-time PCR assays, the CAN1 transcript levels were normalized to tubulin (TUB3) mRNA for each strain. The transcript levels in the Northern blot experiments were normalized to 28S rRNA. Data in the table are mean values from five Northern blot and seven RT-PCR experiments.
Absence of TFIIS had little effect on the abundance of can1–100 transcripts. Although we observed a small increase in can1–100 transcript levels for the dst1Δ strain compared with the isogenic DST1 strain in three of five Northern blot experiments, the difference was within the experimental error. Indeed, it may not be possible to detect such a minor difference in transcript levels with a high level of confidence. In contrast, the can1–100 transcript was reproducibly more abundant in the rpb9Δ background. This result supports the hypothesis that a population of can1–100 transcripts was stabilized in rpb9Δ cells by transcriptional errors that eliminated the stop codon and/or reduced the signal for NMD.
Error Frequency of Rpb9-Deficient Pol II.
To compare the frequency of transcriptional errors in the rpb9Δ and RPB9 yeast, we sequenced individual cDNAs synthesized from RNA obtained from these two strain backgrounds. We tabulated all errors observed within an ≈450-base segment of the CAN1 RNA. We found a significantly higher incidence of both substitution and insertion errors for the rpb9Δ strains, consistent with the proposed decrease in transcriptional accuracy (Table 2). Both types of errors were widely distributed, with no pronounced hotspots (Fig. 5, which is published as supporting information on the PNAS web site).
Table 2.
Types and frequencies of sequence errors in cDNAs from RPB9 and rpb9 Δ yeast
| Strain | Base substitutions | Fraction transversions | No. of deletions | No. of insertions | Total errors | No. of nucleotides sequenced | Substitution frequency (×10−3) | Insertion frequency (×10−3) | Overall error frequency (×10−3) |
|---|---|---|---|---|---|---|---|---|---|
| RPB9 | 97 | 0.16 | 4 | 6 | 107 | 73,632 | 1.3 | 0.08 | 1.4 |
| rpb9Δ | 156 | 0.15 | 4 | 30 | 190 | 92,933 | 1.7 | 0.32 | 2.0 |
cDNAs were prepared from total RNA isolated from the indicated yeast strains and cloned into a bacterial plasmid. DNA from individual bacterial colonies was sequenced. Statistical analysis of the data showed that the probabilities that the observed differences in the substitution and insertion frequencies were not due to chance were 97% and >99%, respectively.
We observed an overall frequency of 1.3 × 10−3 misincorporations per base pair for cDNAs prepared from RNAs isolated from the RPB9 strain. This value, although consistent with some previous estimates of RNA polymerase accuracy (25, 26), is likely to be an overestimate of the true error frequency of wild-type Pol II, because the number includes the unknown background of errors introduced during double-stranded cDNA synthesis. Although the reported error frequencies for the reverse transcriptase and DNA polymerase used to prepare the cDNA are more than two orders of magnitude lower than the value we observed (27, 28), we have no way to determine the actual error rate in our experiments. However, if we assume that errors introduced during cDNA synthesis occurred at a similar frequency for both strains, then the observed difference in misincorporation frequency of the two strains (four substitutions for every 10,000 nucleotides incorporated) can be attributed to the loss of Rpb9.
Although Pol II made fewer frameshift errors than base substitutions in both strain backgrounds (Table 2), insertions constituted a significantly higher proportion of the total errors in the strains lacking Rpb9 (30 of 190 or ≈16% compared with 6 of 107 or ≈6% for the RPB9 strain). In this respect, the rpb9Δ Pol II resembles DNA polymerases depleted of their proofreading exonuclease activities. An increase in the ratio of frameshift-to-substitution errors upon loss of proofreading capability appears to be a general property of DNA polymerases, whereas the distribution of frameshifts between deletions and insertions can vary with different DNA polymerases (1).
Our finding that the number of substitution and frameshift errors was significantly increased in the rpb9Δ strain is consistent with the proposal that transcriptional errors that interfered with NMD were responsible for the higher steady-state level of can1–100 RNA observed for the same strain. NMD could be inhibited either by base substitutions in important sequences, including the nonsense codon itself, or by frameshift errors upstream of the nonsense codon, although the latter class of transcripts would not be expected to produce active Can1.
