

Abstract
The laboratory mouse (Mus musculus) has become one of the best model animal species in biomedical research today because of its abundant genetic/genomic information, and easy mutagenesis using transgenic and gene knockout technology. Genetically engineered mice have become essential tools in both mechanistic studies and drug development. In this article I will review recent topics in gastrointestinal cancer model mice, with emphasis on the results obtained in our laboratory. They include: (i) mouse models for familial adenomatous polyposis (Apc mutant mice; modifier genes of Apc intestinal polyposis; stabilizing β‐catenin mutant mice); (ii) mouse models for colon cancer (mouse models for hereditary non‐polyposis colon cancer; additional mutations in Apc mutant mice; models with mutations in other genes; models for colon cancer associated with inflammatory bowel diseases); and (iii) mouse models for gastric cancer. (Cancer Sci 2006; 97: 355 –361)
Abbreviations:
- AA
arachidonic acid
- ACF
aberrant crypt foci
- APC
adenomatous polyposis coli
- Chr
(mouse) chromosome
- cM
centimorgan(s)
- COX
cyclooxygenase
- FAP
familial adenomatous polyposis
- GI
gastrointestinal
- GSK
glycogen synthase kinase
- IL
interleukin
- K19
cytokeratin 19
- LOH
loss of heterozygosity
- mPGES‐1
microsomal prostaglandin E synthase‐1
- MSI
microsatellite instability
- NSAID
non‐steroidal anti‐inflammatory drug
- PG
prostaglandin
- PI
phosphoinositide
- PLA2
phospholipase A2
- SPEM
spasmolytic polypeptide/trefoil factor 2‐expressing metaplasia
- TGF‐β
transforming growth factor‐β
- TNF‐α
tumour necrosis factor‐α.
The mortality by gastrointestinal cancers ranks at the top of all cancer deaths in many countries. Different from lung cancer, in which the majority of cases could be prevented by elimination of tobacco smoke, there are few environmental factors that can prevent colon carcinogenesis. However, a strong epidemiological link is known between gastric cancer and Helicobacter pylori infection. To establish therapeutic and preventive strategies, it would be very important to develop workable mouse models for gastrointestinal carcinogenesis.
Based on its genetic origin, colon cancer can be divided into two classes, polyposis colon cancer and non‐polyposis colon cancer. Genes whose mutations are responsible for colon carcinogenesis have been discovered through molecular genetic studies of hereditary cancer predisposition syndromes such as FAP, and hereditary non‐polyposis colon cancer. Induced mutations of these genes in mice have provided mouse models that are similar to human colon cancer and polyposis, even though they may not be identical to the human diseases. However, there have been no workable mouse models for gastric cancer that is initiated by Helicobacter infection. Recently, we constructed a transgenic mouse strain that mimics early inflammatory changes in Helicobacter infection, and develops hyperplastic gastric tumors. In this review, I will describe recent progress in the field with some emphasis on the work from my own laboratory.
Mouse models for FAP
FAP is a hereditary disease with dominant inheritance that causes numerous colon polyps. Although the polyps are benign adenomas, some of them will eventually develop into malignant adenocarcinomas, if left untreated. The APC gene was identified on 5q as one of the genes commonly deleted in some FAP kindreds.( 1 , 2 ) It encodes a huge protein of approximately 2850 amino acid residues, forms a complex with axin and helps GSK3β to phosphorylate N‐terminal serine/threonine residues of β‐catenin, accelerating its rapid degradation through ubiquitination.( 3 ) If the APC gene is mutated, GSK3β cannot phosphorylate β‐catenin. Unphosphorylated and therefore stabilized β‐catenin accumulates in the cytoplasm, moves to the nucleus where it activates TCF/LEF transcription factors, inducing a set of new genes: Wnt target genes.( 4 ) Activation of the canonical Wnt pathway in the colonic epithelium appears to be one of the key events in the polyp initiation process( 5 ) (Fig. 1).
Figure 1.
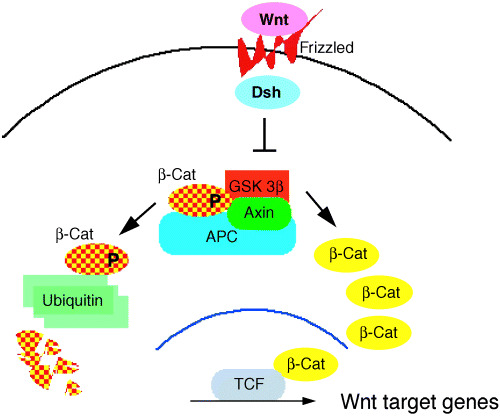
Schematic presentation of the canonical Wnt signal pathway. The left side shows the normal adult tissues where phosphorylation of β‐catenin target serine/threonine residues are phosphorylated and degraded rapidly by ubiquitination. The right side indicates the transcriptional activation of the Wnt target genes by unphosphorylated and therefore stabilized β‐catenin. APC, adenomatous polyposis coli.
