

Abstract
We previously detected HIV-1 Gag-specific CD4+ T cells recognizing reference strain viral epitopes in subjects with progressive, chronic infection. To test whether these CD4+ T cells persist in vivo by failing to recognize autologous HIV-1 epitopes, we compared autologous plasma HIV-1 p24 nucleotide sequences with targeted HXB.2 strain Gag p24 CD4+ T cell epitopes in nine chronically-infected, untreated subjects. In five responding subjects, 10 of 26 HXB.2 strain p24 peptides targeted by CD4+ T cells exactly matched autologous plasma viral sequences. Four subjects with plasma viral loads >100,000 copies/mL had no measurable p24-specific CD4+ T cell responses despite carrying HIV-1 strains that matched HXB.2 sequences at predicted epitopes. These results show that HIV-1-specific CD4+ T cells can persist in chronic HIV-1 infection despite recognition of epitopes present in vivo. However, with high level in vivo HIV-1 replication, CD4+ T cells targeting autologous HIV-1 may be non-responsive or absent.
Keywords: Human immunodeficiency virus type 1, Gag protein, CD4+ T cells, Epitopes, Mutations
Introduction
The failure of most untreated subjects to suppress human immunodeficiency virus type 1 (HIV-1) replication during the chronic phase of infection may result from a defect in HIV-1-specific CD4+ T cell help. This defect may be due in part to the selective depletion of HIV-1-specific CD4+ T cells early in the disease course, an anticipated consequence of the preferential infection of these cells with HIV-1 (Douek et al., 2002). Nevertheless, HIV-1-specific, gamma interferon (IFN-γ)-producing CD4+ T cells were found to persist in some subjects with chronic, progressive disease at levels similar to those found in long-term non-progressors (Pitcher et al., 1999, Wilson et al., 2000). Subsequently, the role of HIV-1-specific CD4+ T cell functional abnormalities in the pathogenesis of progressive HIV-1 disease has been investigated. Abnormalities identified in viremic subjects to date include poor antigen-induced proliferation under standard culture conditions (McNeil et al., 2001, Palmer et al., 2002), as well as a proliferation-limiting lack of IL-2 production (Boaz et al., 2002, Harari et al., 2004, Iyasere et al., 2003, Palmer, Boritz, and Wilson, 2004, Younes et al., 2003) and an associated skewing towards expression of “effector memory” surface markers (Harari et al., 2002, Palmer, Boritz, and Wilson, 2004, Younes et al., 2003). Diminished IL-2 production by HIV-1-specific CD4+ T cells in subjects with chronic HIV-1 replication could limit not just the proliferation of these cells, but also the help they provide to HIV-1-specific cytotoxic T lymphocytes (CTL). The defective antigen-induced proliferation of HIV-1-specific CD8+ T cells during progressive HIV-1 disease (Lichterfeld et al., 2004, Migueles et al., 2002) supports this possibility.
Another possible explanation for the apparent failure of HIV-1-specific, IFN-γ-producing CD4+ T cells to help control HIV-1 disease progression during the chronic phase is that these cells fail to recognize the HIV-1 epitope sequences present in vivo. Given the high mutation rate of replicating HIV-1 (Coffin, 1992), some viral epitopes that stimulate specific CD4+ T cell responses during early infection probably evolve to unrecognizable forms under pressure from specific CTL (Barouch et al., 2002, Birk et al., 1998, Cao et al., 2003, Goulder et al., 1997, Jamieson et al., 2003, Kelleher et al., 2001, Leslie et al., 2004, Soudeyns et al., 1999, Wilson et al., 1999, Yang et al., 2003), neutralizing antibodies (Frost et al., 2005, Wang et al., 2002), the CD4+ T cells themselves (Ciurea et al., 2001), or other selective forces. Although inactive against autologous HIV-1 strains present during advanced infection, memory CD4+ T cells primed by such rapidly-evolving epitopes might nevertheless recognize reference strain HIV-1 antigens in vitro. Over time, CD4+ T cells that do not target emerging HIV-1 variants might persist by remaining quiescent and thus avoiding preferential infection and depletion. In other words, among CD4+ T cells targeting reference strain HIV-1 epitopes, the cells that do not recognize viral epitopes present in vivo may have a survival advantage over those that do.