The sequences required for NMD-mediated degradation of can1–100 transcripts have not been determined, and our sequencing analysis did not address this issue. As discussed above, we cannot be certain which substitution errors were introduced by Pol II rather than by the other polymerases used in preparation of the cDNAs. In addition, we cannot assume that the sequenced cDNAs represented only the stabilized population of can1–100 mRNAs. We cloned relatively short cDNAs representing an internal region of the ORF, and it is possible that transcripts in the process of being degraded were able to serve as templates for cDNA synthesis. Nevertheless, this analysis has definitively shown that the lack of Rpb9 leads to an increase in the types of errors that would be expected to interfere with NMD.
Distinct Roles of Rpb9 and TFIIS.
These experiments provide strong evidence that the Rpb9 subunit of Pol II has an important role in assuring the fidelity of transcription in vivo. In contrast, TFIIS appeared to be less important, although the small increases in canavanine sensitivity and transcript levels observed for the dst1Δ strain may represent a real decrease in transcriptional fidelity compared with the wild-type strain.
The NMD pathway is thought to mitigate deleterious effects of defective transcripts, which might code for dominant-negative proteins or interfere with other cellular processes (21). A general loss of transcriptional fidelity would be expected to increase the production of defective transcripts subject to NMD surveillance, making that pathway more important to cell viability. Consistent with that hypothesis, we observed that an RPB9 deletion reduced the viability of nmd2Δ yeast on minimal media (Figs. 2b and 4). In contrast, deletion of the DST1 gene did not decrease the viability of nmd2Δ yeast strains (Fig. 4), supporting the conclusion that, under normal growth conditions, Rpb9 has a more important role than TFIIS in maintaining accurate transcription.
Fig. 4.
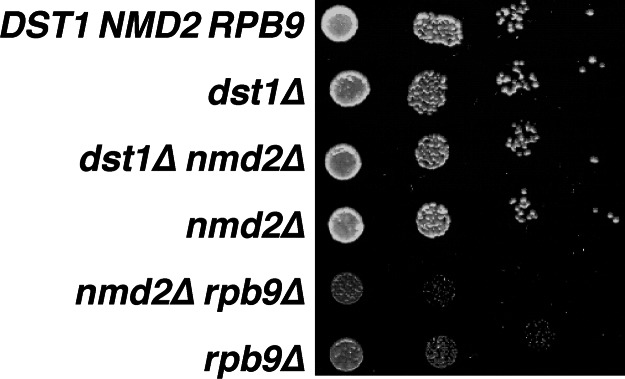
Effect of absence of the NMD pathway on growth of yeast lacking TFIIS or Rpb9. Mid-log phase cultures of the indicated yeast strains were serially diluted and spotted onto synthetic complete medium. Growth was evaluated after 2 days.
That interpretation is also consistent with conclusions of two previous studies addressing the role of TFIIS in transcriptional proofreading in S. cerevisiae. In one case, Reines and coworkers (18) used a nonsense codon in the luciferase gene to assess transcriptional fidelity but found that the background from translational errors limited the ability to detect a low frequency of transcriptional errors. They concluded that any role of TFIIS must be relatively small, in that it could not be detected above the background. In the other study, Koyama et al. (19) found that the absence of TFIIS increased the production of β-galactosidase from a nonsense allele of the lacZ gene under conditions of oxidative stress. They concluded that TFIIS has a role in excising oxidized nucleotides from the nascent transcript. In the absence of oxidative stress, the contribution of TFIIS to lacZ production in this assay was minimal. Consistent with that finding, chromatin immunoprecipitation analysis has shown that TFIIS becomes widely associated with transcribed sequences only under stress conditions (29), suggesting a relatively specialized role for TFIIS in vivo.
The previous lack of an in vivo assay for Pol II transcriptional fidelity has resulted in a reliance on biochemical studies in vitro to identify possible mechanisms for maintaining accuracy. The well characterized role of TFIIS in stimulating the nuclease activity of Pol II (reviewed in ref. 2) suggested that TFIIS could increase fidelity by stimulating a proofreading nuclease that selectively removes mismatched nucleotides from the nascent transcript. Indeed, we have shown that, in vitro, the TFIIS-stimulated nuclease activity can effectively proofread under conditions of rapid chain elongation (7).
