

Abstract
HIV type I (HIV-1) can cause G2 cell cycle arrest and death of CD4+ T lymphocytes in vitro and inexorable depletion of these cells in vivo. However, the molecular mechanism of viral cytopathicity has not been satisfactorily elucidated. Previously, we showed that HIV-1 kills T cells by a necrotic form of cell death that requires high level expression of an integrated provirus but not the env or nef genes. To determine which viral protein(s) are required for cell death, we systematically mutated, alone and in combination, the ORFs of the NL4-3 strain of HIV-1. We found that the elimination of the viral functions encoded by gag-pol and vpu, tat, and rev did not mitigate cytopathicity. However, elimination of the vif and vpr accessory genes together, but not individually, renders the virus incapable of causing cell death and G2 cell cycle blockade. We thus identify vif and vpr as necessary for T cell cytopathic effects induced by HIV-1. These findings may provide an important insight into the molecular mechanism of viral pathogenesis in AIDS.
Keywords: AIDS, apoptosis, lymphocytes, necrosis, NL4, 3
In the absence of antiviral drug therapy, infection with HIV-1 leads to a progressive decline in CD4+ T cell number. This cell depletion produces an immunodeficiency state resulting in the opportunistic infections characteristic of AIDS (reviewed in ref. 1). Mathematical modeling suggested that the majority of HIV-infected cells die with a half-life of ≈2 days (2–4). However, the number of infected cells is reported to be approximately constant (3), suggesting that large numbers of infected cells are produced and destroyed each day. T cell production does not fully balance destruction, leading to the eventual depletion of CD4+ T cells. Recent studies suggest that direct viral infection causes the massive destruction of memory CD4+ T cells in various tissues within 2–3 weeks after acute infection (5–8). In macaques infected with simian immunodeficiency virus, the majority of CD4+ T cells are lost in a period of only 4 days (7, 8). The loss of CD4+ T lymphocytes could be due to direct cytopathic effects or possibly to CD8+ cytolytic T cell responses that kill virally infected cells.
Viral pathogenesis is a multifactorial process, involving host and virus factors that trigger multiple pathologic mechanisms. Cytopathic effects could derive from direct cell injury by the virus or immunopathology as a result of the host’s immune response. Accumulating evidence suggests that significant cell death during HIV-1 infection can be attributed to direct viral killing of infected cells in cultures and in peripheral blood mononuclear cells from AIDS patients (9–11). However, the molecular basis of the viral cytopathic effect, including the specific viral and host components involved, is not yet completely defined.
To understand viral cytopathicity, we have used a single-cell analysis system that distinguishes between infected and uninfected cells by monitoring the expression of a mouse heat-stable antigen (HSA)/CD24 marker engineered in the NL4-3 stain of HIV-1 (12, 13). Our previous quantitative measurements of cell death in infected versus uninfected cells with the NL4-3HSA strain by using flow cytometry showed that only infected cells were killed (14). Moreover, the death of T cells was necrotic rather than apoptotic in nature (12–14). Previous work has suggested that the nef and env genes may have important roles in viral pathogenesis (15–17, reviewed in refs. 18–20). However, direct CD4+ T cell death induced by HIV-1 infection is independent of Env and Nef proteins (12, 13). Thus, the viral genes that account for direct CD4+ T cell necrotic death have not been conclusively determined.
Vpr, an accessory protein of uncertain function in the HIV-1 life cycle, has been implicated in cytopathicity and G2 cell cycle arrest (21–25). Mutational analyses indicated a correlation between cell-cycle arrest and virally induced cell death (25–27). However, abrogation of the G2-phase arrest did not prevent Vpr-induced cell death in other studies (24, 28–31), raising the possibility that cell cycle arrest is not the cause of Vpr-mediated cell death. It is important to ascertain the role Vpr plays in the processes of cell cycle arrest and cell death, vis-à-vis other HIV-1 genes, during infection in CD4+ T lymphocytes.
Vif is an accessory protein that can promote HIV-1 replication in certain restrictive cell types but whose role in viral pathogenesis is controversial (reviewed in ref. 32; refs. 33–38). The first well-described function of Vif came from recent studies that revealed an interaction between Vif and an innate antiviral factor, APOBEC 3G (39). Vif prevents packaging of APOBEC3G, a cytidine deaminase and viral DNA modifier, into virus particles by targeting it for proteasomal degradation (reviewed in ref. 32; 33–35). APOBEC3G expression is limited to primary cells and certain cell lines, including H9 and CEM, in which Vif is required for productive infection by HIV (39). This observation solved the long-standing mystery of why Vif function is cell-type-dependent. However, a possible contribution of Vif to viral pathogenesis and cytopathicity, besides its role in productive infection, has not been clearly delineated.
To identify a viral genes essential for cytopathicity, we created a null mutation in each HIV-1 gene of a laboratory strain, NL4-3, and examined the ability of each mutant virus to kill CD4+ T cells during a single round of infection. We observed the loss in cytopathic capacity if both Vif and Vpr were absent from the virus. Our results indicate that these accessory proteins play central roles in the HIV-1 direct killing of T cells. Intriguingly, proliferation of highly infected cells was observed only in the absence of both Vif and Vpr. Detailed analyses of DNA content in infected cells indicated that Vif and Vpr exclusively and independently cause cell cycle arrest at the G2 phase. Our results suggest a link between HIV-1 cytopathicity and cell cycle arrest and identify two HIV-1 genes responsible for these effects.
