

Abstract
The camphor monoxygenase cytochrome P450cam (CYP101) requires potassium ion (K+) to drive formation of the characteristic high-spin state of the heme Fe+3 upon substrate binding. Amide 1H, 15N correlations in perdeuterated [U-15N] CYP101 were monitored as a function of K+ concentration by 2D-TROSY-HSQC in both camphor-bound oxidized (CYP-S) and camphor-and CO-bound reduced CYP101 (CYP-S-CO). In both forms, K+-induced spectral perturbations are detected in the vicinity of the K+ binding site proposed from crystallographic structures, but are larger and more widespread structurally in CYP-S than in CYP-S-CO. In CYP-S-CO, K+-induced perturbations occur primarily near the proposed K+ binding site in the B-B’ loop and B’ helix, which are also perturbed by binding of effector, putidaredoxin (Pdx). The spectral effects of K+ binding in CYP-S-CO oppose those observed upon Pdxr titration. However, Pdxr titration of CYP-S-CO in the absence of K+ results in multiple conformations. The spin-state equilibrium in the L358P mutant of CYP101 is more sensitive to K+ concentration than WT CYP101, consistent with a hypothesis that L358P preferentially populates conformations enforced by Pdx binding in WT CYP101. Thallium (I), a K+ mimic, minimizes the effects of Pdx titration on the NMR spectrum of CYP-S-CO, but is competent to replace K+ in driving the formation of high-spin CYP-S. These observations suggest that the role of K+ is to stabilize conformers of CYP-S that drive the spin state change prior to the first electron transfer, and that K+ stabilizes the CYP-S-CO conformer that interacts with Pdx. However, upon binding of Pdx, further conformational changes occur that disfavor K+ binding.
Keywords: CYP101, putidaredoxin, NMR, HSQC, electron transfer complex, TROSY, thallium
ABBREVIATIONS: CYP101, cytochrome P450cam; CYP-S, oxidized camphor-bound CYP101; CYP-S-CO, reduced camphor- and carbonmonoxy-bound CYP101; IPTGm, isopropyl β-D-thiogalactoside; LB, Luria-Bertani media; NMR, nuclear magnetic resonance; HSQC, heteronuclear single-quantum correlation; M9, minimal growth medium; OD600, optical density at 600 nm; PdR, putidaredoxin reductase; Pdx, putidaredoxin; Pdxr, reduced putidaredoxin; TROSY, transverse relaxation optimized spectroscopy
Introduction
Enzymes are dynamic, and must access multiple conformations in order to accomplish different and apparently contradictory tasks (i.e., selective binding of substrates, stabilization of transition state(s), release of products) in the course of turnover (1). We are interested in how required protein conformations are stabilized at different points in the reaction cycle of cytochrome P450cam (CYP101). CYP101 is a heme-containing monooxygenase from the soil bacterium Pseudomonas putida that catalyzes the 5-exo hydroxylation of camphor (2). Camphor binding to resting state CYP101 results in a nearly complete shift from a low-spin (S = 1/2, λmax = 417 nm) to a high-spin heme Fe+3 (S = 5/2, λmax = 391 nm) with binding of non-native substrates resulting in less complete shifts to high spin (3, 4). This spin state shift changes the reduction potential of the heme and permits single electron transfer from the reduced iron-sulfur protein putidaredoxin (Pdxr) (2). It has been shown that at least 50 mM potassium ion, K+, is necessary for maximum conversion to the high spin form upon camphor binding, and K+ also stabilizes CYP101 against conversion to the inactive P420 form (5, 6). A K+ binding site has been proposed from X-ray crystallographic structures of CYP101 that includes the backbone carbonyl oxygens of Glu 84, Gly 93, Glu 94 and Tyr 96, and two ordered water molecules (7). Three of these residues, Gly 93, Glu 94 and Tyr 96, are part of the B’ helix (residues 90–96) which is strongly perturbed by binding of reduced putidaredoxin (Pdxr) to reduced substrate- and CO-bound CYP101 (CYP-S-CO) as determined by multidimensional NMR methods (8). Pdx, a Cys4Fe2S2 ferredoxin, is the in vivo effector and reductant of CYP101, and the complex between Pdxr and reduced O2- and camphor-bound CYP101 is the catalytically competent species for camphor hydroxylation (9, 10). We have found that perturbations in CYP-S-CO structure upon Pdxr binding take place not only in the proposed Pdx binding site on the proximal face of CYP101 near the C helix and axial heme Fe ligand (Cys 357) loop, but also in regions remote from the interface between the two proteins, including the B’, F and G helices and portions of β sheets 1, 3 and 5 (8, 11). Residues on the B’, F and G helices and FG loop are implicated in substrate access to and orientation within the active site, suggesting to us that the primary effector role of Pdx is to prevent loss of substrate and/or intermediates and enforce the correct orientation of the substrate for reaction prior to the second electron transfer step (12). These perturbations are also observed, albeit to a lesser extent, when Pdx is replaced by cytochrome b5, a non-physiological effector of camphor hydroxylase activity by CYP101 (11). We have found evidence for a high-barrier conformational change that takes place in CYP-S-CO upon binding of Pdxr that results in reorientation of substrate into the correct geometry for hydroxylation, and occurs on the same time scale as the other perturbations observed upon Pdxr binding to CYP-S-CO (12). Based on these observations as well as the location of the likely K+ binding site involving residues on the B’ helix, we decided to investigate the structural and functional role of K+ binding using the same methodology and to determine what if any relationship might exist between K+ binding and Pdx binding by CYP101.
The effects of K+ on enzyme-substrate interactions in CYP101 have been studied by numerous methods (13, 14), and the K+ binding site has been presumed to be defined by the octahedral ligation sphere surrounding WAT 515 in the 3CCP crystal structure of CYP-S-CO (15). Using a series of uniformly perdeuterated and 15N-labeled samples of CYP101, we have performed one-dimensional (1D) 1H and two-dimensional (2D) 1H-15N TROSY-HSQC NMR experiments to observe the perturbations in CYP101 structure as a function of K+ concentration, effector binding and the oxidation state of the enzyme. We can now confirm that the primary perturbations resulting from K+ binding are indeed largest for residues in the B-B’ loop (Glu 84 to Ile 88) and the B’ helix (residues Arg 90 to Tyr 96) in both oxidation states, although the effects of K+ binding are both more widespread and more profound in oxidized CYP-S than in reduced CYP-S-CO. We also observe that in the absence of K+, titration of CYP-S-CO with Pdxr results in at least two spectroscopically distinguishable conformations in CYP-S-CO. Furthermore, resonances affected by both Pdxr and K+ binding move in opposite directions in Pdxr and K+ titrations, suggesting that the binding of Pdxr and K+ by CYP-S-CO are antagonistic. We find that monovalent thallium, Tl+, which is similar to K+ in size and hydration properties and typically shows high affinity for K+-specific sites, is as effective in driving the spin state change as K+ in CYP-S, but essentially nullifies the spectral effects of Pdxr titration on CYP-S-CO.