A compelling model for the molecular mechanism of TFIIS action was provided by the crystal structure of the Pol II–TFIIS complex (3), in which a zinc ribbon domain of TFIIS inserts into the active site of Pol II. This domain includes two acidic amino acids that are thought to coordinate a Mg2+ ion and position a water molecule to serve as the nucleophile in RNA hydrolysis.
Our finding that TFIIS plays at most a minor role in transcription accuracy under favorable growth conditions suggests that, although it can perform a proofreading function in vitro, it generally does not do so in vivo. One possible explanation is that TFIIS does not associate with the elongation complex in vivo to a sufficient extent or with fast enough kinetics to be useful for proofreading simple mismatches. Perhaps the intracellular concentration of TFIIS is too low or the elongation complex with a mismatched nucleotide is not an optimal substrate for TFIIS binding or activity. In vitro, TFIIS can promote transcript cleavage in elongation complexes stalled for a variety of reasons, but transcripts associated with arrested elongation complexes appear to be most rapidly cleaved. Arrest is a conformational rearrangement of the elongation complex in which the polymerase slides upstream along both the nascent transcript and the DNA, displacing the 3′ end of the RNA from the catalytic site (30, 31). Although arrest might occur after incorporation of a mismatched nucleotide, the arrest propensity at a particular position would be expected to be sequence-specific (32). We have previously shown that human Pol II did not arrest after incorporating a mismatched nucleotide at the sites that we investigated in vitro (7).
In the absence of arrest, the intrinsic nuclease activity of Pol II might be better suited to remove the 3′ nucleotide. Consistent with that idea, we have found that Rpb9-deficient yeast strains have enhanced sensitivity to cordycepin (3′-deoxyadenosine) (data not shown), which is converted into a chain-terminating nucleoside triphosphate in yeast (33). RNA elongation after incorporation of this chain terminator requires nucleolytic excision, and the enhanced sensitivity may be another indicator of defective proofreading activity. Interestingly, TFIIS-deficient strains show no increase in cordycepin sensitivity relative to wild-type yeast (data not shown).
An in vitro analysis of a potential role for Rpb9 in transcriptional fidelity has not been reported. However, it has been shown that Rpb9 is required for optimal stimulation of cleavage activity by TFIIS (10). Crystal structures of Pol II alone or assembled in an elongation complex (34, 35) provide little basis for deducing a potential role for Rpb9 in either the TFIIS-stimulated or intrinsic nuclease activity. Although parts of both proteins interact with Pol II in the structural feature known as the “jaw,” there is no apparent direct interaction between TFIIS and Rpb9. Consistent with that conclusion, the affinity of the association of TFIIS with Pol II is not altered by the absence of Rpb9 (10). Furthermore, there is no direct presence of any part of Rpb9 near the Pol II active site, nor does Rpb9 appear to interact directly with the DNA template or RNA transcript. It does bind, by means of its C-terminal zinc ribbon domain, to residues on the exterior surface of a structural feature called the funnel. The funnel surrounds a pore through which the 3′ end of the RNA is thought to extrude when Pol II arrests. TFIIS infiltrates the catalytic site through this pore (3).
Mechanisms by Which Rpb9 May Affect Transcriptional Fidelity.
We have considered three potential mechanisms by which Rpb9 might enhance transcriptional fidelity. First, Rpb9 may affect discrimination of the correct nucleotide during chain elongation. Such discrimination could be related to the relative rates of incorporation of the templated nucleotide versus an incorrect nucleotide. We have shown that these rates can differ by ≈500-fold in vitro (7). It is possible that Rpb9 has a significant impact on the overall geometry of the active site, although the crystal structures do not provide any insight into how this might occur. Alternatively, discrimination could relate to rates of chain extension after incorporation of a correct versus an incorrect nucleotide. The rate of extension from a correctly templated nucleotide is ≈15- to 20-fold faster than when the 3′ end of the nascent transcript is mismatched (7). Presumably, this slower rate provides time for proofreading before elongation. The observed increased overall rate of elongation of Rpb9-deficient Pol II in vitro (10) could be consistent with decreased kinetic preferences for incorporation of correct versus incorrect nucleotides or for extension of base-paired versus mismatched 3′ ends.