Apc Min mice, Apc Δ716 mice, Apc 1638N mice and other Apc mutant mice
The first mouse mutant in the Apc gene (the mouse homolog of human APC on mouse chromosome 18) was found from a colony of randomly mutagenized mice.( 6 ) This mutant, Min (multiple intestinal neoplasia) was found to carry a truncation mutation at codon 850 of the Apc gene,( 7 ) hence Apc Min. In the C57BL/6 background, the heterozygote develops ∼30 polyps in the small intestine. With gene knockout technology, several Apc mutations have been constructed. Apc Δ716 contains truncating mutation at codon 716, whereas Apc 1638N at codon 1638.( 8 , 9 ) Like in Apc Min mice, both knockout mutants develop polyps mainly in the small intestine. Histologically, all these Apc mutants form polyp adenomas indistinguishable from each other. Interestingly, however, the polyp numbers are very different, even in the same C57BL/6 J background. Namely, Apc Δ716 develops usually ∼300 polyps, whereas Apc 1638N forms only ∼3 polyps. Despite the numerous polyps developing in the small intestine in the Apc Δ716 (as well as in Apc Min) mice, only a few polyps are formed in the colon, although the penetrance is complete. We have recently constructed a mutant mouse strain in which numerous polyps develop in the distal colon; see below.( 10 ) Using Apc Δ716 mice, it was demonstrated that polyp formation is initiated by LOH at the Apc locus in the proliferative zone cells,( 9 ) followed by formation of an outpocket in the intestinal crypt.( 5 ) The results strongly suggested that APC protein is essential for the proliferative zone cells to migrate along the crypt‐villus axis.
Several other Apc gene knockout mice have been reported. Their histopathology appears essentially the same as in Apc Min and Apc Δ716 with the only difference being the number of intestinal polyps.
Modifier genes of Apc intestinal polyposis
Introduction of COX‐2 gene mutation dramatically decreases the polyp number in Apc polyposis mice.( 11 , 12 , 13 ) Likewise, COX‐1 mutation also reduces the polyp multiplicity in Apc polyposis mice (14) (Fig. 2). We first determined expression of COX‐2 protein in intestinal polyps of various sizes, and found that COX‐2 was expressed from a very early stage of polyp formation.( 13 ) We then introduced a COX‐2 gene (Ptgs2) knockout mutation into the Apc Δ716 mice, and discovered that both the number and size of polyps were reduced dramatically in the compound mutant mice, in a mutated gene dose‐dependent manner. To confirm the results with a pharmaceutical compound, we further dosed Apc Δ716 mice with COX‐2 inhibitors, and demonstrated that the polyp number can be reduced in a dose‐dependent manner.( 13 , 15 ) These results gave the rationale to treat human FAP patients with COX‐2 inhibitors such as celecoxib or rofecoxib, and clinical trials confirmed the results of animal experiments.( 16 ) Following these experiments, a number of reports have been published dosing the Apc mutant mice with various drugs or drug candidates. These experiments were summarized in a review.( 17 ) It should be noted that the constitutive isozyme COX‐1 also plays a significant role in polyposis. Introduction of COX‐1 mutation into Apc Min mice reduces the number and size of intestinal polyps by ∼80%, similar to the effect by COX‐2 mutation.( 14 ) In fact, COX‐1 and COX‐2 cooperate in polyp formation by supplying PGE2 that stimulates polyp angiogenesis.( 18 ) Recently, it has been reported that COX‐2 inhibitors are associated with cardiovascular side‐effects in patients participating in sporadic polyposis prevention trials.( 19 , 20 ) Accordingly, COX‐2 inhibitors, as well as regular NSAIDs, must be prescribed with caution to patients who are at high risk with cardiovascular accidents. Polyp prevention studies with young FAP patients without such risks can be pursued with proper monitoring.
Figure 2.
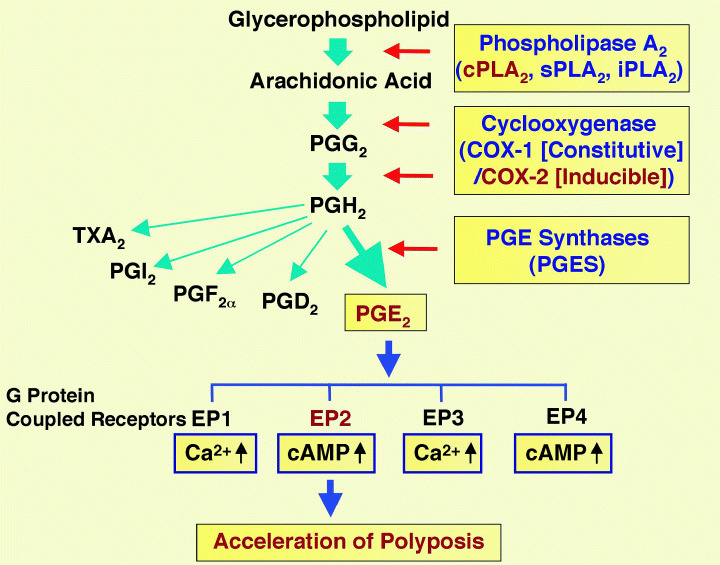
Schematic presentation of arachidonic acid metabolism in the context of intestinal polyposis. COX, cyclooxygenase; PG, prostaglandin; TX, thromboxane.