Testing this hypothesis requires assays sufficiently sensitive to detect low-frequency CD4+ T cell populations targeting individual HIV-1 epitopes. Particularly in subjects with chronic, progressive infection, few assays are sufficiently sensitive to accomplish this (Scriba et al., 2005, Seth et al., 2005). To address this difficulty, we have investigated how the defective proliferation of HIV-1-specific CD4+ T cells from such subjects might be improved, thus allowing in vitro expansion of these cells for easier detection. We showed in a previous study that depleting PBMC of CD8+ cells before antigen stimulation allowed Gag p24 antigen-specific CD4+ T cells to proliferate in culture (Boritz, Palmer, and Wilson, 2004). These cells could then be detected by IFN-γ production in enzyme-linked immunospot (ELISpot) assays. Responses in the “expanded ELISPot” assay were often detectable from subjects showing weak or absent HIV-1-specific CD4+ T cell IFN-γ production in fresh PBMC, demonstrating the high sensitivity of the expanded ELISpot for low-frequency CD4+ T cell populations. Of note, subjects with HIV-1 RNA levels >100,000 copies/mL lacked detectable p24-specific CD4+ T cell responses even in the expanded ELISpot, suggesting that proliferation-competent HIV-1-specific CD4+ T cells may be lost during the chronic phase of disease only at the very highest levels of in vivo HIV-1 replication.
In the present study, we used the expanded ELISpot technique to detect HXB.2 strain p24 peptide-specific CD4+ T cell responses for comparison with autologous plasma HIV-1 p24 amino acid sequences. We aimed to test the hypothesis that HIV-1 antigen-specific, IFN-γ-producing CD4+ T cells detected during progressive HIV-1 disease persist despite chronic HIV-1 replication by failing to recognize the HIV-1 epitopes present in vivo, thus avoiding preferential infection and deletion. If this hypothesis were correct, we expected to observe significant amino acid sequence differences between HXB.2 strain CD4+ T cell target peptides and corresponding autologous HIV-1 sequences. Instead, several subjects had CD4+ T cells recognizing p24 amino acid sequences present in autologous virus. Despite this, we found little evidence that these CD4+ T cells had selected for escape mutations within their target sequences. Furthermore, because plasma HIV-1 molecular clones from several subjects with the highest HIV-1 loads encoded p24 sequences predicted to be CD4+ T cell targets, we could not attribute the lack of p24-specific CD4+ T cell responses in these subjects to sequence differences between the reference HXB.2 strain and autologous virus.
Results
Study Subjects
Nine antiretroviral therapy-naïve adults with chronic HIV-1 infection, based on history and physical exam, HIV-1 ELISA, and fully evolved Western Blot, were recruited for this study. Table I shows that all of these subjects had significant viremia at the time of study, with plasma HIV-1 RNA loads ranging from 15,379 to greater than 750,000 copies/mL (median 150,000 copies/mL). Their peripheral blood CD4+ T cell counts ranged from 6 to 456 cells/μL (median 182 cells/uL), and 5 of 9 met criteria for AIDS with CD4 counts below 200 cells/μL. As no subject was suspected to have been infected during the 6 months leading up to study, these low CD4 counts (normal range, 500-1600 cells/μL) were taken as signs of chronic, progressive HIV-1 infection. Finally, as major histocompatibility complex (MHC) class II molecules restrict epitope presentation to CD4+ T cells and thus help determine which epitopes may become CD4+ T cell targets, we determined the HLA-DRB1 alleles expressed by each subject. As expected, a variety of different alleles were expressed among the 9 subjects, although 4 subjects shared expression of HLA-DRB1*13 alleles(see Table I). Of note, although DRB1*13 alleles have been associated with non-progressive HIV-1 infection, the four subjects in this study bearing these alleles had among the highest plasma viral loads in the group (Malhotra et al., 2001).
Table I.