Results
To identify the HIV-1 gene or genes essential for cytopathicity in infected CD4+ T cells, we created various mutant versions of the laboratory strain, NL4-3HSA possessing combinations of “knockout” mutations in different coding regions of the viral genome (Fig. 1A; see also Table 1, which is published as supporting information on the PNAS web site). The mutations were nonsense substitutions designed to generate premature stops in the ORFs (Table 1). We introduced stop codons and microdeletions to inactivate ORFs, rather than the deletions of entire genes to prevent the destabilization of viral genomes due to large deletions.
Fig. 1.

Locations of mutations in HIV-1NL4-3 and corresponding loss of protein expression due to mutations. (A) NL4-3HSA strains contained various individual or multiple mutations in the ORFs that alter the indicated amino acids shown in the standard single letter code. The mutations either prevent (ATG mutations) or prematurely terminate (stop codons) protein translation. Mouse heat stable antigen (HSA/CD24) replaced the nef gene coding sequence (21). To verify the expected loss of protein expression, lysates from 293T cells transfected with mutant NL4-3HSA strains were analyzed by Western blotting. Mock transfected 293T cells served as a negative control, and the expression of β-actin was used as a loading control. (B and C) The Western blot was probed with HIV-1-infected patient sera to examine Gag and Pol expression (B) or antibodies specifically directed against Vif or Vpr (C).
For a Gag-deficient strain, elimination of the start codon resulted in internal translation starting at M142 (data not shown). Therefore, we generated two additional stop codons in tandem at R150 and L152 to eliminate spurious internal translation initiations. A Western blot of lysates from 293T cells transfected with a plasmid encoding the Gag-Pol-deficient virus confirmed the expected absence of the Pr160 Gag-Pro-Pol polyprotein and Pr55 Gag (Fig. 1B). Similarly, stop codons were used to eliminate Vif and Vpr, which was also confirmed by Western blotting (Fig. 1C). A virus containing a Vpu deletion was produced by using a construct generated and validated in refs. 40 and 41. All of the mutant strains were constructed on a Env−Nef−HSA+ version of NL4-3 described in refs. 12, 13, and 21. This construct harbors a frameshift mutation in the env gene to restrict replication to a single round of infection by using VSV-G-pseudotyped virus (12, 13, 21). Moreover, the nef gene is replaced by the mouse heat stable antigen (HSA/CD24), which serves as a cell surface marker for infection that is readily detected by flow cytometry (12, 13, 21).
We found that infection of Jurkat cells with Env− strains of NL4-3HSA with an multiplicity of infection (MOI) of 5 led to death of infected cells within 4 days (Fig. 2Ac). Substantial death also occurred in an infected population at an MOI of 1 or 3, manifesting 3% and 13% viability, respectively, although there was greater cell survival at each time point as the MOI decreased (Fig. 2B). These results demonstrated the profound cytopathicity of the NL4-3HSAEnv− strain in these culture conditions as shown in refs. 12–14. In a typical experiment, very few HSAhi, i.e., infected, cells remained alive after 4 and 8 days at a MOI of 5 and 3, respectively. As a control, Jurkat cells were infected with virus particles carrying a GFP-encoding genome devoid of all HIV proteins except for Tat and Rev. We found that even at a high MOI, this virus produced no significant decrease in cell viability (Fig. 2Ab). Thus, proviral expression of NL4-3 genes are necessary for cytopathicity under these culture conditions as shown in refs. 12 and 14.
Fig. 2.
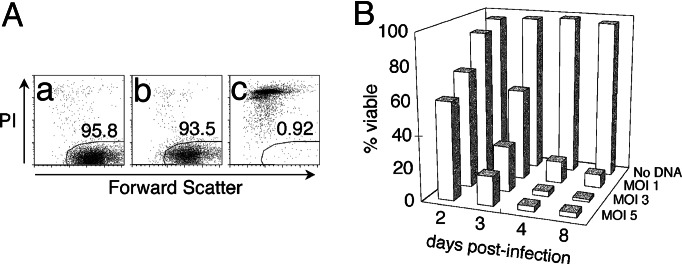
The NL4-3HSA strain of HIV-1 causes cytopathicity in Jurkat cells. (A) Jurkat cells were mock infected (a) or infected with NL-EGFP (b) or NL4-3HSA Env− (c) at a MOI of 5. The viability of infected cells was analyzed by flow cytometry by using forward light scatter (FSC; x axis) and staining with PI (y axis). Each panel represents 10,000 total events on day 4 after the start of infection. The polygonal gate indicates the percent of viable, PI-negative cells. (B) Jurkat cells were infected with the NL4-3HSA Env− strain at various MOI, and viability was monitored by flow cytometry over time. Mock-infected Jurkat cells were at least 95% viable on day 4 based on PI staining. Viability of infected (HSA-positive) cells over time was plotted on the graph.