To assess the role of the AA/COX‐2/PGE2 pathway further, we constructed several additional compound mutants of Apc Δ716 mice with other genes in the pathway. Although COX is the rate‐limiting enzyme in the pathway, its substrate AA is not freely available in vivo, but supplied on demand by the activity of phospholipases that cleave AA from membrane phospholipids. One of the key enzymes in this activity is cytosolic phospholipase A2 (cPLA2). We introduced a knockout gene for the enzyme into Apc Δ716 mice, and demonstrated that polyp expansion is reduced significantly.( 21 ) In the downstream of the pathway, however, the direct metabolite of AA by COX is PGH2 that is converted further to various prostanoids as PGA2, D2, F 2α, E2, I2, and thromboxane A2 (TXA2). Of these, PGE2 appears to play the major role in polyposis by binding to four G‐protein‐coupled cell surface receptors, EP1, EP2, EP3 and/or EP4. To determine which particular EP receptor is involved in the polyposis phenotype, we introduced knockout mutations for the receptors into Apc Δ716 mice, and scored the polyp number and size. The results clearly showed a significant suppression of the polyposis phenotype only in the compound mutant mice with EP2 mutation.( 22 ) Moreover, we found that induction of COX‐2 itself, as well as other phenotypes associated with COX‐2 induction such as angiogenesis and basement membrane changes, are also mediated by PGE2 and EP2 receptor. The former phenotype indicates that a positive feedback signal mediated through EP2 and intracellular cyclic AMP regulates COX‐2 expression.( 22 )
Expression of COX‐2 has been observed not only in adenomatous polyps, but also in hamartomatous polyps and malignant cancers as well. For example, COX‐2 expression has been reported in Peutz–Jeghers gastrointestinal hamartomas.( 23 ) We have recently investigated expression of COX‐2 in hamartomas of three models, namely, in mutant mice of Smad4, Cdx2 and Lkb1, respectively, and found significant expression levels in all models.( 24 ) These results indicate that COX‐2 induction is a general phenomenon common to most tumors.
Other possible targets have been proposed for the antitumor activities of NSAIDs,( 25 ) but these pathways remain to be investigated further by genetic means, that is, crossing of the target gene knockout mice with the polyposis mice.
Another avenue of similar experiments is to study the effects of various diets and food additives, in attempts to identify the epidemiologic effects on cancer initiation and progression. For example, we demonstrated that docosahexaenoic acid reduces intestinal polyp development, although the effect is moderate and found only in female mice,( 26 ) whereas feeding Apc Δ716 mice with a high‐fat diet increases polyp numbers significantly.( 27 ) Likewise, a Western‐style diet (high fat and low calcium) accelerates tumor formation in Apc 1638N mice.( 28 ) It is also interesting to point out that calorie intake restriction by 40% reduces intestinal polyps in Apc Min mice by ∼60%, suggesting that dietary interventions can partially offset genetic susceptibility to intestinal carcinogenesis.( 29 )
Many other genes have been introduced into Apc polyposis mice, and some turned out to increase the number of polyps. For example, the DNA helicase gene mutation that is responsible for Bloom syndrome increases the number of intestinal polyps in Apc Min mice.( 30 , 31 ) However, the mutation for the telomerase RNA gene caused a complex phenotype in Apc Min mice. The polyp numbers increased in several generations, then decreased thereafter.( 32 )
Introduction of homozygous mutation in p21, a cyclin inhibitor, into Apc 1638N mice increases tumor multiplicity approximately two‐fold, and affects goblet cell differentiation.( 33 )
One of the matrix metalloproteinases, matrilysin, is implicated in cancer invasion and metastasis, and its mutation reduces the polyp number in Apc Min mice, indicating that the enzyme is involved in the formation of adenomatous polyps.( 34 )
Introduction of homozygous mutation for the inducible nitric oxide synthase gene reduced the polyp number in Apc Min mice by less than half in the small intestine, and to about 10% of the control in the colon, suggesting a possibility in colonic polyp prevention with inducible nitric oxide synthase‐selective inhibitors.( 35 )
Accumulating evidence indicates that the promoter regions of many cancer related genes are silenced by methylation of the cytidine residues. However, the effects on intestinal polyposis by knockout mutations in DNA methylation enzymes have been complex and confusing. Mutations in the DNA methyltransferase gene (Dnmt1), in conjunction with an enzyme inhibitor, reduced the polyp number in Apc Min mice from over 100 to less than two, suggesting suppressive effects of DNA hypomethylation on intestinal polyposis.( 36 ) However, introduction of Mbd2 homozygous mutation in Apc Min mice also reduced the polyp numbers by approximately 90%. Because Mbd2 encodes methyl‐CpG binding repressor that recruits co‐repressor complexes to methylated DNA, these results indicate that DNA methylation‐mediated gene silencing is integral to the Apc Min polyposis.( 37 )
The multidrug resistance 1 (Mdr1) gene promoter contains the TCF‐LEF binding site, and is activated by Wnt signaling. In Mdr1‐deficient Apc Min mice (Apc Min/+ Mdr1a/b −/–), the polyp number was reduced to almost half that of Apc Min mice, although the clinical relevance of these results is yet to be investigated.( 38 )
As described above, all Apc mutant mice develop adenomatous polyps in the small intestine, rather than in the colon. However, recent studies show that an additional mutation in the Cdx2 gene in Apc Δ716 mice reverses the polyp localization, shifting most polyps to the colon, as in human FAP.( 10 ) Interestingly, the dramatic increase in the colon polyp number is caused by the increased frequency of Apc LOH, caused by chromosomal instability. The latter results from activation of the mTOR pathway and acceleration of the G1 to S phase transition in the cell cycle.( 10 ) These results present a new mechanism for chromosomal instability, and suggest a possibility of treatment and prevention of colon cancer with chromosomal instability.