Finally, we have also investigated K+ binding by L358P CYP101. This mutant has been shown to have a higher affinity for Pdx than WT CYP101 (16), and so might be expected to be perturbed structurally in regions affected by Pdx binding. We find that spin-state changes in this mutant are more sensitive to K+ concentration than the WT enzyme, and furthermore we show that the orientation of camphor in the active site of reduced CO-bound L358P in the absence of Pdxr largely matches that observed in the WT complex between Pdxr and CYP-S-CO.
Materials and Methods
Mutagenesis of CYP101
The L358P mutant of CYP101 was generated in plasmid pDNC334A that encodes for C334A CYP101. Cys 334 is a solvent-exposed cysteine, and was replaced by alanine in order to prevent dimerization at concentrations used for NMR experiments. This mutant has been shown to be identical in activity to wild-type CYP101 (17), and for convenience in comparing C334A CYP101 with L358P CYP101, C334A will be referred to as WT.
A four primer-method mutagenesis method was used for introducing the L358P mutation. This method uses two side primers and two middle mutagenic primers. The side primers introduce appropriate restriction sites at the 5’ and 3’ termini of the gene, and the middle primers introduce the desired mutation. Primers used were as follows: CYP101 side primers, 5’-TTTCACACAGGAAACAGACCATATGACGAC-3’ and 5’-CCAAAACAGCCAAGCTTT CAGCTACTTATAC-3’; mutagenic primers, 5’-CATCTGTGCCCTGGCCAGCACC-3’ and 5’-GGTGCTGGCCAGGGCACAGATG-3’. The side primers include engineered NdeI and HindIII restriction sites. Experimental methods used for four-primer mutagenesis were described previously (18).
Expression and Purification of WT and L358P CYP101
The expression and purification of isotopically labeled CYP101 has been described previously (11). Purification of L358P CYP101 was identical to that of WT, as were isotopic labeling procedures. The purity of CYP101 was determined spectroscopically. Fractions with an absorption ratio A391/A280 greater than 1.4 were used for experiments described below.
Spin state measurements on WT and L358P CYP101
Optical spectroscopy was used to monitor the Soret bands at 391 and 417 nm as indicators of the high-spin (HS) and low-spin (LS) ferric forms, respectively. All spectra were obtained using an Agilent 8453 UV/visible spectrophotometer. Oxidized camphor-bound WT and L358P CYP101 was added to 50 mM Tris-HCl pH 7.4, 1 mM camphor with different K+ concentrations varying from 0 to 50, 100, 200, 400 mM in septum-sealed cuvettes. The second derivatives of the absorbance spectra were employed for determining relative HS/LS concentrations. This method allowed direct observation of the peaks at both wavelengths with near baseline resolution (19). Fractional concentrations of HS and LS forms were determined from relative peak areas. Calculations are uncorrected for differences in extinction coefficients of HS and LS forms.
NMR Spectroscopy
Perdeuterated [U-15N] CYP101 was isolated and purified from NCM533 using the same procedure described above with the exception that 0.3% d8-glycerol (CIL) was used as the carbon source in the D2O-based M9 medium. 15NH4Cl (CIL) was used as sole nitrogen source and all inorganic salts were perdeuterated by lyophilization from D2O followed by solution in D2O. CYP101 samples for all NMR experiments were 0.2–0.5 mM in the NMR buffer 90% H2O 10% D2O, pH 7.4, 50 mM Tris-HCl, 2 mM D-camphor. All CYP101 samples for reduction were placed in a carbon monoxide (CO) atmosphere for 10 min before and 2 min after the addition of aliquots of 0.25 M Na2S2O4 (prepared in filtered and degassed 1 M Tris-HCl, pH 8.0). The reduced CO- and camphor-bound CYP-S-CO samples were then transferred in an anaerobic chamber to an NMR tube (Shigemi, Inc., Allison Park, PA). Perdeuterated Pdx was expressed and purified according to published methods (20). All NMR experiments were performed at 25°C on a Varian Inova 600 MHz spectrometer operating at 599.702 and 60.774 MHz for 1H and 15N, respectively.
NMR titrations of CYP-S-CO with K+
A series of 1H, 15N TROSY-HSQC spectra of perdeuterated 15N-labeled CYP-S-CO were acquired as a function of K+ concentration at 25 °C. Potassium ion concentration (as KCl) was varied between 0, 50, and 100 mM. All spectra were acquired with 596 (1H) × 128 (15N) complex points, 1H sweep width of 8000 Hz, and 15N sweep width of 2200 Hz. A similar experiment was performed in which 100 mM NaCl replaced KCl in order to differentiate between K+-specific and general salt effects. All 1H chemical shifts are reported in parts per million (d) relative to trimethylsilylpropionic acid sodium salt. 15N chemical shifts are reported relative to liquid ammonia. All NMR spectra were processed and analyzed using the Topspin software package (Bruker Biospin, Inc.).
The titration of CYP101 with perdeuterated Pdxr in presence of 100 mM KCl has been described previously (11). Another Pdxr titration series was performed at 25 °C in the absence of K+ following the same procedure. 1D 1H spectra of samples were recorded immediately prior to acquisition of 2D spectra in order to determine camphor orientation. A third Pdxr titration was performed in the presence of Tl+ (as 100 mM TlNO3). For this sample, Tris-HCl was replaced with Tris-HNO3 to prevent precipitation of relatively insoluble TlCl. A 2D HNCO dataset was acquired using 1020 (1H) × 256 (13C) complex points, a 1H sweep width of 10000 Hz, and 13C sweep width of 3770 Hz to detect perturbations due to Tl+ using a 2H, 13C, 15N labeled CYP-S-CO sample in 50 mM TlNO3. Appropriate safety and waste disposal precautions were exercised in the use of the high toxic thallium salt.
Results
Spin-state dependence on K+ concentration in oxidized WT and L358P CYP-S
As shown in Figure 1 and Table 1, the fractions of high spin (HS) heme Fe+3 increases in oxidized WT CYP-S with increasing K+ concentration, with 100 mM KCl yielding close to maximum high spin, in agreement with previous studies (13, 21). On the other hand, oxidized L358P CYP-S exhibits a lower fraction of HS at any given K+ concentration than WT at the same camphor concentration, although camphor appears to be completely bound to L358P upon reduction (vide infra). At 100 mM K+, where WT CYP-S is 87% HS as measured from peak areas in second derivative spectra, L358P CYP-S is only 65% HS. Furthermore, as K+ ion concentration is increased beyond 100 mM, WT CYP-S reaches maximum fraction of high spin at 200 mM KCl, with no change in the HS/LS ratio past this point, while the L358P CYP-S continues to increase in % HS up to 400 mM KCl, the highest salt concentration at which measurements were made.
Figure 1.

Left) Second-derivative UV-visible spectra of oxidized WT CYP-S showing the effect of K+ concentration on spin state equilibrium. Black curve was obtained at 0 mM KCl, red curve at 50 mM KCl and blue curve in 100 mM KCl. All spectra were recorded at 25°C with 6 μM CYP101 in 50 mM Tris-HCl, 1 mM camphor, pH 7.4. Right) Second-derivative UV-visible spectra of oxidized L358P CYP-S. Black curve was obtained at 0 mM KCl, red curve at 50 mM KCl and blue curve in 100 mM KCl. Spectra were recorded at 25°C with 3 μM L358P CYP101 in 50 mM Tris-HCl, 1 mM camphor, pH 7.4.