A second possible role of Rpb9 in transcriptional fidelity is in mediating interaction between Pol II and other components of the transcription machinery. Lack of an in vivo assay has precluded the use of genetic screens to identify genes that affect fidelity, so almost nothing is known about what factors might be involved. It is possible that chromatin structure or elongation factors may play a major role, and the position of Rpb9 at the leading edge of the elongating polymerase hints that interactions between Pol II and other proteins may occur through Rpb9. Consistent with this general idea, deletion of RPB9 is lethal when combined with deletions of genes encoding some components of the chromatin remodeling complex SAGA (Spt-Ada-Gcn5 acetyltransferase) and the elongation complex Elongator (36), and the Spt7 component of SAGA is a two-hybrid partner with Rpb9 (37).
A third mechanism by which Rpb9 could enhance fidelity is by contributing to an intrinsic Pol II proofreading nuclease. We argue that this possibility is the most parsimonious view at present, based on what is known about the properties and functions of Rpb9 and the related subunits of Pol III (Rpc11) and Pol I (Rpa12). We have previously shown that the intrinsic nuclease activity of human Pol II is capable of postinsertion error correction of a misincorporated nucleotide in vitro (7). Rpb9 has not yet been demonstrated to contribute to the intrinsic nuclease activity of Pol II, but it clearly is required for optimal function of TFIIS (10). Rpb9 was reported to be dispensable for the intrinsic nuclease activity, because depletion of Rpb9 was shown not to reduce the transcript cleavage observed in Pol II complexes at elevated pH (10, 38). However, it is possible that the elevated pH altered the mechanism of cleavage in these studies, because the hydroxyl ions may catalyze the reaction directly (38).
The Rpc11 subunit of Pol III has been shown to be essential for the intrinsic nuclease activity of that polymerase (13), and mutational analysis has suggested a functional relationship between loss of nuclease activity and other observed behaviors of the polymerase, some of which are shared by Pol II lacking Rpb9. For example, Pol III lacking Rpc11 responded inefficiently to normal transcriptional pause sites in vitro (13), a property also observed for Pol II lacking Rpb9 (10). In addition, an rpc11 mutation in S. pombe that impaired the nuclease activity of Pol III conferred hypersensitivity to 6-azauracil (14), as did deletion of the S. cerevisiae rpa12 gene encoding the homologous subunit for Pol I (39). Enhanced sensitivity to 6-azauracil and mycophenolic acid, which inhibit de novo nucleotide biosynthesis and deplete nucleotide pools, is also characteristic of strains with deletions in RPB9 and DST1 (37, 40, 41). Intriguingly, the absence of wild-type Rpc11 led to the production of transcripts that were one or more nucleotides longer than those synthesized by the Rpc11-containing enzyme both in vivo (14) and in side-by-side reactions in vitro (13, 14). This behavior, which was also correlated with the loss of nuclease activity, may be related to the increased frequency of transcription insertion errors observed in the rpb9Δ yeast strains.
Considering these phenotypic similarities, as well as their structural similarities, there is currently no reason to suppose that Rpb9 and the related subunits of Pol I and Pol III have completely different roles in their respective enzymes, although some functional differences between the RNA polymerases, reflecting their specific roles in the cell, are expected. The three Rpb9-like subunits show significant sequence similarity in both the N-and C-terminal domains, and, based on that information, as well as a variety of structural studies, all three have been proposed to occupy similar positions on their RNA polymerases (3, 13, 39, 42). Based on current knowledge, it is difficult to understand how these subunits contribute to an activity that is thought to occur in the catalytic active site, which is >30 Å from the closest approach of Rpb9 in the existing crystal structures. It is possible that the action is truly effected at a distance, but it is also possible that the RNA polymerases undergo a conformational shift that brings some portion of the subunit closer to the 3′ end of the RNA.
Taken together, these observations support the idea that the decreased Pol II transcription fidelity in rpb9Δ yeast was due to an altered nuclease activity that affected transcriptional proofreading.
Materials and Methods
Yeast Strains.
Yeast strains used in this study are listed in Table 3, which is published as supporting information on the PNAS web site. The strains shown in Fig. 4 are derivatives of YPH499. The parent of all other strains was a trp1 deletion derivative of BY4742 (Research Genetics) constructed in the laboratory of George Sprague (University of Oregon). The can1–100 allele was introduced into the BY4742 background by transformation of a PCR product derived from a can1–100 strain. The presence of can1–100 was verified in the transformants by sequencing DNA derived from PCR amplification of the CAN1 locus.