Stabilizing β‐catenin mutant mice
Because APC forms a complex with other proteins that mediates the Wnt signaling pathway, it is reasonable to ask whether mutations in other components of the complex can also cause polyps in the mouse intestines. In fact, stabilizing mutations in the serine/threonine residues of β‐catenin have been identified in a subpopulation of colon cancer that do not carry APC mutations. To test such a possibility experimentally, conditional stabilizing β‐catenin mutations have been introduced that are specifically expressed in the intestines. When expression of stabilized β‐catenin is induced from calbindin promoter, mice developed only a few polyps in the small intestine.( 39 ) In contrast, expression of Cre recombinase driven by K19 or fatty acid binding protein gene promoter to introduce floxed stabilizing mutation in the β‐catenin gene, caused formation of 700–3000 polyps in the small intestine.( 40 ) These results confirm the role of Wnt signaling activation in polyp formation, and indicate that polyps are initiated essentially in the rapidly multiplying cells in the proliferative zone. The floxed β‐catenin mutant mice have been used in several other organ systems, providing evidence for the roles of Wnt signaling in prostate tumorigenesis,( 41 ) and embryonic and immune system development.( 42 , 43 )
Mouse models for colon cancer
All Apc mutant mice develop adenomatous polyps, but they do not progress into invasive or metastatic adenocarcinomas at a significant frequency. Because of the heavy tumor load in the small intestine, most Apc mutant mice die young (4–5 months) due to anemia and cachexia, and some of them by intestinal intussusception. If additional mutations are introduced into these mice, however, the intestinal polyposis phenotypes are modified, and sometimes markedly malignant adenocarcinomas develop.
Mouse models for hereditary non‐polyposis colon cancer
Single homozygous mutations in the DNA mismatch repair genes rarely cause adenocarcinomas in mice. Interestingly, compound mutant mice of some of these genes can cause adenocarcinomas of the intestines. However, their histopathology is much milder than human colorectal cancer, and no metastasis to the liver or lymph nodes is observed.
Additional mutations in Apc Min, Apc Δ716 or Apc 1638N mice
For example, we have introduced Smad4 mutation (see the section ‘Modifier genes of Apc intestinal polyposis’) into Apc Δ716 polyposis mice, and constructed a model for malignant adenocarcinoma.( 44 ) Although human homologs SMAD4 and APC are on separate chromosomes, the mouse genes are both found on mouse chromosome 18, approximately 30 cM apart. Because polyps are initiated by Apc LOH in Apc Δ716 intestines, and because this LOH is caused by loss of the entire chromosome 18 due to recombination at the ribosomal DNA locus near the centromere, LOH of Apc also results in LOH at Smad4. Taking advantage of this fact, we have constructed mice that carried Apc Δ716 and Smad4 mutations on the same chromosome in the cis‐ configuration. In the intestinal polyps, both Apc and Smad4 loci are lost, resulting in homozygous mutant cells for both loci. Importantly, the intestinal polyps in these mice progress rapidly into very invasive adenocarcinomas.( 44 ) Interestingly, however, these adenocarcinomas do not metastasize during the short lifespan of these mice. The histopathology is somewhat similar to that of the right side colon cancer in human that is often caused by mutations in the type II receptor for TGF‐β. In fact, this model verifies tumor progression by sequential mutations in multiple genes. Moreover, some of these mice also develop adenocarcinomas at the duodenal papilla of Vater, which is one of the complications in human FAP after colectomy.