Table 1.
Percent high-spin in CYP-S as a function of salt concentration in WT and L358P CYP101 as measured by second-derivative UV-visible spectroscopy. CYP101 concentration was 5 μM in 50 mM Tris HCl or Tris HNO3 (for TlNO3 measurements) pH 7.4 with 2 mM D-camphor. Relative concentrations were calculated from peak areas uncorrected for extinction coefficients.
| Conc. KCl or TlNO3 (mM) | % HS, WT CYP-S, K+ (Tl+) | %HS, L358P, K+ |
|---|---|---|
| 0 | 36 (45) | 16 |
| 50 | 82 (82) | 57 |
| 100 | 88 (87) | 65 |
| 200 | 93 | 69 |
| 400 | 93 | 73 |
Structural perturbations in CYP-S-CO and CYP-S due to K+ binding
We have recently described progress in the assignment of backbone 1H, 15N and 13C resonances of CYP-S-CO (11). We have now identified nearly all of the amide 1H, 15N correlations in the vicinity of the proposed K+ binding site in CYP-S-CO using multidimensional NMR methods, as well as Cα and CO 13C backbone resonances. These assignments include the resonances of Ser 83, Glu 84, Cys 85, Phe 87 and Ile 88, as well as the complete B’ helix, Arg 90-Tyr 96. Residues 86 and 89 are both prolines and hence do not contribute correlations to NH-detected 2D and 3D NMR experiments, although Cα and Cβ correlations are available for these residues through the HNCA, HN(CO)CA and HNCACB data sets. Combined with previous assignments, we can comprehensively survey the effects of K+ binding on secondary structural features in CYP101. We observe that the effect of K+ is not general in CYP-S-CO, and most residues are unperturbed by changing K+ concentration. The largest perturbations are observed for the amide 1H,15N correlations of Glu 84, Cys 85 and Glu 94, which shift by more than 30 Hz in either the 1H or 15N dimensions between 0 and 100 mM KCl at 600 MHz 1H frequency (see Table 2 and Figure 2). All of these residues are in the B-B’ loop and B’ helix, in the vicinity of the crystallographically defined K+ binding site. Other residues in this region undergo smaller shifts (30 Hz > δmax >10 Hz), including Ser 83, Phe 87, Ile 88, Arg 90, Glu 91, Ala 92, Ala 95 and Tyr 96. Shifts are also observed for Glu 40 (A helix), several residues in the β1 sheet, G helix, and Gly 248 in the I helix. However, a comparison of effects between samples containing 100 mM NaCl and 100 mM KCl suggest that, with the exception of Ala 194 in the G helix, perturbations in regions other than the B-B’ loop and B’ helix of CYP-S-CO are due to generalized salt effects rather than K+ specific (Table 2). The spatial distribution of perturbed shifts in the CYP-S-CO structure are shown in Figure 3.
Table 2.
Magnitudes of 15N and 1H chemical shift perturbations in the presence of 100 mM KCl and 100 mM NaCl relative to no alkali salt in reduced camphor- and CO-bound WT CYP101 (CYP-S-CO) and oxidized camphor-bound WT CYP101 (CYP-S). Absolute values of Δδ are given in Hz. All buffers are 50 mM Tris HCl, pH 7.4. Spectra were obtained at 25 °C. Secondary structural features are labeled according to the scheme of Raag and Poulos (15). Correlations listed as “<10” show less than 10 Hz perturbations in either 15N or 1H. “2X” indicate doubling of resonances.
| Residue | Location in secondary structure | CYP-S-CO Δδ (15N, 1H) (Hz) (100-0) mM K+ | CYP-S-CO Δδ (15N,1H) (Hz) (100-0) mM Na+ | CYP-S Δδ (15N,1H) (Hz) (100-0) mM K+ | CYP-S Δδ (15N, 1H) (Hz) (100-0) mM Na+ |
|---|---|---|---|---|---|
| Cys 85* | B-B’ loop | 83, 42 | 30, 12 | Missing in no salt | Missing in no salt |
| Glu 94* | B’ | 65, 32 | 56, 29 | c | c |
| Ala 95* | B’ | 17, 10 | <10 | Missing in no salt | Missing in no salt |
| Asp 97* | B’ | a. | a. | b | b |
| Glu 20 | 2, 9 | 2, 7 | 3, 12 | 3, 12 | |
| Leu 22 | 1, 10 | 2, 7 | 10, 36 | 7, 36 | |
| Val 23 | 0, 9 | 0, 5 | 4, 18 | 2, 18 | |
| Phe 26 | 9, 18 | 4, 11 | b | b | |
| Glu 40 | A | 3, 18 | 4, 18 | 17, 42 | 12, 36 |
| Glu 47 | 2, 12 | 2, 10 | 2, 36 | 0. 30 | |
| Asp 52 | β1 | 3, 14 | 3, 11 | 6, 30 | 4, 24 |
| Leu 53 | β1 | 4, 18 | 3, 15 | 15, 48 | 11, 36 |
| Val 54 | β1 | 0, 4 | 5, 5 | 1, 30 | 3, 30 |
| Trp 55 | β1 | 0, 17 | <10 | 22, 59 | 17, 39 |
| Asn 59 | β1 | 2, 11 | 2, 11 | Missing in no salt | Missing in no salt |
| His 62 | β1 | 1, 10 | 1, 6 | 16, 6 | 11, 12 |
| Ile 64 | β1 | 1, 24 | 3, 24 | Missing in no salt | Missing in no salt |
| Thr 66 | 4, 2 | 2, 2 | Missing in no salt | 2X, 100 mM Na+ | |
| Gly 68 | B | 4, 10 | 1, 10 | Missing in no salt | Missing in no salt |
| Gln 69 | B | Not assigned | Not assigned | Missing in no salt | Missing in no salt |
| Asp 77 | B | 0, 9 | 0, 5 | 6, 30 | 5, 18 |
| Ser 83 | B-B’ loop | 3, 26 | <10 | Missing in no salt | Missing in no salt |
| Glu 84 | B-B’ loop | 9, 48 | 1, 26 | c. | c. |
| Phe 87 | B-B’ loop | 2, 23 | <10 | b | b |
| Ile 88 | B-B’ loop | 7, 21 | <10 | b | b |
| Arg 90 | B’ | 13, 10 | <10 | c. | c. |
| Glu 91 | B’ | 18, 52 | 10, 31 | Missing in no salt | Missing in no salt |
| Ala 92 | B’ | 2X in no salt | 2X in no salt | Missing in no salt | Missing in no salt |
| Gly 93 | B’ | 13, 17 | 5, 10 | Missing in no salt | Missing in no salt |
| Tyr 96 | B’ | c. | c. | c. | c. |
| Phe 98 | B’ | c. | c. | c. | c. |
| Ile 99 | c. | c. | c. | c. | |
| Thr 101 | <10 | <10 | c. | c. | |
| Glu 107 | C | <10 | <10 | Missing in no salt | Missing in no salt, 100 mM Na+ |
| Gln 108 | C | 0. 17 | <10 | 2X in no salt | 2X in no salt 100 mM Na+ |
| Glu 152 | E | 2, 12 | <10 | Missing in no salt | Missing in no salt, 100 mM Na+ |
| Ala 167 | E | c. | c. | 2X in no salt | 2X in no salt |