Because of their well described biochemical functions, we initially hypothesized that one of the enzymatic or structural proteins, such as protease, would be responsible for the cytopathic effect of HIV-1. We, therefore, initially disrupted the gag-pol reading frame (Fig. 1A and Table 1). We prepared infectious virions harboring HIV-1 genomes lacking structural and/or enzymatic genes by cotransfecting a complementing helper plasmid, ΔR8.2ΔVpr into the 293T producer cells (23). This encapsidation-deficient NL4-3 helper plasmid provides in trans the wild-type proteins necessary for packaging and virus production. Surprisingly, we found that the Gag−Pol− strain still killed infected Jurkat cells at a rate comparable to that of the NL4-3HSA Env− parental strain (Fig. 3A). We therefore tested combinations of mutations in accessory genes together with those in structural and enzymatic genes. A frameshift mutation in vif gene in the Gag−Pol− background retarded the onset of cell death by 2 days; however, it did not reduce the eventual overall level of cell death. Removal of Vpr in the Gag−Pol− backbone also diminished direct cell killing such that ≈25% of HSAhi cells were still alive after 8 days of infection (Fig. 3 A and B). By contrast, little change in cytopathicity was detected with Gag−Pol− and with Vpu− strain of NL4-3HSA. The higher viability in cultures exposed to virus with mutations in Gag, Pol, and Vpr reflected outgrowth of uninfected cells and the survival of infected cells. The fraction of HSA-expressing cells became smaller at day 8 compared to earlier days because of uninfected, rather than infected, cells increasing in the culture (Fig. 3B). The outgrowth of uninfected cells was usually observed with Env− strains because a single round of infection was never 100% efficient.
Fig. 3.

Cytopathicity of mutant NL4-3HSA strains in the absence of Gag and Pol. Mutant virus stocks were generated in the presence of ΔR8.2ΔVpr helper plasmid in 293T producer cells and used to infect Jurkat cells at a MOI of 5. Cytopathicity of the total population (A) and fractions of infected cells (B) were monitored by flow cytometry. Cell viability was analyzed as a function of time after the initiation of infection by propidium iodide exclusion staining as shown in Fig. 2A. The fraction of infected cells was monitored by HSA staining. Viability associated with virus expression is shown on the y axis, and time in days is shown for each row of samples.
Next, we investigated the properties of viruses with multiple mutations in accessory genes and to Gag and Pol mutations. Cells infected with a Gag−Pol−Vpr−Vpu− strain showed 68% viability and 37% HSAhi cells at 8 days after infection (Fig. 3 A and B). However, the most impressive results were obtained with Gag−Pol−Vif−Vpr− and Gag−Pol−Vif−Vpr−Vpu− mutant strains at MOI of 5, which showed 67% and 78% viability at 8 days of infection, respectively, with 65–85% of these cells being HSAhi (Fig. 3 A and B). Hence, cell death was clearly reduced when both Vif and Vpr were absent. No other combination of gene mutations produced a comparable attenuation of cytopathicity and survival of highly infected cells.
To directly examine the contribution of Vif and Vpr to cytopathicity, we created the same mutations in the Gag+Pol+ background of NL4-3HSAEnv−. We first investigated the phenotypes of mutant NL4-3 strains lacking only one of the accessory genes, vif, vpr, or vpu. We found these mutant strains to be as potently cytopathic as NL4-3HSAEnv− at an MOI of 5, despite minor variations in the initial killing rate (Fig. 4A and B). The combined loss of Vif and Vpu or Vpr and Vpu expression did not significantly alter cytopathicity. By contrast, when both Vif and Vpr were disrupted, killing was clearly diminished (Fig. 4 A and B; see also Fig. 7, which is published as supporting information on the PNAS web site). The effect was even more prominent at an MOI of 3, at which 80% of the cells survived 8 days after infection with 71% HSAhi cells (Fig. 4A; see also Fig. 8, which is published as supporting information on the PNAS web site). The HSAhi cells were still viable and proliferating 25 days after infection (Fig. 7B). These results indicated important roles for the Vif and Vpr accessory proteins in HIV-1 cytopathicity.
Fig. 4.

Cytopathicity of mutant HIV-1NL4-3 strains. Jurkat T cells were mock-infected or infected with various strains of NL4-3HSA at a MOI of 3 (A) and 5 (B). The viability of the total population was measured as described in Fig. 2A. The unpaired Student’s t test for mutant vs. the parental NL4-3HSA Env− strain on day 7 indicated P < 0.05 for the viability of cells infected with Vif−Vpr− and Vif−Vpr−Vpu− strains.
We found that the expression of vpu contributes modestly to direct cell killing when infection is performed at a higher MOI (Fig. 4B and 8B). In cells infected with Vif−Vpr− strain, high HSA expression was detected at day 8 in 72% and 78% of live cells at MOI of 3 and 5, respectively (Fig. 8). However, the cytopathicity of this Vpu expressing virus was severely compromised at an MOI of 3, indicating that Vif and Vpr play the dominant cytopathic role. Additionally, cell death due to Vif−Vpr− and Vif−Vpr−Vpu− strains was the same when infection was performed at an MOI of 3 (Fig. 4A). Together, these data indicate that the effect of Vpu on direct cell killing is not substantial, especially at low levels of infection.
We also examined the phenotypes of the mutant strains in peripheral CD4+ T cells, a MOLT-4 T cell line, and a SupT1 T cell line to extend our results to other transformed and nontransformed T cells. Peripheral CD4+ T cells were infected with mutant NL4-3HSA strains expressing Env to increase infectivity. In infected cultures, 80–90% of the cells were HSA-positive (data not shown). The removal of both Vif and Vpr significantly alleviated HIV killing among these infected cells, permitting 57% viability in HSAhi cells as opposed to 15–21% viability in Vif and/or Vpr-expressing infected cells (Fig. 5A). Vif killing was also observed in the peripheral CD4+ T cells, which expresses APOBEC3G. Therefore, Vif-induced cytopathicity in Jurkat cells is unlikely to be a result of overexpression/hyperactivation of Vif in the absence of its natural substrate APOBEC3G.