Colon cancer models with mutations in other genes
Other colon cancer models have been reported that are caused by mutations in various other genes. For example, a knockout mutation in the TGF‐β1 gene (Tgfb1) introduced into Rag2 mutant mice causes adenocarcinomas with strong local invasions.( 45 ) However, it has been reported that the homozygous mutant in Smad3, encoding another cellular signaling molecule in the TGF‐β pathway and a partner of SMAD4, develops colon cancer that metastasizes to the draining lymph nodes.( 46 ) However, the penetrance of this phenotype appears to be low, and lymph node metastasis has not been found in all affected mice. Furthermore, the metastatic phenotype has not been observed in any similar knockout mutants of Smad4 constructed by other groups.( 47 )
With the use of the promoter for the villin gene whose expression is specific to the intestinal epithelium, transgenic mice have been constructed that express activated mutant of K‐ras (K‐ras V12G). Most transgenic mice develop single or multiple lesions, ranging from microadenomas to invasive adenocarcinomas, without mutations in Apc. ( 48 ) Similar to the cis‐ compound Apc Δ716 Smad4 mutant mice, none of the adenocarcinomas in this model metastasize to distant loci, although the tumors are highly invasive locally.
Homozygous mutation in the Muc2 gene that encodes the most abundant secreted gastrointestinal mucin causes adenomas and adenocarcinomas in the intestines.( 49 ) Although the incidence and multiplicity are low, the adenocarcinoma is locally invasive without distant metastasis.
Mouse models for colon cancer associated with inflammatory bowel disease
Colon cancer associated with inflammatory bowel disease is somewhat different from regular (i.e., non‐inflammatory) colon cancer. Malignant cancer develops after long and sustained inflammation in the intestines. The etiology of the inflammation is mostly autoimmune reactions, and alterations in various immune cells and mediators have been reported. As models for this type of colon cancer, several mutant mouse strains have been constructed. For example, IL‐10 deficient mice produce aberrant cytokines, especially γ‐interferon, and invasive adenocarcinoma develop in the colon in 60% of mice by 6 months of age.( 50 ) Likewise, compound mutant mice for IL‐2 and β2‐microglobulin genes also develop ulcerative colitis‐like disease, and approximately one‐third of them produces adenocarcinomas in the colon.( 51 ) Interestingly, null mutant mice for one of G proteins, Gαi2, also develop similar colitis and adenocarcinoma of the colon.( 52 ) However, introduction of dominant negative N‐cadherin causes inflammatory bowel disease and adenomas, but no carcinomas.( 53 )
Mouse models for gastric cancer
Gastric cancer is still one of the most deadly cancers in many countries.( 54 ) Although the epidemiologic correlation has been established between H. pylori infection and gastric carcinogenesis,( 55 ) the molecular mechanism that leads to gastric cancer remains to be elucidated. One of the problems with the direct mechanism hypothesis, for example, that one of the H. pyroli gene products directly causes gastric cancer, is the fact that only a small fraction of the infected patients develop cancer, and only after many years. Accordingly, it is rather more reasonable to assume that sustained infection with H. pylori causes a precancerous condition that can lead to carcinogenesis after additional insults. Based on such an ‘indirect’ hypothesis, we have focused on the role of prostaglandin E2 in earlier stages of gastric carcinogenesis. It is known that Helicobacter infection induces COX‐2 and mPGES‐1 in the gastric mucosa.( 56 , 57 ) COX‐2 is induced also in gastric cancer tissues.( 58 ) As a model for H. pyroli infected inflammation in the stomach, we constructed transgenic mice that simultaneously express COX‐2 and mPGES‐1 in the gastric mucosa, using K19 gene promoter: Tg.K19‐C2mE mice.( 57 ) With the induction of both COX‐2 and mPGES‐1, PGE2 production is increased efficiently as seen in the Helicobacter‐infected gastric mucosa. Interestingly, the transgenic mice developed metaplasia, and hyperplastic tumors in the glandular stomach, with heavy macrophage infiltrations. Although gastric bacterial counts in the transgenic mice were within the normal range, treatment with antibiotics significantly suppressed activation of the macrophages and hyperplastic tumors. Importantly, the antibiotic treatment did not affect the macrophage accumulation. Notably, treatment of the transgenic mice with lipopolysacchrides induced proinflammatory cytokines through Toll‐like receptor 4 in the gastric epithelial cells. These results indicate that an increased level of PGE2 enhances macrophage infiltration, and that they are activated through epithelial cells by the gastric flora, resulting in gastric metaplasia and hyperplastic tumors.
To further investigate the gastric hyperplastic tumors in the transgenic mice, we determined the levels of various cytokines and growth factors, and found that TNF‐α and IL‐1β were significantly increased. We then introduced knockout mutations for TNF‐α (Tnf) and IL‐1 receptor α chain (Il1r1) genes. Interestingly, only the compound mutant Tnf (–/–) Tg.K19‐C2mE mice showed significant suppression of hyperplastic tumors. Importantly, SPEM was also suppressed in the Tnf (–/–)Tg.K19‐C2mE mice, indicating that TNF‐α‐dependent inflammation was responsible for SPEM development (Fig. 3). Because gastric metaplasia to the SPEM lineage is considered as a preneoplastic lesion of gastric cancer, it is possible that inhibition of TNF‐α‐dependent inflammation, together with eradication of Helicobacter, can be an effective prevention strategy for gastric cancer.( 59 )
Figure 3.