| Gly 168 | E | <10 | <10 | Missing in no salt | Missing in no salt, 100 mM Na+ |
| Glu 171 | E-F loop | <10 | <10 | 8, 30 | 7, 24 |
| Gly 189 | E-F loop | 0, 10 | <10 | 14, 18 | 10, 18 |
| Thr 192 | G | 3, 13 | 3, 11 | 12, 48 | 11, 36 |
| Ala 194 | G | 9, 53 | 5, 36 | 1, 48 | 2, 42 |
| Lys 197 | G | c. | c. | Missing in no salt | Missing in no salt, 100 mM Na+ |
| Ile 207 | G | 0, 10 | <10 | 22, 2 | 18, 2 |
| Glu 209 | G | 0, 17 | 0, 13 | Missing in no salt | Missing in no salt |
| Gln 213 | G | 0, 10 | 0, 10 | Missing in no salt | Missing in no salt |
| Gly 216 | G-H loop | <10 | <10 | 6, 30 | 6, 24 |
| Ala 224 | H | Not assigned | Not assigned | Missing in no salt | Missing in no salt |
| Gly 226 | β2 | 0, 12 | <10 | 23, 36 | 17, 30 |
| Val 228 | β2 | 4, 10 | 2, 10 | 3, 48 | 2, 36 |
| Asn 229 | β2 | <10 | <10 | 31, 6 | 23, 12 |
| Ile 233 | β2 | <10 | <10 | 7, 24 | 9, 12 |
| Thr 234 | I | <10 | 0, 10 | 3, 30 | 6, 19 |
| Ser 267 | I | <10 | <10 | 6, 18 | 4, 6 |
| Glu 306 | β4 | 2, 10 | <10 | 4, 17 | 4, 17 |
| Phe 307 | β4 | <10 | <10 | 10, 18 | 10, 12 |
| His 308 | β4 | 2, 11 | <10 | 19, 2 | 19, 2 |
| Val 310 | β4 | <10 | 2, 10 | 2X in no salt | 2X in no salt |
| Leu 312 | β4 | <10 | <10 | 4, 30 | 6, 30 |
| Asp 339 | <10 | <10 | 8, 12 | 7, 18 | |
| Asp 380 | 0, 10 | <10 | 5, 24 | 3, 24 | |
| Ala 384 | β5 | 0, 10 | <10 | 2, 18 | 2, 18 |
| His 391 | β5 | 0, 13 | 0, 10 | 11, 54 | 9, 42 |
| Gly 398 | β5 | <10 | <10 | 17, 12 | 14, 6 |
| Gln 400 | β5 | 0, 14 | 0, 14 | 31, 72 | 26, 60 |
| Leu 402 | β5 | 2, 16 | 2, 14 | 7, 36 | 5, 24 |
| Ala 409 | <10 | <10 | 1, 24 | 1, 24 | |
| Thr 410 | <10 | <10 | 2X in no salt | 2X in no salt | |
| Val 414 | <10 | <10 | 7, 30 | 7, 24 |
denotes NH groups bonded to proposed carbonyl ligands of K+. a) The NH correlation of Thr 97 has been identified in spectra obtained at 800 MHz, but could not be detected above noise level at 600 MHz. b) Signals missing due to paramagnetism in CYP-S. c) Overlapped signals prevent determination of Δδ.
Figure 2.

Comparison of K+ and Na+ salt effects on 1H, 15N correlations in TROSY-HSQC spectra of reduced CYP-S-CO (upper spectra) and oxidized CYP-S (lower spectra). Spectra on the left show the amide NH correlation of Cys 85, adjacent to carbonyl of Glu 84, proposed to ligate K+ (7). Upper left is reduced, lower left is oxidized. No correlation for Cys 85 is observed in the absence of alkali chloride salt in CYP-S (lower left). Spectra on the right show differential effects in reduced CYP-S-CO and oxidized CYP-S for Gly 226, located in the β2 sheet. All spectra were obtained at 25 °C, 600 MHz in 50 mM Tris-HCl, 2 mM camphor, pH 7.4.
Figure 3.

NMR spectral perturbations induced by K+ titration in CYP-S-CO and CYP-S mapped onto the CYP101 structure. Both figures are based on the 3CPP structure of Raag and Poulos (15). WAT 515, presumed to be K+ in the 3CPP structure, is indicated by a red sphere in both structures. Heme and camphor are in salmon. Left) Reduced CYP-S-CO. Backbone positions marked in red are perturbed and include the B-B’ loop and B’ helix including K+ binding residues Glu 84, Gly 93, Glu 94 and Tyr 96. Those in dark blue are perturbed by >10 Hz in either 15N or 1H, and include portions of the β1 and β5 strands. See Table 2 for details. Right) Oxidized CYP-S. Backbone positions marked in red indicate residues that are either doubled or missing in the absence of K+. These include portions of the β1, β4 and β5 strands, the B-B’ loop, B’ helix, C helix, E helix, F-G loop and G helix. See Table 2 for details. Residues in white indicate paramagnetic bleaching in the oxidized CYP-S for which no data is available. Figures were generated using MOLSCRIPT (35).
The NH correlations of the Glu 84-Cys 85 and Gly 93-Glu 94 amide groups show unusually large 15N perturbations due to K+ binding in CYP-S-CO (Table 2). The carbonyls of these amides are proposed to act as ligands to K+, so this data supports the identity of the proposed K+ binding site. The 15N, 1H correlation of the Glu 94-Ala 95 peptide, another proposed ligand, shows a smaller but still substantial 15N shift. The NH correlation of the Tyr 96-Thr 97 peptide bond, the fourth proposed ligand, while clearly identified in spectra obtained at 800 MHz, is not detected at 600 MHz, for unknown reasons.
In the oxidized CYP-S, assignments are not as comprehensive as in the reduced CYP-S-CO due primarily to paramagnetic relaxation of resonances within ~14 Å of the heme. However, sufficient assignments have been made so that we can we can monitor effects of K+ binding over much of the protein structure. We find that the spectral perturbations are more widespread in CYP-S than in CYP-S-CO, and in general, are also larger in magnitude for a given residue in CYP-S than in CYP-S-CO (Figure 2). Although many correlations in the B-B’ loop and B’ helix are lost in CYP-S due to paramagnetic effects, this region is strongly affected by changes in K+ concentration, and Cys 85, Glu 91 and Ala 92 are not detected in the absence of K+, suggesting either a very large chemical shift change, or a conformational equilibrium that is at intermediate exchange on the 1H chemical shift time scale (Table 2). Other residues that can only be detected in CYP-S in the presence of K+ are found in the E, G and I helices, and K+ dependent perturbations are also found in the A, B, and H helices and portions of beta-sheets 1, 2, 4 and 5 (Table 2). The spatial distribution of K+-induced perturbations in the CYP-S structure is also shown in Figure 3.