Fig. 5.
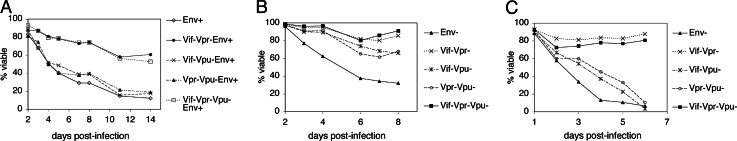
Time course measurements of the viability of CD4+ T cell lines infected with NL4-3HSAEnv+ strains carrying mutations in accessory genes. (A) Peripheral CD4+ T cells after 3 days of stimulation and infection with VSV-G pseudotyped viruses at a MOI of 30. The extent of infection and viability were monitored by flow cytometry by staining for the HSA marker and with PI staining, respectively, as described in Fig. 2. The percentage of PI-negative infected cells is shown in the graph. (B and C) MOLT-4 (B) and SupT1 (C) T cell lines were infected with mutant strains of NL4-3HSA, and the viability of infected cells was monitored by flow cytometry.
MOLT-4 and SupT1 cells were similarly infected with the mutant NL4-3HSA Env− strains used for Jurkat infections (Fig. 5 B and C). MOLT-4 showed the highest viability of all infected cells examined. However, only 30–40% of MOLT-4 cells were HSAhi compared to 80–90% for the other cell types, which probably accounts for the apparent increase in viability among these cells (Fig. 5B). Nevertheless, infection of either MOLT-4 or SupT1 cells with Vif−Vpr− and Vif−Vpr−Vpu− viruses resulted in viability comparable to that seen among uninfected cells, in contrast to the 40% reduction in viability observed when vif and vpr genes were intact. Hence, the data from multiple T cell lines and normal nontransformed T cells reveal a principal role for both the Vpr and Vif accessory proteins, but not other viral proteins, in HIV-1 mediated cytopathicity.
During these experiments, we noticed that cells infected with Vif−Vpr− strains underwent cell division and maintained high HSA expression for extended periods of time. However, the proliferation of infected cells was not observed with the other strains, even those lacking Vpr, if Vif was intact. The HSAhi fraction of these cells appeared to diminish as a consequence of a higher rate of proliferation among uninfected cells as well as cytopathicity (Fig. 7). This observation was surprising because cell cycle arrest is typically believed to require the presence of Vpr. An intriguing possibility was that Vif also caused cell cycle arrest. We therefore examined the DNA content of cells infected with various mutant viruses. The majority of uninfected Jurkat cells or those infected with the GFP-only virus were at the G1 phase of the cell cycle (Fig. 6; see also Fig. 9 a and b, which is published as supporting information on the PNAS web site). As expected, the DNA profile shifted from the G1 phase to the G2 phase in cells infected with Env− virus, which contained intact Vpr, indicating cell cycle arrest (c). Surprisingly, we found that viruses lacking Vpr also showed arrest at the G2 phase (f and j). However, G2 blockade was abolished if Vif was eliminated from the Vpr− virus (d, h, and k). Infected peripheral CD4+ T cells also showed similar cell cycle profiles (Fig. 10, which is published as supporting information on the PNAS web site), suggesting that APOBEC3G does not affect Vif-induced cell cycle arrest.
Fig. 6.

Cell cycle profiles of Jurkat cells infected with various mutant NL4-3HSA strains. Infected Jurkat cells were examined for DNA content (x axis) vs. cell number (y axis) 2 days after infection to assess their position in the cell cycle.
To confirm Vif-induced cell cycle arrest, we infected Jurkat cells that were synchronized at the border of G1 and S phases and examined the progression of cell cycle in infected cells (42). We pretreated cells with G1/S blocker aphidicolin for 16 h, and after a 10-h release, we infected the cells in the presence of the drug. Then, infected cells were again released from G1/S phase blockade after 16 h of the drug treatment. Cells were analyzed for HSA expression and DNA content at 0, 10, 20, and 40 h after the release by flow cytometry. At the time of release (16 h after infection), infected cells were at G1 phase (Fig. 11, which is published as supporting information on the PNAS web site). Ten hours after release, infected cells clearly accumulated at the G2 phase in the presence of Vif and Vpr either alone or in combination (Fig 11 A, C, and D), confirming cycle arrest by Vif and Vpr. However, in the absence of Vif and Vpr, infected cells did not show the characteristic G2 arrest profile even at 40 h after infection (Fig. 11B), supporting G2 cell cycle blockade independently by Vif and Vpr.