Virological and immunological characteristics of study subjects.
| Subject | HIV-1 RNA (copies/mL) | CD4 count (cells/uL) | HLA-DRβ1 alleles |
|---|---|---|---|
| UH156 | 15,379 | 299 | *0405, *0701 |
| UH168 | 46,704 | 162 | *0301, *0401 |
| UH178 | >750,000a | 143 | *1101, *1301 |
| UH185 | 150,000 | 456 | *0901, *1202 |
| UH200 | 90,400 | 182 | *1302, *1503 |
| UH202 | 58,900 | 378 | *0701, *1601 |
| UH208 | 177,000 | 408 | *1101, *1303 |
| UH210 | 460,313 | 88 | *1101, *1301 |
| UH212 | >750,000 | 6 | *0407, *1601 |
Value exceeded the quantitative range of the assay.
HIV-1 p24 peptide-specific CD4+ T cell responses
As shown in Table II, CD4+ T cell epitope recognition patterns were variable among the 9 study subjects. Four subjects had CD4+ T cells recognizing multiple p24 epitopes, ranging from 4 peptides targeted in UH168 to 10 in UH200. Of note, both UH168 and UH200 had CD4 counts < 200 cell/μL and thus met CD4 cell count criteria for AIDS. By contrast, UH185 responded to a only single peptide, and 4 subjects (UH178, UH208, UH210, and UH212) showed no p24 peptide-specific CD4+ T cell responses at all. The level of HIV-1 replication in vivo predicted the breadth of p24-specific CD4+ T cell responses, as plasma HIV-1 RNA levels were inversely correlated with the number of p24 peptides targeted (Figure 1). This association remained significant using “minimum” breadth estimates in which paired responses to adjacent p24 peptides overlapping by 10 residues were assumed to represent a single epitope-specific response (Spearman R = -0.804, p = 0.014). No significant association was found between maximum breadth of epitope recognition and peripheral CD4 T cell count (Spearman R = 0.38, p = 0.3).
Table II.
HIV-1 p24 peptide-specific CD4+ T cell responses of treatment-naive study subjects determined in expanded IFN-γ ELISpot assays.
| p24 peptidea | CD4+ T cell response (Net IFN-γ SFCs/106 CD3+CD4+ lymphocytes)b |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| UH156 | UH168 | UH178 | UH185 | UH200 | UH202 | UH208 | UH210 | UH212 | |
| Pool | 1778 | 823 | – | 3448 | 8704 | NAc | – | – | – |
| 131-150 | – | – | – | – | 2593 | 383 | – | – | – |
| 141-160 | – | – | – | – | – | – | – | – | – |
| 151-170 | 333 | – | – | – | 1481 | 1571 | – | – | – |
| 161-180 | – | – | – | – | – | 421 | – | – | – |
| 171-190 | – | – | – | – | 1296 | – | – | – | – |
| 181-195 | – | – | – | – | 3519 | – | – | – | – |
| 185-199 | – | – | – | – | – | – | – | – | – |
| 189-203 | – | – | – | – | – | – | – | – | – |
| 191-210 | – | – | – | – | 1111 | – | – | – | – |
| 201-220 | 556 | – | – | – | 926 | – | – | – | – |
| 211-230 | 611 | 412 | – | 1778 | – | 613 | – | – | – |
| 221-240 | – | 494 | – | – | – | – | – | – | – |
| 231-250 | 389 | 2963 | – | – | 1111 | – | – | – | – |
| 241-260 | – | – | – | – | – | – | – | – | – |
| 251-270 | – | – | – | – | 1667 | – | – | – | – |
| 261-280 | – | – | – | – | 6296 | – | – | – | – |
| 271-290 | – | – | – | – | – | 1264 | – | – | |
| 281-300 | – | – | – | – | – | – | – | – | – |
| 291-310 | 500 | 412 | – | – | 3981 | 1609 | – | – | – |
| 301-320 | – | – | – | – | – | – | – | – | – |
| 311-330 | – | – | – | – | – | – | – | – | – |
| 321-340 | – | – | – | – | – | – | – | – | – |
| 331-350 | – | – | – | – | – | – | – | – | – |
| 341-360 | – | – | – | – | – | – | – | – | – |
| 349-363 | – | – | – | – | – | – | – | – | – |
| # targetsd | 5 (4) | 4 (3) | 0 | 1 | 10 (7) | 6 (5) | 0 | 0 | 0 |
HIV-1 Gag p24 peptides designated by amino acid residue positions within the HXB.2 strain sequence of Gag. “Pool,” results from stimulation with a pool of all 25 peptides shown.