Structural perturbations in potassium-free CYP-S-CO due to Pdxr binding
Two-dimensional 1H-15N TROSY-HSQC NMR experiments were also used to monitor the titration of perdeuterated [U-15N] CYP-S-CO with perdeuterated Pdxr in the presence and absence of K+. At 25 °C in 100 mM K+, Pdxr-induced chemical shift perturbations in CYP-S-CO matched those observed previously (8, 11). Interestingly, those residues that are perturbed both by changes in Pdxr concentration and K+ concentration are affected in the opposite sense by the two variables, that is, the direction of the shift (in both 1H and 15N dimensions of the HSQC) with increasing K+ concentration is opposite to that observed with increasing concentration of Pdxr (See Figure 4).
Figure 4.

Comparison of the effects of K+ and Pdxr binding in the presence of K+ to 0.3 mM CYP-S-CO (90/10 H2O/D2O, 50 mM Tris-HCl pH 7.4, 2 mM camphor, 25 °C) on 1H,15N TROSY-HSCQ correlations obtained at 600 MHz 1H. Spectra shown are typical. Titration of K+-bound CYP-S-CO with Pdxr moves correlations affected by K+ binding in the direction of the K+-free form in both 15N and 1H dimensions.
In the absence of K+, spectral perturbations in CYP-S-CO due to Pdxr titration were larger for many NH correlations than those observed in the presence of K+, suggesting tighter binding. However, we were unable to obtain a reasonable fit for Kd of the Pdxr-CYP-S-CO complex in the absence of K+ using a standard two-sites model, although such fits yielded reasonable values for Kd of the Pdxr-CYP-S-CO in K+-containing solutions (8, 11). We also observed that, in the absence of K+, many resonances split into two peaks with the increasing Pdxr concentration (Figure 5). This indicates that at least two spectroscopically distinguishable CYP-S-CO conformers exist in the presence of Pdx, and it may be that only one of these conformers is competent to bind Pdxr.
Figure 5.

Evidence for two conformers at slow exchange in CYP-S-CO in the presence of Pdxr and the absence of K+. Addition of 1 eq. of Pdxr to 0.5 mM K+-free CYP-S-CO (90/10 H2O/D2O, 50 mM Tris-HCl pH 7.4, 2 mM camphor, 25 °C) splits the NH correlations of Glu 40 and Glu 91 in 1H,15N TROSY-HSCQ correlations obtained at 600 MHz 1H. For comparison, the addition of 1 eq. of Pdxr to 0.3 mM CYP-S-CO in the presence of K+ is shown for Glu 91 in the bottom panel (90/10 H2O/D2O, 100 mM KCl, 50 mM Tris-HCl pH 7.4, 2 mM camphor, 25 °C).
Perturbations at bound camphor due to the L358P mutation and K+
We have recently reported a high barrier conformational shift observed in the active site of CYP-S-CO as a function of added Pdxr (12). This shift is detected as a slow exchange perturbation to the chemical shift of the 8-CH3 1H signal of the camphor bound in the CYP101 active site. All three methyl signals (the bridgehead 10-CH3, and the geminal 8-CH3 and 9-CH3) of enzyme-bound camphor are observed in the upfield 1H NMR spectrum of highly perduterated CYP-S-CO (Figure 6). Upon titration with Pdxr, the 9-methyl resonance titrates in an upfield direction, while the 8-methyl undergoes a slow exchange shift to a new downfield position (see Ref. (12) and Fig. 6 for more details). We now find that in the L358P mutant, even in the absence of Pdxr, the 8-methyl 1H resonance is shifted downfield close to the position observed for the 8-methyl in the Pdxr-CYP-S-CO (WT) complex, indicating that the high-barrier conformational shift that is forced by Pdxr binding to WT CYP-S-CO is essentially complete in L358P CYP-S-CO even in the absence of Pdx (Figure 6). Camphor appears to be stoichiometrically bound to L358P CYP-S-CO; that is, comparison of signal intensities indicate that substrate-free L358P CYP-CO is not a significant component of the sample.
Figure 6.
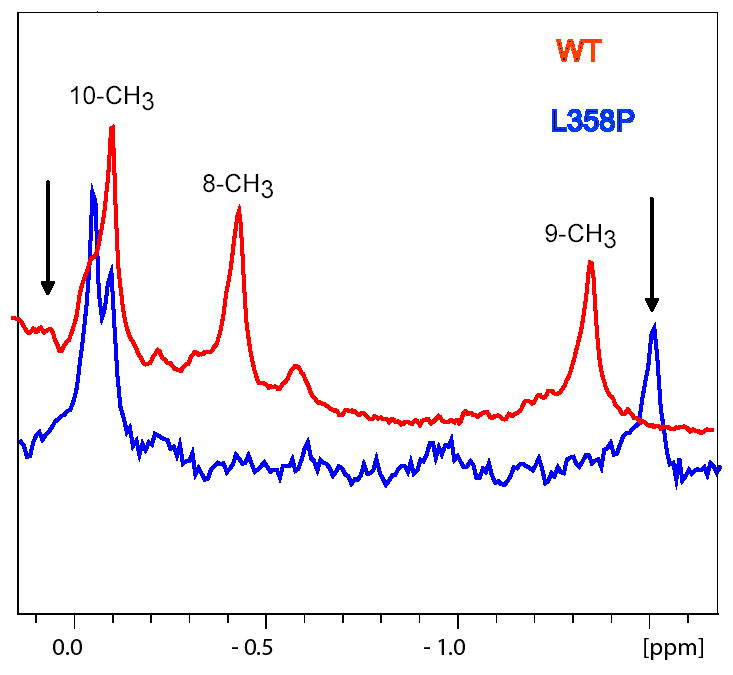
Upfield regions of the 600 MHz 1H NMR spectra of perdeuterated WT (red) and L358P (blue) CYP-S-CO showing the three methyl resonances of enzyme-bound camphor (12). Black arrows indicate the position of the 8-methyl (left, downfield) and 9-methyl (right, upfield) resonances in WT CYP-S-CO in the presence of 3 eq. of Pdxr. Sample conditions are 90/10 H2O/D2O, 50 mM Tris-HCl pH 7.4, 2 mM camphor, 25 °C.
On the other hand, K+ effects are relatively small on the 1H spectrum of bound camphor in either the WT or L358P CYP-S-CO complexes. In WT, the 9-CH3 and 10-CH3 1H resonances differ only slightly in the presence of 100 mM K+ from those observed with no K+ present (Δδ = 4 and 15 Hz at 600 MHz 1H, respectively). The 8-CH3 resonance shows a slightly larger chemical shift change (Δδ = 40 Hz at 600 MHz 1H) going from 0 to 100 mM K+. In L358P, 8-CH3 also had a larger shift (Δδ = 21 Hz) compared to the 9-CH3 (1.8 Hz) and 10-CH3 methyl groups (4.2 Hz). As such, it is unlikely that the high-barrier conformational change that differentiates the Pdx-bound versus Pdx-free forms of CYP-S-CO is directly related to the presence or absence of K+.