Because flow cytometric analyses of DNA content do not distinguish between G2 and M phases, we also examined the presence or absence of a mitotic marker by Western blotting. Lysates from infected Jurkat cells were analyzed for the presence or absence of phosphorylation of histone H3 at serine 28. The phosphorylation of histone H3 at Ser-28 occurs during mitotic chromosomal condensation and, thus, serves as an early mitotic marker (43, 44). Ser-28 phosphorylation of histone H3 was detected in mock-infected cells, indicating the presence of mitotic cells in culture (Fig. 12, lane 1, which is published as supporting information on the PNAS web site). The amount of Ser-28-phosphorylated H3 was greatly diminished in cells infected with Vif or Vpr expressing viruses (Fig. 12, lanes 2, 4, and 5), suggesting that cell cycle arrest occurs at the G2 rather than the M phase of cell cycle. By contrast, phosphorylated H3 at Ser-28 was restored in cells infected with Vif−Vpr− virus (Fig. 12, lane 3), suggesting that the mutant virus does not cause G2 cell cycle arrest. Thus, Vif, in addition to Vpr, independently causes cell cycle arrest at the G2 phase in infected cells. Hence, both genes that play a major role in cytopathicity share the property of cell cycle inhibition at the G2 stage.
Discussion
Since discovery of the in vitro cytopathic effect of poliovirus by Enders and coworkers (45), viral cytopathicity has been believed to be a key element of viral pathogenesis in vivo. In vitro, HIV-1 is a highly cytopathic virus that causes CD4+ T cell death shortly after infection. Nevertheless, the importance of this event for AIDS pathogenesis and the mechanism(s) by which it comes about have been unresolved after extensive investigation. Recent work has emphasized that direct viral killing of infected T cells, especially memory T cells, makes an important contribution to AIDS pathogenesis during acute infection (7, 8). We previously demonstrated that necrotic death occurs in CD4+ T cells during a single round of infection with the NL4-3HSA laboratory strain of HIV-1 (12–14). We also observed that this cytopathic effect could be attributed to direct killing rather than killing of bystander cells (14). Therefore, in the present study, we sought to define the viral molecules responsible for the direct killing of CD4+ T cells. In particular, we attempted to attribute a lethal effect to specific viral molecules independent of any role in viral infectivity. We systematically created various combinations of mutations that prevented the expression of various viral proteins in the NL4-3HSA Env− strain and examined cytopathicity. Although we initially hypothesized that one or more of the structural proteins or enzymes encoded in the HIV genome would cause cell death, this conjecture was not correct. The cytopathicity of the Gag−Pol− strain of NL4-3HSA was unchanged from that of the Env− strain, resulting in >80% cell death in 6 days (Fig. 3).
By contrast, disruption of both the Vif and Vpr coding regions substantially ameliorated virus-induced cell death, indicating that either of these gene products could independently render a viral cytopathic effect. The abrogation of cell death did not diminish and, therefore, was independent of infectivity at least in this in vitro culture system. Jurkat cells highly infected with the NL4-3HSAVif−Vpr− or NL4-3HSAVif−Vpr−Vpu− strain were still viable 25 days after infection (Fig. 7B). Viability of peripheral blood lymphocytes and other T cell lines was sustained if they were infected with the NL4-3HSAVif−Vpr− or NL4-3HSAVif−Vpr−Vpu− strain (Fig. 5). Although all of these results were derived from in vitro tissue culture with an MOI >1, the requirement for either Vif and Vpr for cytopathic effects suggests that these genes could be important in HIV pathogenesis beyond their role in virus infectivity.
Could there be viral components besides Vif and Vpr involved? It was evident that a low level of cell death could be instigated by viruses devoid of Vif or Vpr. However, it was difficult to ascribe this effect to any particular viral molecule, and, therefore, the question remains open. Interestingly, although Gag and Pol themselves did not contribute significantly to cytopathicity, their removal enhanced the protective effect of eliminating Vpr. For example, a Vpr− strain caused ≈88% death in the presence of Gag and Pol, but Gag−Pol−Vpr− mutant strain led to only 55% cell death (Figs. 3 and 4). Furthermore, strains lacking Vpr and Vpu killed 90% of cells in 8 days, whereas cells infected with a Gag−Pol−Vpr−Vpu− strain killed only 32% of the cells (Figs. 3 and 4). The decrease in cytopathicity in Gag−Pol− strains was evident only when coupled with the Vpr mutation. Thus, the Gag-Pol polyprotein apparently enhances killing by Vif, which causes cytopathicity in the vpr mutant strain. Vif is the least stable HIV-1 protein (46), and Gag-Pol may act to stabilize the protein levels of Vif (K.S. and M.J.L., unpublished observations).
Unexpectedly, we observed cell cycle arrest at the G2 phase in infected cells in the absence of Vpr and determined that Vif was responsible. It is possible that HIV-1 cytopathicity and cell cycle arrest might be correlated; both properties have been previously ascribed to Vpr (reviewed in refs. 22, 47, and 48). Other studies reported an increase in LTR activity at the G2 phase and suggested that this increase contributes to viral killing (42, 49, 50). However, the HSA marker was driven by the LTR in our NL4-3HSA vector, and we observed no increased HSA expression in the presence of Vif and Vpr.
Agents that cause G2 cell cycle blockade such as etoposide, vincristine, and nocodazole are known to cause cell death (39, 40, 51). Hence, it may be that, unlike the G1 phase, arrest in G2, in which replication has doubled the DNA content, is not a natural stopping point in the cell cycle that is tolerated by T cells. Increased ploidy can lead to untoward effects such as chromosome instability (52). In conclusion, our study sheds light on the components of HIV-1 required for cell cycle arrest and cell death and sets the stage for determining the interrelationship of the two in viral pathogenesis.
Materials and Methods
Virus Stock Production.