The median net response to the pool of p24 peptides among the 10 negative control subjects was 16 SFCs/106 CD3+CD4+ lymphocytes, or less than 1 SFC/well. Net responses of 4 SFCs/well greater than this negative control value were considered positive. Negative responses are shown as dashes.
NA, not attempted.
Number of individual peptides stimulating positive responses. Numbers in parentheses represent minimum number of positive epitope-specific responses if contiguous positive peptides are assumed to represent a single epitope-specific response.
Figure 1.
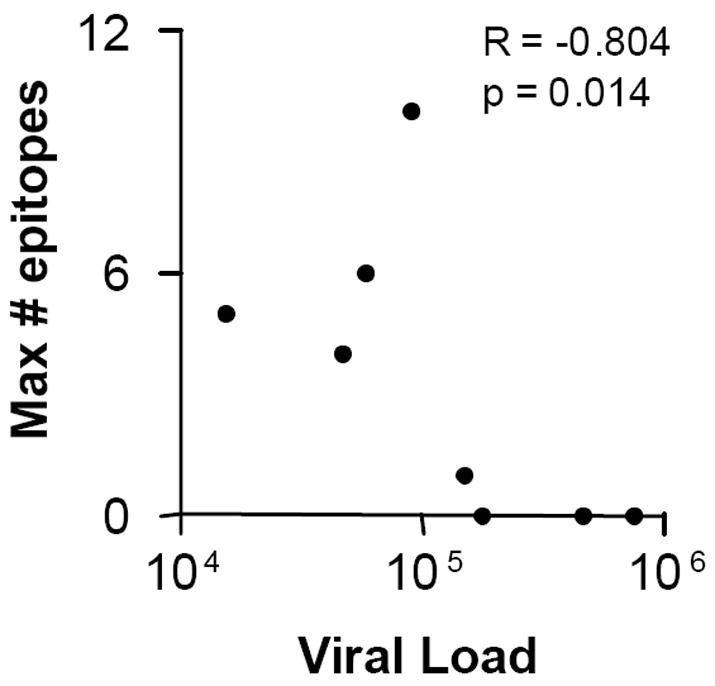
Significant inverse correlation of plasma HIV-1 RNA level with number of p24-specific CD4+ T cell target peptides, as shown for each study subject in Table II. Spearman R and p values are shown.
RT-PCR and sequencing of plasma HIV-1 p24 genes
Figure 2 is a neighbor-joining phylogenetic tree demonstrating the relationships among all 63 p24 nucleotide sequences from the study subjects. It shows that 59 of 63 sequences were unique. All sequences were different from the HXB.2 reference strain upon which synthetic peptides were based, with genetic distances ranging from approximately 2.5% nucleotide sequence non-identity in UH208 to over 10% non-identity in UH156 (a subject who contracted HIV-1 in Ethiopia). Finally, p24 nucleotide sequences from each subject were more closely related to one another than to any of the other sequences. This confirmed that samples had not been contaminated with extraneous nucleic acid before or during RT-PCR. Alignments of amino acid sequences deduced from the p24 molecular clones of individual study subjects are provided as supplemental data.
Figure 2.