Tl+ effects on spin state equilibrium in CYP-S and Tl+-induced spectral perturbations in CYP-S-CO
Thallium has two common oxidation states in aqueous media, Tl(I) and Tl(III). Tl(I) salts are often used in biophysical studies as K+ mimics, and Tl+ often binds more tightly to K+-selective sites than K+ itself. Indeed, the extreme toxicity of Tl+ is due in large part to its ability to inhibit K+-dependent processes in vivo (22). In line with this, we find Tl+ to be as effective as K+ on a molar basis in shifting the spin state equilibrium of CYP-S towards the high-spin form (see Table 1). We also compared the K+- and Tl+-saturated forms of CYP-S-CO using a two-dimensional version of TROSY-HNCO resolved in the 1H and 13C dimensions. This experiment provides correlations between the amide 1H of residue i with the carbonyl 13C of residue i-1. The 2D-TROSY-HNCO spectra of perdeuterated 13C, 15N-labeled CYP-S-CO show differences for all four of the carbonyl carbons proposed to ligate K+ depending on whether 100 mM KCl or 50 mM TlNO3 is present. The carbonyl of Glu 94 is absent in the presence of Tl+, although it is readily identified in the presence of K+. The carbonyl of Glu 84 is shifted by 116 Hz between the two spectra, and the carbonyls of Gly 93 and Tyr 96 are shifted by 20 Hz and 15 Hz respectively. Because both naturally occurring isotopes of Tl, 203Tl and 205Tl are spin ½ nuclei, we hoped to see splitting of the 13C resonances of the ligating carbonyls, as two-bond 205Tl-13C couplings (~270 Hz) have been observed for Tl+3-carbonyl complexes (23). However, such splitting was not observed, suggesting that Tl+ exchange from the site is fast enough to decouple the two spins.
The replacement of 100 mM KCl with 100 mM ThNO3 nearly completely suppresses the NMR spectral perturbations normally associated with the titration of CYP-S-CO by Pdxr (8, 11). While it is tempting to interpret this observation as indicating that the tight binding of Tl+ to the K+ binding site interferes with the conformational selection that normally occurs upon binding of Pdxr to CYP-S-CO, we cannot show that the inhibition does not result from other unrelated interactions between Pdxr and Tl+. However, treatment of Pdxr solutions with 100 mM Tl+ does not result in UV/visible spectrophotometric changes or precipitation, indicating that at least Tl+ is not denaturing Pdxr. We also observe NADH oxidation in the CYP101-Pdx-PdR system reconstituted in Tl+-containing buffers, but turnover measurements in this system are as yet inconclusive.
Discussion
Localization and oxidation state dependence of K+ binding to CYP101
The specific effects of potassium ion binding upon substrate affinity (Kd) and the position of spin-state equilibrium (Kspin) in oxidized substrate-bound CYP101 are well-characterized (13, 21, 24). The advent of sequence-specific backbone resonance NMR assignments for both oxidized and reduced CYP101 allows us to now confirm the locale of K+ interaction with CYP101 and to extend these observations to other regions of the enzyme. In both CYP-S and CYP-S-CO, the NMR data is consistent with K+ binding at the site formed by the carbonyl oxygens of residues Glu 84, Gly 93, Glu 94 and Tyr 96. In CYP-S-CO, the largest confirmed K+-induced perturbations are observed to 15N resonances of Cys 85 and Glu 94, each of which are directly bonded to proposed ligand carbonyl groups. The carbonyl 13C of Glu 84 shows the largest confirmed perturbation of any signal upon replacement of K+ by Tl+ (Δδ = 116 Hz). However, while the carbonyl 13C resonance of Glu 94 could not be detected in the HNCO spectrum of Tl+-perturbed CYP-S-CO, it can be detected in 100 mM KCl, and so is strongly perturbed as well. With absolute shifts of 20 and 15 Hz, respectively, the Gly 93 and Tyr 96 carbonyl 13C resonances are also perturbed by Tl+ replacement of K+, although not as strongly as those of Glu 84 and Glu 94.
One conclusion that can be reached from the current work is that potassium binding effects in wild type CYP101 are more far-reaching in oxidized form than in the reduced protein. Evidence from other laboratories shows that K+ stabilizes the CYP101 structure and renders the protein less susceptible to pressure-induced denaturation, as evidenced by the concentration of the inactivated form P420 after reduction and CO binding (5, 6). However, we did not see any direct correlation between the potassium-dependent high-spin/low-spin ratio observed in the oxidized substrate-bound enzyme and the amount of P420 present after reduction. Nor did reduction of substrate-bound CYP101 in the absence of K+ result in significantly more P420 under the conditions of our experiments.
Antagonistic behavior in K+ and effector binding by CYP-S-CO
As was shown previously (9), the B’ helix (residues 90–96) is one of the most uniformly perturbed secondary structures in CYP-S-CO upon binding of Pdxr, and it also contains three of the four backbone carbonyls that form the K+-binding site in the 3CPP structure, those of Gly 93, Glu 94 and Tyr 96. We have also now identified the backbone NH correlations for residues Glu 84-Ile 88 in the B-B’ loop, with Glu 84 providing the fourth carbonyl oxygen ligand for the cationic binding site. All of these residues are perturbed both by K+ and Pdxr. As noted above, for residues affected by both K+ and Pdxr, the direction of both 15N and 1H shifts upon titration of CYP-S-CO with Pdxr are opposite to those observed upon titration with K+ (see Figs. 4 and 5). This suggests that in CYP-S-CO, K+ binding is weaker in the Pdxr-enforced conformation. While we could not obtain a reasonable fit for the Kd for the Pdxr-CYP-S-CO complex in the absence of K+ using a two-sites model due to the presence of at least two conformers of CYP-S-CO at slow exchange in the presence of Pdxr (Figure 5), qualitatively we note that spectral perturbations in CYP-S-CO due to Pdxr in the absence of K+ are in general larger than those observed when K+ is present. Taken collectively, these observations suggest that K+ stabilizes one conformation of CYP-S-CO that, upon binding of Pdxr, shows lower affinity for K+, i.e., antagonistic binding. In the absence of K+, multiple conformers are present that bind with different affinities for Pdxr, giving rise to complex behavior and splitting of resonances in the HSQC experiment. Such allosteric regulation has been observed in other enzymes, including IMPDH (25, 26), pyruvate kinase (27–29) and dialkylglycine decarboxylase (30, 31).
Unlike the situation with K+, the introduction of thallium (Tl+) ion in the form of TlNO3 appears to almost completely prevent the spectral changes induced by Pdxr in CYP-S-CO as monitored by NMR titration. One possible interpretation of this is that Tl+ binds to the K+ binding site tightly enough to inhibit the conformational change that accompanies Pdx binding and is predicted by the present model to weaken potassium ion binding. However, we cannot rule out inhibition of Pdxr-CYP-S-CO interaction by Tl+ by direct interaction between Pdxr and Tl+ (e.g., binding of Tl+ to the carboxylates of Asp 34 and Asp 38, which play a significant role in the interactions between CYP101 and Pdx (32, 33)). Still, we did not observe any UV/visible spectral changes for Pdxr upon addition of 100 mM TlNO3, indicating that at least Pdxr is stable to the presence of thallium ion, and the Fe-S cluster is not significantly affected by presence of Tl+.