RNA Preparation.
Total RNA was extracted from cells grown in 30 ml of yeast extract peptone dextrose complete medium to A600 of 0.4–0.6 by using the acid phenol method (43). RNA samples were then treated with RNase-Free DNase (Stratagene) according to the manufacturer's protocol, extracted with phenol/chloroform, and precipitated with ethanol. RNA was resuspended in 0.2-μm-filtered water, and the concentration was determined by spectrophotometry. The purity and integrity of the RNA were confirmed by agarose gel electrophoresis. Single-use aliquots of RNA were stored at −80°C.
Northern Blot Analysis.
Total RNA (10 μg) was resolved on a 1.2% (wt/vol) agarose/6% formaldehyde gel and blotted onto magna nylon transfer membrane (Osmonics, Minnetonka, MN). Filters were stained with methylene blue, and digital images of the rRNA bands were stored to normalize for equal loading of the RNA. The template for the CAN1 DNA probe was generated by PCR amplification of genomic DNA. The forward primer (5′-GCATATGTACAATGAGCCGGT) and reverse primer (5′-TGGGTTTCTCCAATAACGGA) cover, respectively, positions 39–59 and 779–798 of the CAN1 gene. The 720-bp amplicon was 32P-labeled by random priming (Klenow exo−, New England Biolabs), and the radioactively labeled probe was hybridized to the filter at 65°C overnight in CHURCH buffer (0.5 M NaPO4, pH 7.5/1 mM EDTA/7% SDS). The membrane was then exposed to a phosphor screen (Molecular Dynamics) and scanned with a phosphoimager (STORM 860, Molecular Dynamics).
cDNA Synthesis.
cDNAs were synthesized from total RNA by using the SuperScript II RNaseH reverse transcriptase system (Invitrogen) and then incubated with RNase H. For real-time PCR, cDNA synthesis was primed with random hexamers. For cloning and DNA sequencing, a CAN1 primer hybridizing to positions 826–846 of the ORF was used. Double-stranded DNA corresponding to ORF nucleotides 9–798 was synthesized by one round of PCR with High Fidelity polymerase (Roche).
Real-Time PCR.
primer express software was used to design all primer pairs for real-time PCR. These sets include a forward and reverse primer for the CAN1 gene (5′-CACTTTTGCCCTGGAACTTAGTG and 5′-TGCCGGCAGTGGAACTTT, respectively) and a pair of primers for the detection of TUB3 cDNA levels (forward, 5′-CCTGCGCCTCAATTGTCTACT; reverse, 5′-TTCCAGGGTGGTATGCGTG). Quantitative real-time PCR was done in 96-well plates by using the automated ABI PRISM 7900HT sequence detection system. RT-PCR mixtures were prepared in bulk as a master mix, and then aliquots were pipetted into the individual wells. Each well was loaded with a 25-μl reaction mixture containing 75 ng of template cDNA, forward and reverse primers at 500 nM, and 12.5 μl of 2× SYBRGreen PCR Master Mix. A “no template” control was run in parallel with every reaction. Fluorescence emissions were recorded in real time at the specified wavelengths, and data were analyzed automatically with sds 2.0 software. The expression data obtained represent average values from a minimum of three replicate experiments. Unless otherwise stated, all reagents were purchased from Applied Biosystems.
Error Rate Determination.
Double-stranded cDNAs were ligated into HindIII/EcoRI sites in pBluescriptII ks+ (Stratagene). Plasmid DNA was transformed into E. coli and then isolated from individual bacterial colonies and sequenced at the Institute of Molecular Biology Biotechnology Laboratory (Eugene, OR). The sequencing software program (Beckman CEQ 8000 Genetic Analysis System sequence investigator) was used to evaluate the quality of sequencing data for each possible misincorporation. Regions of sequence with confidence limits <0.98 were rejected; within accepted regions, sequence changes with confidence limits deviating by >0.05 from the neighboring positions were not included. To determine the significance of the differences in the observed error frequencies, we calculated the z-ratio and one-tail probability for the difference in independent proportions.
Supplementary Material
Acknowledgments
We thank Adam Peterson for construction of can1Δ strains, Thomas Redditt for technical help, and Frank Stahl for discussions of statistical methods. This work was supported by National Institutes of Health Grant GM59644 (to D.K.H.).