All virus stocks were produced in 293T cells and pseudotyped by using VSV-G to replace Env, as described in ref. 51. Briefly, 2 μg of HIV-1 plasmid was cotransfected with 1 μg each of pLVSV-G (National Institutes of Allergy and Infectious Diseases AIDS Research and Reference Reagent Program) and pCMVΔR8.2ΔVpr (23) into 293T cells in a six-well plate by using 13.2 μl of ExGen 500 reagent according to the manufacturer’s instructions (Fermentas, Hanover, MD). The virus was collected 48 h after transfection and stored at −80°C. Virus titers were determined by infecting Jurkat T cells with various volumes of virus stocks and assessing the MOI based on the Poisson distribution as described in ref. 13. For cytopathicity studies, 106 Jurkat cells were infected at a MOI of 3 or 5 in the presence of 5 μg/ml polybrene (Sigma) in a 12-well plate, followed by 30 min of centrifugation at 800 × g at room temperature.
For CD4+ T cell infection, peripheral blood lymphocytes were prepared from buffy coats by Ficoll-Paque Plus gradient separation (Amersham Pharmacia Biosciences), and CD4+ T cells were isolated by positive selection by using MACS CD4 microbeads (Miltenyi Biotec, Auburn, CA). After the isolation, CD4+ T cells were immediately stimulated with 3 μg/ml of plate-bound CD3 and 2 μg/ml CD28 antibodies for 3 days before infection. Infection was performed at a MOI of 30 as described above except that 15 μg/ml polybrene and 100 units/ml of IL-2 were added at the time of infection.
Viability and Infectivity Assay.
Cell viability and infectivity were assessed by flow cytometry on FACS Calibur (Becton Dickinson). Cells were harvested every 24 h and stained with propidium iodide (PI) for viability and with fluorescein-conjugated CD24 antibody (HSA, BD Pharmingen) for infectivity.
Construction of Mutant NL4-3HSA Strains.
Various mutations were introduced into pNL4-3HSA Env− by site-directed mutagenesis or subcloning of mutant fragments. Details are described in Supporting Text, which is published as supporting information on the PNAS web site. The mutations were confirmed by DNA sequencing.
Cell Cycle Analysis.
To analyze cell cycle arrest induced by HIV-1, the DNA content of infected Jurkat cells and peripheral CD4+ T cells was examined by flow cytometry. Jurkat cells stained for the surface expression of HSA (13) were first incubated in 1% paraformaldehyde for 10 min at room temperature, washed with 1× PBS, and fixed with 70% ethanol at −20°C. Subsequently, the fixed cells were washed and stained with DNA staining solution (4 μg/ml PI/50 μg/ml RNase/0.45 mg/ml sodium citrate in 1× PBS). Peripheral CD4+ T cells were stained with 20 μM cell-permeable DNA dye, DRAQ5 (Axxora, San Diego), for 5 min at 37°C according to the manufacturer’s instructions after HSA staining. Stained cells were examined on a FACS Calibur flow cytometer and analyzed by flowjo software (Tree Star, Ashland, OR).
Cell Synchronization and Infection.
Jurkat cells were synchronized with a G1/S phase blocker, aphidicolin, as described in ref. 42. Briefly, cells were treated with aphidicolin for 16 h and washed for three times to remove the drug. After 10 h of release, cells were infected with NL4-3HSA wild-type or mutant strains in the presence or absence of aphidicolin. Cells were harvested 0, 10, 20, and 40 h after the release. Viability, fractions of infected cells, and DNA content were analyzed by flow cytometry as described above by using PI, HSA, and DRAQ5 staining, respectively. Cell cycle status was also examined at 20 h after the release by Western blotting as described below.
Western Blot Analyses.
To verify the loss of protein expression by site-directed mutagenesis, infected Jurkat cells were lysed with 2% SDS lysis buffer (30 mM Tris·Cl, pH 8.0/150 mM NaCl/2% SDS/1 mM EDTA), and protein concentration in lysates was estimated by a bicinchoninic acid assay (Pierce). Equal amount of proteins were loaded onto a SDS/PAGE, and the loss of protein expression was examined by Western blotting. Patient sera were used to detect Gag and Pol, and Vif and Vpr were probed with specific antibodies. Vif monoclonal antibody (no. 319) was obtained from Michael H. Malim (National Institutes of Health) (36, 53, 54). The polyclonal antibody against Vpr was generously provided by K. Strebel (National Institutes of Allergy and Infectious Diseases, National Institutes of Health). For cell cycle profile, lysates were analyzed by a Western blot probed for an early mitotic marker by using anti-phopho-histone H3 (Ser-28) antibody (Upstate Biotechnology, Charlottesville, VA).
Supplementary Material
Acknowledgments
We thank Klaus Strebel for providing us a ΔVpu plasmid and Vpr antibody and for valuable discussions; Stephen Porcella at the Rocky Mountain Laboratory for DNA sequencing support; Diane Bolton, John Coffin, Malcolm Martin, Danny Douek, and the members of the Lenardo laboratory for helpful suggestions; Eugenia Park, Marianne Selkirk, and Melody Duvall for early studies; Nicolas Bidere, Lixin Zhang, Ronald Germain, and Danny Douek for critical reading of the manuscript; Shirley Starnes for manuscript preparation; and Anthony Fauci for the generous availability of his BL-3 facility. K.S. is a Japan Society for the Promotion of Science research fellow for Japanese biomedical and behavioral research at the National Insitutes of Health. This research was supported by the Intramural Research Program of the National Institutes of Allergy and Infectious Diseases, National Institutes of Health.