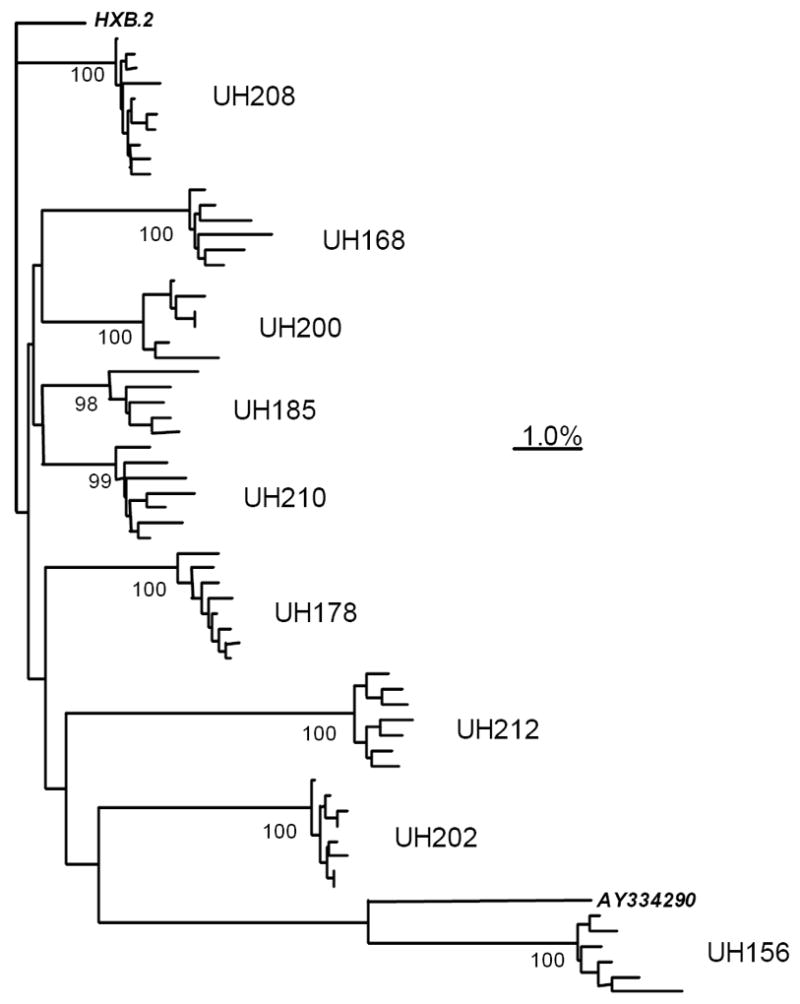
Neighbor-joining phylogenetic tree showing clustering of plasma HIV-1 Gag p24 molecular clones at the nucleotide level. Each terminal branch represents a single molecular clone; clusters of clones from each subject are labeled with subject numbers. Numbers at nodes indicate percentages of 1,000 bootstrap resamplings in which given clusters were supported. Distances between clones are determined by summation of intervening horizontal branch lengths. The scale bar represents a distance of 1.0% nucleotide non-identity between sequences. HXB.2 is the clade B reference strain upon which synthetic peptides were based. The sequence of AY334290, an HIV-1 clade A isolate derived in Ethiopia, was obtained from the Los Alamos HIV Sequence Database and included to show proximity to the sequences from UH156, who was infected with HIV-1 in Ethiopia.
Autologous plasma HIV-1 p24 amino acid sequences corresponding to HXB.2-derived CD4+ T cell target peptides
Table III shows the deduced autologous HIV-1 amino acid sequence data at all HXB.2-based target peptides identified from the 5 subjects with detectable p24-specific CD4+ T cell responses. It shows that all 5 subjects had CD4+ T cells targeting HXB.2 strain peptides that differed from corresponding autologous p24 amino acid sequences. Targeted HXB.2 strain p24 sequences differed from autologous sequences at up to 3 positions in some cases (the 151-170 peptide in UH156, and the 211-230 peptide in UH202). On the other hand, several subjects showed CD4+ T cell recognition of HXB.2 peptides that exactly matched autologous viral clones. For example, 7 of 10 HXB.2 target peptides from UH200 and 1 of 6 from UH202 were identical to HXB.2 in all autologous clones. In UH168, although no HXB.2 target epitope exactly matched every autologous clone, 2 of 4 matched at least 50% of the clones sequenced. Altogether, of 26 HXB.2 strain peptides found to contain CD4+ T cell epitopes, 10 exactly matched some or all corresponding autologous viral sequences from the subjects with responses against them. Furthermore, many amino acid differences between HXB.2 strain target peptides and autologous epitopes were highly conservative. Examples are V215L in UH156, UH168, UH185, and UH200; E203D in UH156; and S310T in UH156, UH168, and UH202.