Increased K+ sensitivity of and evidence for the Pdx-enforced camphor orientation in the L358P mutant of CYP101
Morishima and co-workers first described a mutant of CYP101, L358P, that removes a hydrogen bond observed in the WT protein between the axial Cys 357 ligand thiolate sulfur and the backbone amide proton of Leu 358 (16, 34). Somewhat surprisingly, this mutant also shows upfield 1H NMR chemical shift patterns reminiscent of those observed in Pdx-bound WT CYP-S-CO, and exhibits a higher affinity for Pdxr in the CO-bound reduced form than WT (14). The L358P mutation thus appears to force the selection of subset of solution conformations of the CYP101 structure that includes those conformations selected by Pdxr binding. As a consequence of this selection, the L358P mutant of CYP101 shows some constitutive substrate turnover (i.e., camphor hydroxylation) in the absence of effector. The conformational selection is not complete, since titration of the reduced and CO-bound L358P mutant with Pdxr results in further conformational changes, and improves the efficiency of the enzyme ((16) and B. OuYang, unpublished). However, we can confirm that even in the absence of Pdx, the majority of camphor bound to reduced L358P is in the orientation selected by Pdxr binding to WT CYP-S-CO (Figure 6). That is, the high-barrier conformational shift observed in the active site of CYP-S-CO upon Pdxr binding is essentially complete in the L358P mutant, even in the absence of Pdx (34).
As discussed above, the binding of Pdxr to CYP-S-CO appears to disfavor K+ binding (the two binding events are antagonistic). As shown in Figure 1, K+ binding to L358P CYP101 is also perturbed, and a higher concentration of K+ is needed to shift the spin-state equilibrium in favor of the camphor-bound high spin form than is required in the WT enzyme. At 100 mM K+, the WT enzyme is close to maximum concentration of high-spin form (87%, with 93% at 200 mM K+), while that of the L358P mutant is only 65% high-spin at 100 mM KCl concentration, and the fraction of HS L358P CYP-S continues to increase up to the highest salt concentration measured (400 mM) (see Figure 1 and Table 1). All of this is consistent with the hypothesis that the Pdx-bound form of the enzyme binds K+ less tightly than the Pdx-free form: If the L358P mutation selects conformers that are similar to the Pdx-bound form, one expects that K+ binding would be weaker for this mutant.
Conclusions
From the current results, it is clear that NMR methods are useful for distinguishing specific from non-specific effects of cations on protein structure. Near the proposed K+ binding site of CYP101, particularly the B-B’ loop and B’ helix and adjacent structural features, K+ effects are more pronounced than those induced by Na+ in both oxidation states. In some regions of the enzyme, salt effects appear to be non-specific, and are more likely due to general electric field effects than specific binding. Such regions include residues near the N-terminus (Glu 20-Val 23), C-terminus (Leu 402-Val 414) and portions of the β4 and β5 sheets (Table 2). These features are distinguished by the similarity of effects observed in the presence of either Na+ or K+ ions.
That the oxidized form of the enzyme, CYP-S, is more globally susceptible to both specific and non-specific cationic effects than reduced CYP-S-CO is also readily apparent from the present data. Where direct comparison is possible, the salt-induced perturbations are invariably larger for a given residue in CYP-S than in CYP-S-CO: Indeed, in many cases resonances in CYP-S are undetectable in the absence of salt (Table 2). In some cases, for residues in the C, E and G helices, the phenomenon is K+-specific, that is, no resonance is observed in the absence of K+, even if 100 mM Na+ is present. In other cases, the resonance is doubled in the presence of 100 mM NaCl, indicating that K+ effectively stabilizes a single set of conformers, while Na+ does not.
Finally, although the interactions between K+, CYP-S-CO and the effector Pdxr are complex and need to be more thoroughly investigated, evidence from spectral perturbations in the WT enzyme as well as the evidence that the L358P mutant is less effective at using K+ to effect spin state change suggest that both K+ and Pdx play similar roles, that is, to stabilize functionally important conformations of CYP101 at different stages of its complex reaction cycle.
Acknowledgments
The authors thank Prof. Luet-Lok Wong for the plasmid pDNC334A that expresses CYP101 C334A. We also thank Dr. Lingyun Rui for a gift of perdeuterated 15N, 13C-labeled CYP101, Dr. Rui and Dr. Julie Wei for access to their NMR data, and Prof. Chris Miller for a gift of thallium salts and helpful suggestions.
Footnotes
This work was supported in part by a grant from U.S. Public Health Service (R01-GM44191, TCP). GMP acknowledges previous support from PHS training grant GM007596 and current support from R01-GM067786 (TCP, PI).
References
- 1.Britt BM. For enzymes, bigger is better. Biophys Chem. 1997;69:63–70. doi: 10.1016/s0301-4622(97)00082-3. [DOI] [PubMed] [Google Scholar]
- 2.Mueller EJ, Loida PJ, Sligar SG. Cytochrome P450: Structure, Function and Biochemistry. 1995. pp. 83–124. [Google Scholar]
- 3.Lange R, Bonfils C, Debey P. The low-spin reversible highspin transition equilibrium of camphor-bound cytochrome P-450. Effects of medium and temperature on equilibrium data. Eur J Biochem. 1977;79:623–628. doi: 10.1111/j.1432-1033.1977.tb11847.x. [DOI] [PubMed] [Google Scholar]
- 4.Lange R, Pierre J, Debey P. Visible and ultraviolet spectral transitions of camphor-bound cytochrome P-450. A comprehensive study. Eur J Biochem. 1980;107:441–445. doi: 10.1111/j.1432-1033.1980.tb06049.x. [DOI] [PubMed] [Google Scholar]
- 5.Hui Bon Hoa G, Marden MC. The pressure dependence of the spin equilibrium of camphor-bound cytochrome P450. Eur J Biochem. 1982;124:311–315. doi: 10.1111/j.1432-1033.1982.tb06593.x. [DOI] [PubMed] [Google Scholar]
- 6.Peterson JA. Camphor binding by cytochrome P450cam. Arch Biochem Biophys. 1971;144:678–693. [Google Scholar]
- 7.Poulos TL, Finzel BC, Howard AJ. High-resolution crystal structure of cytochrome P450cam. J Mol Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 8.Pochapsky SS, Pochapsky TC, Wei JW. A model for effector activity in a highly specific biological electron transfer complex: The cytochrome P450cam-putidaredoxin couple. Biochemistry. 2003;42:5649–5656. doi: 10.1021/bi034263s. [DOI] [PubMed] [Google Scholar]