Abbreviations
- HSA
heat-stable antigen
- MOI
multiplicity of infection
- PI
propidium iodide.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Gallo R. C. Immunol. Rev. 2002;185:236–265. doi: 10.1034/j.1600-065x.2002.18520.x. [DOI] [PubMed] [Google Scholar]
- 2.Ho D. D., Neumann A. U., Perelson A. S., Chen W., Leonard J. M., Markowitz M. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 3.Perelson A. S., Neumann A. U., Markowitz M., Leonard J. M., Ho D. D. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 4.Wei X., Ghosh S. K., Taylor M. E., Johnson V. A., Emini E. A., Deutsch P., Lifson J. D., Bonhoeffer S., Nowak M. A., Hahn B. H., et al. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 5.Nishimura Y., Brown C. R., Mattapallil J. J., Igarashi T., Buckler-White A., Lafont B. A., Hirsch V. M., Roederer M., Martin M. A. Proc. Natl. Acad. Sci. USA. 2005;102:8000–8005. doi: 10.1073/pnas.0503233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Douek D. C., Picker L. J., Koup R. A. Annu. Rev. Immunol. 2003;21:265–304. doi: 10.1146/annurev.immunol.21.120601.141053. [DOI] [PubMed] [Google Scholar]
- 7.Mattapallil J. J., Douek D. C., Hill B., Nishimura Y., Martin M., Roederer M. Nature. 2005;434:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 8.Li Q., Duan L., Estes J. D., Ma Z.-M., Rourke T., Wang Y., Reilly C., Carlis J., Miller C. J., Haase A. T. Nature. 2005;434:1148–1152. doi: 10.1038/nature03513. [DOI] [PubMed] [Google Scholar]
- 9.Somasundaran M., Robinson H. L. J. Virol. 1987;61:3114–3119. doi: 10.1128/jvi.61.10.3114-3119.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stevenson M., Meier C., Mann A. M., Chapman N., Wasiak A. Cell. 1988;53:483–496. doi: 10.1016/0092-8674(88)90168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sylwester A., Murphy S., Shutt D., Soll D. R. J. Immun. 1997;158:3996–4007. [PubMed] [Google Scholar]
- 12.Lenardo M. J., Angleman S. B., Bounkeua V., Dimas J., Duvall M. G., Graubard M. B., Hornung F., Selkirk M. C., Speirs C. K., Trageser C., et al. J. Virol. 2002;76:5082–5093. doi: 10.1128/JVI.76.10.5082-5093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolton D. L., Hahn B.-I., Park E. A., Lehnhoff L. L., Hornung F., Lenardo M. J. J. Virol. 2002;76:5094–5107. doi: 10.1128/JVI.76.10.5094-5107.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Speirs C., van Nimwegen E., Bolton D., Zavolan M., Duvall M., Angleman S., Siegel R., Perelson A. A., Lenardo M. J. J. Virol. 2005;79:4025–4032. doi: 10.1128/JVI.79.7.4025-4032.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirchhoff F., Carl S., Sopper S., Sauermann U., Matz-Rensing K., Stahl-Hennig C. Virology. 1999;254:61–70. doi: 10.1006/viro.1998.9522. [DOI] [PubMed] [Google Scholar]
- 16.Kirchhoff F., Easterbrook P. J., Douglas N., Troop M., Greenough T. C., Weber J., Carl S., Sullivan J. L., Daniels R. S. J. Virol. 1999;73:5497–5508. doi: 10.1128/jvi.73.7.5497-5508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao J., Vasir B., Sodroski J. G. J. Virol. 1994;68:4662–4668. doi: 10.1128/jvi.68.7.4662-4668.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roshal M., Zhu Y., Planelles V. Apoptosis. 2001;6:103–116. doi: 10.1023/a:1009636530839. [DOI] [PubMed] [Google Scholar]
- 19.Selliah N., Finkel T. H. Virology. 2001;286:412–421. doi: 10.1006/viro.2001.0994. [DOI] [PubMed] [Google Scholar]
- 20.Cao J., Park I. W., Cooper A., Sodroski J. J. Virol. 1996;70:1340–1354. doi: 10.1128/jvi.70.3.1340-1354.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He J., Choe S., Walker R., Di Marzio P., Morgan D. O., Landau N. R. J. Virol. 1995;69:6705–6711. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jowett J. B., Planelles V., Poon B., Shah N. P., Chen M. L., Chen I. S. J. Virol. 1995;69:6304–6313. doi: 10.1128/jvi.69.10.6304-6313.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stewart S. A., Poon B., Jowett J. B. M., Xie Y., Chen I. S. Y. Proc. Natl. Acad. Sci. USA. 1999;96:12039–12043. doi: 10.1073/pnas.96.21.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart S. A., Poon B., Jowett J. B., Chen I. S. J. Virol. 1997;71:5579–5592. doi: 10.1128/jvi.71.7.5579-5592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao X. J., Mouland A. J., Subbramanian R. A., Forget J., Rougeau N., Bergeron D., Cohen E. A. J. Virol. 1998;72:4686–4693. doi: 10.1128/jvi.72.6.4686-4693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukumori T., Akari H., Yoshida A., Fujita M., Koyama A. H., Kagawa S., Adachi A. Microbes Infect. 2000;2:1011–1017. doi: 10.1016/s1286-4579(00)01255-7. [DOI] [PubMed] [Google Scholar]