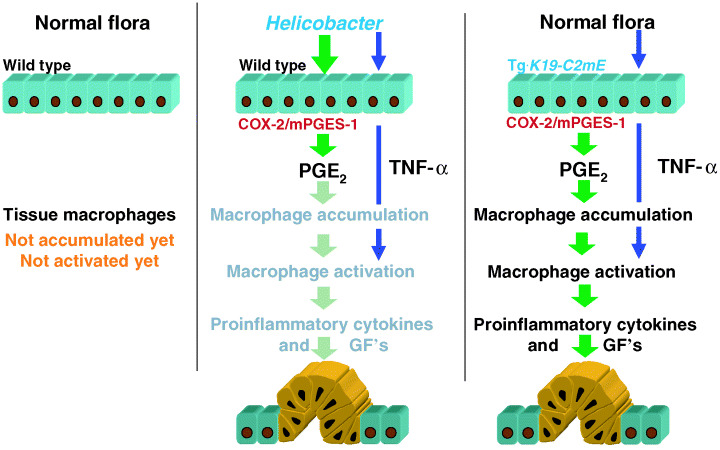
A likely mechanism for hyperplastic tumor development by Helicobacter pylori infection. COX, cyclooxygenase; GF, growth factor; PG, prostaglandin; TNF‐α, tumour necrosis factor‐α.
In conclusion, many mouse models have been established that are useful for investigations of the initiation, expansion and progression of gastrointestinal cancers. They are also valuable tools to evaluate various pharmaceutical and biological agents for prevention and treatment of gastrointestinal cancer. Yet, one of the key outstanding issues in this field of research is to establish practical models of cancer metastasis to the liver, lung and lymph nodes. Such models should greatly help us find novel measures to overcome colon cancer in the 21st century.
Acknowledgments
I am grateful to the members of my laboratory who have contributed to the papers I have cited. The research programs in my laboratory have been supported by grants from MESSC, Japan; OPSR, Japan; University of Tokyo–Banyu Pharmaceutical Co. Joint Fund; Takeda Foundation, and Mitsubishi Foundation.
References
- 1. Groden J, Thliveris A, Samowitz W et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991; 66: 589–600. [DOI] [PubMed] [Google Scholar]
- 2. Kinzler KW, Nilbert MC, Vogelstein B et al. Identification of a gene located at chromosome 5q21 that is mutated in colorectal cancers. Science 1991; 251: 1366–251. [DOI] [PubMed] [Google Scholar]
- 3. Polakis P. The oncogenic activation of β‐catenin. Curr Opin Genet Devel 1999; 9: 15–21. [DOI] [PubMed] [Google Scholar]
- 4. Korinek V, Barker N, Morin PJ et al. Constitutive transcriptional activation by a β‐catenin‐Tcf complex in APC−/– colon carcinoma. Science 1997; 275: 1784–7. [DOI] [PubMed] [Google Scholar]
- 5. Oshima H, Oshima M, Kobayashi M et al. Morphological and molecular processes of polyp formation in ApcΔ716 knockout mice. Cancer Res 1997; 57: 1644–9. [PubMed] [Google Scholar]
- 6. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990; 247: 322–4. [DOI] [PubMed] [Google Scholar]
- 7. Su L‐K, Kinzler KW, Vogelstein B et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992; 256: 668–70. [DOI] [PubMed] [Google Scholar]
- 8. Fodde R, Edelmann W, Yang K et al. A targeted chain‐termination mutation in the mouse Apc gene results in multiple tumors. Proc Natl Acad Sci USA 1994; 91: 8969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oshima M, Oshima H, Kitagawa K et al. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncation Apc gene. Proc Natl Acad Sci USA 1995; 92: 4482–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aoki K, Tamai Y, Horiike S et al. Colonic polyposis caused by mTOR‐mediated chromosomal instability in Apc +/Δ716 Cdx2 +/– compound mutant mice. Nat Genet 2003; 35: 323–30. [DOI] [PubMed] [Google Scholar]
- 11. Taketo MM. Cyclooxygenase‐2 inhibitors in tumorigenesis (Part I). J Natl Cancer Inst 1998; 90: 1529–36. [DOI] [PubMed] [Google Scholar]
- 12. Taketo MM. Cyclooxygenase‐2 inhibitors in tumorigenesis (Part II). J Natl Cancer Inst 1998; 90: 1609–20. [DOI] [PubMed] [Google Scholar]
- 13. Oshima M, Dinchuk JE, Kargman SL et al. Suppression of intestinal polyposis in Apc Δ716 knockout mice by inhibition of cyclooxygenase 2 (COX‐2). Cell 1996; 87: 803–9. [DOI] [PubMed] [Google Scholar]
- 14. Chulada PC, Thompson MB, Mahler JF et al. Genetic disruption of Ptgs‐1, as well as of Ptgs‐2, reduces intestinal tumorigenesis in Min mice. Cancer Res 2000; 60: 4705–8. [PubMed] [Google Scholar]
- 15. Oshima M, Murai (Hata) N, Kargman S et al. Chemoprevention of intestinal polyposis in the Apc Δ716 mouse by rofecoxib, a specific cyclooxygenase‐2 inhibitor. Cancer Res 2001; 61: 1733–40. [PubMed] [Google Scholar]
- 16. Steinbach G, Lynch PM, Phillips RKS et al. The effect of celecoxib, a cyclooxygenase‐2 inhibitor, in familial adenomatous polyposis. New Engl J Med 2000; 342: 1946–52. [DOI] [PubMed] [Google Scholar]