Abbreviations
- NMD
nonsense-mediated decay
- Pol I
RNA polymerase I
- Pol II
RNA polymerase II
- Pol III
RNA polymerase III
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Shcherbakova P. V., Pavlov Y. I., Chilkova O., Rogozin I. B., Johansson E., Kunkel T. A. J. Biol. Chem. 2003;278:43770–43780. doi: 10.1074/jbc.M306893200. [DOI] [PubMed] [Google Scholar]
- 2.Fish R. N., Kane C. M. Biochim. Biophys. Acta. 2002;1577:287–307. doi: 10.1016/s0167-4781(02)00459-1. [DOI] [PubMed] [Google Scholar]
- 3.Kettenberger H., Armache K.-J., Cramer P. Cell. 2003;114:347–357. doi: 10.1016/s0092-8674(03)00598-1. [DOI] [PubMed] [Google Scholar]
- 4.Opalka N., Chlenov M., Chacon P., Rice W. J., Wriggers W., Darst S. A. Cell. 2003;114:335–345. doi: 10.1016/s0092-8674(03)00600-7. [DOI] [PubMed] [Google Scholar]
- 5.Laptenko O., Lee J., Lomakin I., Borukhov S. EMBO J. 2003;22:6322–6334. doi: 10.1093/emboj/cdg610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sosunova E., Sosunov V., Kozlov M., Nikiforov V., Goldfarb A., Mustaev A. Proc. Natl. Acad. Sci. USA. 2003;100:15469–15474. doi: 10.1073/pnas.2536698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas M. J., Platas A. A., Hawley D. K. Cell. 1998;93:627–637. doi: 10.1016/s0092-8674(00)81191-5. [DOI] [PubMed] [Google Scholar]
- 8.Jeon C., Agarwal K. Proc. Natl. Acad. Sci. USA. 1996;93:13677–13682. doi: 10.1073/pnas.93.24.13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erie D. A., Hajiseyedjavadi O., Young M. C., von Hippel P. H. Science. 1993;262:867–873. doi: 10.1126/science.8235608. [DOI] [PubMed] [Google Scholar]
- 10.Awrey D. E., Weilbaecher R. G., Hemming S. A., Orlicky S. M., Kane C. M., Edwards A. M. J. Biol. Chem. 1997;272:14747–14754. doi: 10.1074/jbc.272.23.14747. [DOI] [PubMed] [Google Scholar]
- 11.McKune K., Moore P. A., Hull M. W., Woychik N. A. Mol. Cell. Biol. 1995;15:6895–6900. doi: 10.1128/mcb.15.12.6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison D. A., Mortin M. A., Corces V. G. Mol. Cell. Biol. 1992;12:928–935. doi: 10.1128/mcb.12.3.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chedin S., Riva M., Schultz P., Sentenac A., Carles C. Genes Dev. 1998;12:3857–3871. doi: 10.1101/gad.12.24.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y., Intine R. V., Mozlin A., Hasson S., Maraia R. J. Mol. Cell. Biol. 2005;25:621–636. doi: 10.1128/MCB.25.2.621-636.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hausner W., Lange U., Musfeldt M. J. Biol. Chem. 2000;275:12393–12399. doi: 10.1074/jbc.275.17.12393. [DOI] [PubMed] [Google Scholar]
- 16.Lange U., Hausner W. Mol. Microbiol. 2004;52:1133–1143. doi: 10.1111/j.1365-2958.2004.04039.x. [DOI] [PubMed] [Google Scholar]
- 17.Blank A., Gallant J. A., Burgess R. R., Loeb L. A. Biochemistry. 1986;25:5920–5928. doi: 10.1021/bi00368a013. [DOI] [PubMed] [Google Scholar]
- 18.Shaw R. J., Bonawitz N. D., Reines D. J. Biol. Chem. 2002;277:24420–24426. doi: 10.1074/jbc.M202059200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koyama H., Ito T., Nakanishi T., Kawamura N., Sekimizu K. Genes Cells. 2003;8:779–788. doi: 10.1046/j.1365-2443.2003.00677.x. [DOI] [PubMed] [Google Scholar]
- 20.Hilleren P., Parker R. Annu. Rev. Genet. 1999;33:229–260. doi: 10.1146/annurev.genet.33.1.229. [DOI] [PubMed] [Google Scholar]