- 27.Zhu Y., Gelbard H. A., Roshal M., Pursell S., Jamieson B. D., Planelles V. J. Virol. 2001;75:3791–3801. doi: 10.1128/JVI.75.8.3791-3801.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lum J. J., Cohen O. J., Nie Z., Weaver J. G., Gomez T. S., Yao X. J., Lynch D., Pilon A. A., Hawley N., Kim J. E., et al. J. Invest. 2003;111:1547–1554. doi: 10.1172/JCI16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Somasundaran M., Sharkey M., Brichacek B., Luzuriaga K., Emerman M., Sullivan J. L., Stevenson M. Proc. Natl. Acad. Sci. USA. 2002;99:9503–9508. doi: 10.1073/pnas.142313699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waldhuber M. G., Bateson M., Tan J., Greenway A. L., McPhee D. A. Virology. 2003;313:91–104. doi: 10.1016/s0042-6822(03)00258-7. [DOI] [PubMed] [Google Scholar]
- 31.Nishizawa M., Kamata M., Mojin T., Nakai Y., Aida Y. Virology. 2000;276:16–26. doi: 10.1006/viro.2000.0534. [DOI] [PubMed] [Google Scholar]
- 32.Schrofelbauer B., Yu Q., Landau N. R. AIDS Rev. 2004;6:34–39. [PubMed] [Google Scholar]
- 33.Goff S. P. Cell. 2003;114:281–283. doi: 10.1016/s0092-8674(03)00602-0. [DOI] [PubMed] [Google Scholar]
- 34.Rose K. M., Marin M., Kozak S. L., Kabat D. Trends Mol. Med. 2004;10:291–297. doi: 10.1016/j.molmed.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 35.Navarro F., Landau N. R. Curr. Opin. Immunol. 2004;16:477–482. doi: 10.1016/j.coi.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 36.Fouchier R. A., Simon J. H., Jaffe A. B., Malim M. H. J. Virol. 1996;70:8263–8269. doi: 10.1128/jvi.70.12.8263-8269.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H., Wu X., Newman M., Shaw G. M., Hahn B. H., Kappes J. C. J. Virol. 1995;69:7630–7638. doi: 10.1128/jvi.69.12.7630-7638.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karczewski M. K., Strebel K. J. Virol. 1996;70:494–507. doi: 10.1128/jvi.70.1.494-507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheehy A. M., Gaddis N. C., Choi J. D., Malim M. H. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 40.Akari H., Bour S., Kao S., Adachi A., Strebel K. J. Exp. Med. 2001;194:1299–1311. doi: 10.1084/jem.194.9.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klimkait T., Strebel K., Hoggan M. D., Martin M. A., Orenstein J. M. J. Virol. 1990;64:621–629. doi: 10.1128/jvi.64.2.621-629.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goh W. C., Rogel M. E., Kinsey C. M., Michael S. F., Fultz P. N., Nowak M. A., Hahn B. H., Emerman M. Nat. Med. 1998;4:65–71. doi: 10.1038/nm0198-065. [DOI] [PubMed] [Google Scholar]
- 43.Goto H., Yasui Y., Nigg E. A., Inagaki M. Genes Cells. 2002;7:11–17. doi: 10.1046/j.1356-9597.2001.00498.x. [DOI] [PubMed] [Google Scholar]
- 44.Shibata K., Inagaki M., Ajiro K. Eur. J. Biochem. 1990;192:87–93. doi: 10.1111/j.1432-1033.1990.tb19199.x. [DOI] [PubMed] [Google Scholar]
- 45.Enders J. F. Annu. Rev. Microbiol. 1954;8:473–502. doi: 10.1146/annurev.mi.08.100154.002353. [DOI] [PubMed] [Google Scholar]
- 46.Zhang H., Pomerantz R. J., Dornadula G., Sun Y. J. Virol. 2000;74:8252–8261. doi: 10.1128/jvi.74.18.8252-8261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emerman M. Curr. Biol. 1996;6:1096–1103. doi: 10.1016/s0960-9822(02)00676-0. [DOI] [PubMed] [Google Scholar]
- 48.Re F., Luban J. Prog. Cell Cycle Res. 1997;3:21–27. doi: 10.1007/978-1-4615-5371-7_2. [DOI] [PubMed] [Google Scholar]
- 49.Forget J., Yao X. J., Mercier J., Cohen E. A. J. Mol. Biol. 1998;284:915–923. doi: 10.1006/jmbi.1998.2206. [DOI] [PubMed] [Google Scholar]
- 50.Gummuluru S., Emerman M. J. Virol. 1999;73:5422–5430. doi: 10.1128/jvi.73.7.5422-5430.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bartz S. R., Vodicka M. A. Methods. 1997;12:337–342. doi: 10.1006/meth.1997.0487. [DOI] [PubMed] [Google Scholar]
- 52.Fujisawa T., Bandi M., Nitta M., Ivanova E. V., Bronson R. T., Pellman D. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 53.Simon J. H., Southerling T. E., Peterson J. C., Meyer B. E., Malim M. H. J. Virol. 1995;69:4166–4172. doi: 10.1128/jvi.69.7.4166-4172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simon J. H., Fouchier R. A., Southerling T. E., Guerra C. B., Grant C. K., Malim M. H. J. Virol. 1997;71:5259–5267. doi: 10.1128/jvi.71.7.5259-5267.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.