- 17. Oshima M, Taketo MM. COX selectivity and animal models for colon cancer. Curr Pharmaceut Design 2002; 8: 102–34. [DOI] [PubMed] [Google Scholar]
- 18. Takeda H, Sonoshita M, Sugihara K et al. Cooperation of cyclooxygenase 1 and cyclooxygenase 2 in intestinal polyposis. Cancer Res 2003; 63: 4872–7. [PubMed] [Google Scholar]
- 19. Solomon SD, McMurray JJV, Pfeffer MA et al. Cardiovascular risk assessment with celecoxib in a clinical trial for colorectal adenoma prevention. New Engl J Med 2005; 352: 1071–80. [DOI] [PubMed] [Google Scholar]
- 20. Bresalier RS, Sandler RS, Quan H et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. New Engl J Med 2005; 352: 1092–102. [DOI] [PubMed] [Google Scholar]
- 21. Takaku K, Sonoshita M, Sasaki N et al. Suppression of intestinal polyposis in Apc Δ716 knockout mice by an additional mutation in the cytosolic phospholipase A2 gene. J Biol Chem 2001; 275: 34013–6. [DOI] [PubMed] [Google Scholar]
- 22. Sonoshita M, Takaku K, Sasaki N et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc Δ716 knockout mice. Nat Med 2001; 7: 1048–51. [DOI] [PubMed] [Google Scholar]
- 23. De Leng WW, Westerman AM, De Rooij FW et al. Cyclooxygenase 2 expression and molecular alterations in Peutz–Jeghers hamartomas and carcinomas. Clin Cancer Res 2003; 9: 3065–72. [PubMed] [Google Scholar]
- 24. Takeda H, Miyoshi H, Tamai Y et al. Simultaneous expression of COX‐2 and mPGES‐1 in mouse gastrointestinal hamartomas. Br J Cancer 2004; 90: 701–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DuBois RN. New agents for cancer prevention. J Natl Cancer Inst 2002; 94: 1732–3. [DOI] [PubMed] [Google Scholar]
- 26. Oshima M, Takahashi M, Oshima H et al. Effects of docosahexaenoic acid (DHA) on intestinal polyp development in Apc Δ716 knockout mice. Carcinogenesis 1995; 16: 2605–7. [DOI] [PubMed] [Google Scholar]
- 27. Hioki K, Shivapurkar N, Oshima H et al. Suppression of intestinal polyp development by low‐fat and high‐fiber diet in Apc Δ716 knockout mice. Carcinogenesis 1997; 18: 1863–5. [DOI] [PubMed] [Google Scholar]
- 28. Yang K, Edelman W, Fan K et al. Dietary modulation of carcinoma development in a mouse model for human familial adenomatous polyposis. Cancer Res 1998; 58: 5713–7. [PubMed] [Google Scholar]
- 29. Mai V, Colbert LH, Berrigan D et al. Calorie restriction and diet composition modulate spontaneous intestinal tumorigenesis in Apc Min mice through different mechanisms. Cancer Res 2003; 63: 1752–5. [PubMed] [Google Scholar]
- 30. Luo G, Santoro IM, McDaniel LD et al. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet 2000; 26: 424–9. [DOI] [PubMed] [Google Scholar]
- 31. Heppner Goss K, Risinger MA, Kordich JJ et al. Enhanced tumor formation in mice heterozygous for Blm mutation. Science 2002; 297: 2051–3. [DOI] [PubMed] [Google Scholar]
- 32. Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet 2001; 28: 155–9. [DOI] [PubMed] [Google Scholar]
- 33. Yang WC, Mathew J, Velcich A et al. Targeted inactivation of the p21 WAF/cip1 gene enhances Apc‐initiated tumor formation and the tumor‐promoting activity of a western‐style high‐risk diet by altering cell maturation in the intestinal mucosa. Cancer Res 2001; 61: 565–9. [PubMed] [Google Scholar]
- 34. Wilson CL, Heppner KJ, Labosky PA, Hogan BLM, Matrisian LM. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc Natl Acad Sci USA 1997; 94: 1402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ahn B, Oshima H. Suppression of intestinal polyposis in Apc Min/+ mice by inhibiting nitric oxide production. Cancer Res 2001; 61: 8357–60. [PubMed] [Google Scholar]
- 36. Laird PW, Jackson‐Grusby L, Faseli A et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell 1995; 81: 197–205. [DOI] [PubMed] [Google Scholar]
- 37. Sansom OJ, Berger J, Bishop SM et al. Deficiency of Mbd2 suppresses intestinal tumorigenesis. Nat Genet 2003; 34: 145–7. [DOI] [PubMed] [Google Scholar]
- 38. Yamada T, Mori Y, Hayashi R et al. Suppression of intestinal polyposis in Mdr1‐deficient Apc Min/+ mice. Cancer Res 2003; 63: 895–901. [PubMed] [Google Scholar]