- 9.Lipscomb JD, Sligar SG, Namtvedt MJ, Gunsalus IC. Autooxidation and hydroxylation reactions of oxygenated cytochrome P-450cam. J Biol Chem. 1976;251:1116–1124. [PubMed] [Google Scholar]
- 10.Unno M, Shimada H, Toba Y, Makino R, Ishimura Y. Role of Arg112 of Cytochrome P450cam in the electron transfer from reduced putidaredoxin. Analyses with site-directed mutants. J Biol Chem. 1996;271:17869–17874. doi: 10.1074/jbc.271.30.17869. [DOI] [PubMed] [Google Scholar]
- 11.Rui LY, Pochapsky SS, Pochapsky TC. Comparison of the complexes formed by cytochrome P450(cam) with cytochrome b(5) and putidaredoxin, two effectors of camphor hydroxylase activity. Biochemistry. 2006;45:3887–3897. doi: 10.1021/bi052318f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei JY, Pochapsky TC, Pochapsky SS. Detection of a High-Barrier Conformational Change in the Active Site of Cytochrome P450cam upon Binding of Putidaredoxin. J Am Chem Soc. 2005;127:6974–6976. doi: 10.1021/ja051195j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deprez E, Gill E, Helms V, Wade RC, Hui Bon Hoa G. Specific and non-specific effects of potassium cations on substrate-protein ineractions in cytochromes P450cam and P450lin. J Inorg Biochem. 2002;91:597–606. doi: 10.1016/s0162-0134(02)00467-1. [DOI] [PubMed] [Google Scholar]
- 14.Andrew CG, Westlake CFHC, Jacqueline Donovan, Luet-lok Wong. Mutations of glutamate-84 at the putative potassium-binding site affect camphor binding and oxidation by cytochrome P450cam. Eur J Biochem. 1999;265:929–935. doi: 10.1046/j.1432-1327.1999.00793.x. [DOI] [PubMed] [Google Scholar]
- 15.Raag R, Poulos TL. Crystal structure of the carbon-monoxide substrate cytochrome- P-450cam ternary complex. Biochemistry. 1989;28:7586–7592. doi: 10.1021/bi00445a013. [DOI] [PubMed] [Google Scholar]
- 16.Tosha T, Yoshioka S, Ishimori K, Morishima I. L358P mutation on cytochrome P450cam simulates structural changes upon putidaredoxin binding - The structural changes trigger electron transfer to oxy-P450cam from electron donors. J Biol Chem. 2004;279:42836–42843. doi: 10.1074/jbc.M404216200. [DOI] [PubMed] [Google Scholar]
- 17.Nickerson DP, Wong LL. The dimerization of Pseudomonas Putida cytochrome P450cam: practical consequences and engineering of a monomeric enzyme. Protein Engineering. 1997;10:1357–1361. doi: 10.1093/protein/10.12.1357. [DOI] [PubMed] [Google Scholar]
- 18.Pochapsky TC, Kostic M, Jain N, Pejchal R. Redox-dependent conformational selection in a Cys4Fe2S2 ferredoxin. Biochemistry. 2001;40:5602–5614. doi: 10.1021/bi0028845. [DOI] [PubMed] [Google Scholar]
- 19.Guengerich FP. Oxidation–reduction properties of rat liver cytochromes P-450 and NADPH- cytochrome P-450 reductase related to catalysis in reconstituted systems. Biochemistry. 1983;22:2811–2820. doi: 10.1021/bi00281a007. [DOI] [PubMed] [Google Scholar]
- 20.Ye X, Pochapsky TC, Pochapsky SS. 1H NMR sequential assignments and identification of secondary structural elements in oxidized putidaredoxin, an electron-transfer protein from Pseudomonas. Biochemistry. 1992;31:1961–1968. doi: 10.1021/bi00122a009. [DOI] [PubMed] [Google Scholar]
- 21.E Deprez CDP, Hui Bon Hoa G, Douzou P. Effects of Monovalent cations on Cytochrome P-450 Camphor Evidence for Preferential Binding of Potassium. FEBS Lett. 1994;347:207–210. doi: 10.1016/0014-5793(94)00545-1. [DOI] [PubMed] [Google Scholar]
- 22.Peter ALJ, Viraraghavan T. Thallium: a review of public health and environmental concerns. Environment Intl. 2005;31:493–501. doi: 10.1016/j.envint.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Aramini JM, Krygsman PH, Vogel HJ. Tl-205 And C-13 NMR studies of human serotransferrin and chicken ovotransferrin. Biochemistry. 1994;33:3304–3311. doi: 10.1021/bi00177a022. [DOI] [PubMed] [Google Scholar]
- 24.Westlake ACG, Harford-Cross CF, Donovan J, Wong LL. Mutations of glutamate-84 at the putative potassium-binding site affect camphor binding and oxidation by cytochrome P450(cam) Eur J Biochem. 1999;265:929–935. doi: 10.1046/j.1432-1327.1999.00793.x. [DOI] [PubMed] [Google Scholar]
- 25.Gan L, Petsko GA, Hedstrom L. Crystal structure of a ternary complex of Tritrichomonas foetus inosine 5'-monophosphate dehydrogenase: NAD+ orients the active site loop for catalysis. Biochemistry. 2002;41:13309–17. doi: 10.1021/bi0203785. [DOI] [PubMed] [Google Scholar]
- 26.Kerr KM, Cahoon M, Bosco DA, Hedstrom L. Monovalent cation activation in Escherichia coli inosine 5'-monophosphate dehydrogenase. Arch Biochem Biophys. 2000;37:131–7. doi: 10.1006/abbi.1999.1644. [DOI] [PubMed] [Google Scholar]
- 27.Laughlin LT, Reed GH. The monovalent cation requirement of rabbit muscle pyruvate kinase is eliminated by substitution of lysine for glutamate 117. Arch Biochem Biophys. 1997;348:262–7. doi: 10.1006/abbi.1997.0448. [DOI] [PubMed] [Google Scholar]
- 28.Larsen TM, Benning MM, Wesenberg GE, Rayment I, Reed GH. Ligand-induced domain movement in pyruvate kinase: structure of the enzyme from rabbit muscle with Mg2+, K+, and L-phospholactate at 2.7 resolution. Arch Biochem Biophys. 1997;345(2):199–206. doi: 10.1006/abbi.1997.0257. [DOI] [PubMed] [Google Scholar]
- 29.Larsen TM, Laughlin LT, Holden HM, Rayment I, Reed GH. Structure of rabbit muscle pyruvate kinase complexed with Mn2+, K+, and pyruvate. Biochemistry. 1994;33(20):6301–9. doi: 10.1021/bi00186a033. [DOI] [PubMed] [Google Scholar]
- 30.Zhou X, Jin X, Medhekar R, Chen X, Dieckmann T, Toney MD. Rapid kinetic and isotopic studies on dialkylglycine decarboxylase. Biochemistry. 2001;40(5):1367–77. doi: 10.1021/bi001237a. [DOI] [PubMed] [Google Scholar]
- 31.Zhou X, Kay S, Toney MD. Coexisting kinetically distinguishable forms of dialkylglycine decarboxylase engendered by alkali metal ions. Biochemistry. 1998;37(16):5761–9. doi: 10.1021/bi973010u. [DOI] [PubMed] [Google Scholar]
- 32.Pochapsky TC, Lyons TA, Kazanis S, Arakaki T, Ratnaswamy G. A structure-based model for cytochrome P450(cam)-putidaredoxin interactions. Biochimie. 1996;78:723–733. doi: 10.1016/s0300-9084(97)82530-8. [DOI] [PubMed] [Google Scholar]
- 33.Roitberg AE, Holden MJ, Mayhew MP, Kurnikov IV, Beratan DN, Vilker VL. Binding and electron transfer between putidaredoxin and cytochrome P450cam. Theory and experiments. J Am Chem Soc. 1998;120:8927–8932. [Google Scholar]
- 34.Nagano S, Tosha T, Ishimori K, Morishima I, Poulos TL. Crystal structure of the cytochrome P450cam mutant that exhibits the same spectral perturbations induced by putidaredoxin binding. J Biol Chem. 2004;279:42844–42849. doi: 10.1074/jbc.M404217200. [DOI] [PubMed] [Google Scholar]
- 35.Kraulis PJ. Molscript - a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]