- 21.Baker K. E., Parker R. Curr. Opin. Cell Biol. 2004;16:293–299. doi: 10.1016/j.ceb.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 22.Hoffmann W. J. Biol. Chem. 1985;260:11831–11837. [PubMed] [Google Scholar]
- 23.Maderazo A. B., He F., Mangus D. A., Jacobson A. Mol. Cell. Biol. 2000;20:4591–4603. doi: 10.1128/mcb.20.13.4591-4603.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hemming S. A., Jansma D. B., Macgregor P. F., Goryachev A., Friesen J. D., Edwards A. M. J. Biol. Chem. 2000;275:35506–35511. doi: 10.1074/jbc.M004721200. [DOI] [PubMed] [Google Scholar]
- 25.Erie D. A., Yager T. D., von Hippel P. H. Annu. Rev. Biophys. Biomol. Struct. 1992;21:379–415. doi: 10.1146/annurev.bb.21.060192.002115. [DOI] [PubMed] [Google Scholar]
- 26.de Mercoyrol L., Corda Y., Job C., Job D. Eur. J. Biochem. 1992;206:49–58. doi: 10.1111/j.1432-1033.1992.tb16900.x. [DOI] [PubMed] [Google Scholar]
- 27.Roberts J. D., Preston B. D., Johnston L. A., Soni A., Loeb L. A., Kunkel T. A. Mol. Cell. Biol. 1989;9:469–476. doi: 10.1128/mcb.9.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pavlov A. R., Pavlova N. V., Kozyavkin S. A., Slesarev A. I. Trends Biotechnol. 2004;22:253–260. doi: 10.1016/j.tibtech.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Pokholok D. K., Hannett N. M., Young R. A. Mol. Cell. 2002;9:799–809. doi: 10.1016/s1097-2765(02)00502-6. [DOI] [PubMed] [Google Scholar]
- 30.Reeder T. C., Hawley D. K. Cell. 1996;87:767–777. doi: 10.1016/s0092-8674(00)81395-1. [DOI] [PubMed] [Google Scholar]
- 31.Komissarova N., Kashlev M. Proc. Natl. Acad. Sci. USA. 1997;94:1755–1760. doi: 10.1073/pnas.94.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nudler E., Mustaev A., Lukhtanov E., Goldfarb A. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- 33.Anderson J. M., Roth R. J. Bacteriol. 1976;128:689–691. doi: 10.1128/jb.128.2.689-691.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cramer P., Bushnell D. A., Kornberg R. D. Science. 2001;292:1863–1876. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- 35.Gnatt A. L., Cramer P., Fu J., Bushnell D. A., Kornberg R. D. Science. 2001;292:1876–1882. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- 36.Van Mullem V., Wery M., Werner M., Vandenhaute J., Thuriaux P. J. Biol. Chem. 2002;277:10220–10225. doi: 10.1074/jbc.M107207200. [DOI] [PubMed] [Google Scholar]
- 37.Wery M., Shematorova E., Van Driessche B., Vandenhaute J., Thuriaux P., Van Mullem V. EMBO J. 2004;23:4232–4242. doi: 10.1038/sj.emboj.7600326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weilbaecher R. G., Awrey D. E., Edwards A. M., Kane C. M. J. Biol. Chem. 2003;278:24189–24199. doi: 10.1074/jbc.M211197200. [DOI] [PubMed] [Google Scholar]
- 39.Van Mullem V., Landrieux E., Vandenhaute J., Thuriaux P. Mol. Microbiol. 2002;43:1105–1113. doi: 10.1046/j.1365-2958.2002.02824.x. [DOI] [PubMed] [Google Scholar]
- 40.Hemming S. A., Edwards A. M. J. Biol. Chem. 2000;275:2288–2294. doi: 10.1074/jbc.275.4.2288. [DOI] [PubMed] [Google Scholar]
- 41.Exinger F., Lacroute F. Curr. Genet. 1992;22:9–11. doi: 10.1007/BF00351735. [DOI] [PubMed] [Google Scholar]
- 42.Bischler N., Brino L., Carles C., Riva M., Tschochner H., Mallouh V., Schultz P. EMBO J. 2002;21:4136–4144. doi: 10.1093/emboj/cdf392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ausubel F. M., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K. Short Protocols in Molecular Biology. 4th Ed. New York: Wiley; 1999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.