- 39. Romagnolo B, Berrebi D, Saadi‐Keddoucci S et al. Intestinal dysplasia and adenoma in transgenic mice after overexpression of an activated β‐catenin. Cancer Res 1999; 59: 3875–9. [PubMed] [Google Scholar]
- 40. Harada N, Tamai Y, Ishikawa T et al. Intestinal polyposis in mice with a dominant stable mutation of the β‐catenin gene. EMBO J 1999; 18: 5931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gounari F, Signoretti S, Bronson R et al. Stabilization of β‐catenin induces lesions reminiscent of prostatic intraepithelial neoplasia, but terminal squamous transdifferentiation of other secretory epithelia. Oncogene 2002; 21: 4099–107. [DOI] [PubMed] [Google Scholar]
- 42. Lickert H, Domon C, Huls G et al. Wnt/β‐catenin signaling regulates the expression of the homeobox gene Cdx1 in embryonic intestine. Development 2000; 127: 3805–13. [DOI] [PubMed] [Google Scholar]
- 43. Gounari F, Aifantis I, Khazaie K et al. Somatic activation of β‐catenin bypasses pre‐TCR signaling and TCR selection in thymocyte development. Nat Immunol 2001; 2: 863–9. [DOI] [PubMed] [Google Scholar]
- 44. Takaku K, Oshima M, Miyoshi H et al. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 1998; 92: 645–56. [DOI] [PubMed] [Google Scholar]
- 45. Engle SJ, Hoying JB, Boivin GP et al. Transforming growth factor β1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res 1999; 59: 3379–86. [PubMed] [Google Scholar]
- 46. Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell 1998; 94: 703–14. [DOI] [PubMed] [Google Scholar]
- 47. Yang X, Letterio JJ, Lechleider RJ et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF‐β. EMBO J 1999; 18: 1280–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Janssen K‐P, El Marjou F, Pinto D et al. Targeted expression of oncogenic K‐ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Targeted expression of oncogenic K‐ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterol 2002; 123: 492–504. [DOI] [PubMed] [Google Scholar]
- 49. Velcich A, Yang W, Heyer J et al. Colorectal cancer in mice genetically deficient in the mucin Muc2 . Science 2002; 295: 1726–9. [DOI] [PubMed] [Google Scholar]
- 50. Berg DJ, Davidson N, Kühn R et al. Enterocolitis and colon cancer in interleukin‐10‐deficient mice are associated with aberrant cytokine production and CD4+ TH1‐like responses. J Clin Invest 1996; 98: 1010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shah SA, Simplson SJ, Brown LF et al. Development of colonic adenocarcinomas in a mouse model of ulcerative colitis. Inflamm Bowel Dis 1998; 4: 196–202. [DOI] [PubMed] [Google Scholar]
- 52. Rudolph U, Finegold MJ, Rich SS et al. Ulcerative colitis and adenocarcinoma of the colon in Gαi2‐deficient mice. Nat Genet 1995; 10: 143–50. [DOI] [PubMed] [Google Scholar]
- 53. Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N‐cadherin. Science 1995; 270: 1203–7. [DOI] [PubMed] [Google Scholar]
- 54. Ushijima T, Sasako M. Focus on gastric cancer. Cancer Cell 2004; 5: 121–5. [DOI] [PubMed] [Google Scholar]
- 55. Correa P. Helicobacter pylori infection and gastric cancer. Cancer Epidemiol Biomarkers Prev 2003; 12: 238s–241s. [PubMed] [Google Scholar]
- 56. Sung JJY, Leung WK, Go MYY et al. Cyclooxygenase‐2 expression in Helicobacter pylori‐associated premalignant and malignant gastric lesions. Am J Pathol 2000; 157: 729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Oshima H, Oshima M, Inaba K, Taketo MM. Hyperplastic gastric tumors induced by activated macrophages in COX‐2/mPGES‐1 transgenic mice. EMBO J 2004; 23: 1669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ristimaki A, Honkanen N, Jankala H et al. Expression of cyclooxygenase‐2 in human gastric carcinoma. Cancer Res 1997; 57: 1276–80. [PubMed] [Google Scholar]
- 59. Oshima M, Oshima H, Matsunaga A, Taketo MM. Hyperplastic gastric tumors with spasmolytic peptide‐expressing metaplasia (SPEM) caused by TNF‐α‐dependent inflammation in COX‐2/mPGES‐1 transgenic mice. Cancer Res 2005; 65: 9147–51. [DOI] [PubMed] [Google Scholar]