Table III.
Autologous plasma HIV-1 p24 amino acid sequences corresponding to HXB.2 strain CD4+ T cell target peptides.
| Subject | Targeted HXB.2 peptides | Autologous plasma HIV-1 | ||
|---|---|---|---|---|
| Position | Sequence | Variant sequencesa | % HXB.2b | |
| UH156 | 151-170 | TLNAWVKVVEEKAFSPEVIP | V159 I, V168 I, P170 S (1/6) | 0% |
| 201-220 | LKETINEEAAEWDRVHPVHA | E203 D, V215 L | 0% | |
| 211-230 | EWDRVHPVHAGPIAPGQMRE | V215 L, I223 V | 0% | |
| 231-250 | PRGSDIAGTTSTLQEQIGWM | G248 A | 0% | |
| 291-310 | EPFRDYVDRFYKTLRAEQAS | Y301 F, S310 T | 0% | |
| UH168 | 211-230 | EWDRVHPVHAGPIAPGQMRE | V215 L, I223 V (3/6) | 0% |
| 221-240 | GPIAPGQMREPRGSDIAGTT | I223 V (3/6) | 50% | |
| 231-250 | PRGSDIAGTTSTLQEQIGWM | I247 V, G248 R (1/6) | 0% | |
| 291-310 | EPFRDYVDRFYKTLRAEQAS | D295 G (1/6), S310 T (1/6) | 67% | |
| UH185 | 211-230 | EWDRVHPVHAGPIAPGQMRE | V215 L | 0% |
| UH200 | 131-150 | NYPIVQNIQGQMVHQAISPR | I138 M | 0% |
| 151-170 | TLNAWVKVVEEKAFSPEVIP | None | 100% | |
| 171-190 | MFSALSEGATPQDLNTMLNT | None | 100% | |
| 181-195 | PQDLNTMLNTVGGHQ | None | 100% | |
| 191-210 | VGGHQAAMQMLKETINEEAA | None | 100% | |
| 201-220 | LKETINEEAAEWDRVHPVHA | V215 L | 0% | |
| 231-250 | PRGSDIAGTTSTLQEQIGWM | None | 100% | |
| 251-270 | TNNPPIPVGEIYKRWIILGL | None | 100% | |
| 261-280 | IYKRWIILGLNKIVRMYSPT | None | 100% | |
| 291-310 | EPFRDYVDRFYKTLRAEQAS | T303 V | 0% | |
| UH202 | 131-150 | NYPIVQNIQGQMVHQAISPR | I138 L (6/8), I147 L | 0% |
| 151-170 | TLNAWVKVVEEKAFSPEVIP | V159 I | 0% | |
| 161-180 | EKAFSPEVIPMFSALSEGAT | None | 100% | |
| 211-230 | EWDRVHPVHAGPIAPGQMRE | V215 M, H219 Q, M228 I | 0% | |
| 271-290 | NKIVRMYSPTSILDIRQGPK | T280 V | 0% | |
| 291-310 | EPFRDYVDRFYKTLRAEQAS | S310 T | 0% | |
First letters refer to the HXB.2 sequence, numbers refer to residue positions, and second letters refer to the subjects’ autologous variants. The proportion of the molecular clones in which each variant occurred is shown in parentheses; variants with no proportions specified occurred in all molecular clones.
Percentage of the molecular clones from each subject encoding amino acid sequences identical to the HXB.2 sequence at these positions.
Non-synonymous genetic variation with p24-specific CD4+ T cell target regions
To determine whether the p24 epitope-specific CD4+ T cells might select for escape mutations at their target epitopes, we examined whether the ratio of non-synonymous to synonymous nucleotide substitutions (dN/dS) was greater within CD4+ T cell target epitopes than in untargeted regions of p24. Table IV shows that of 9 subjects studied, only UH212 had a group of plasma p24 clones with no non-synonymous variation among them (i.e., all amino acid sequences were identical). At least 2 different deduced p24 amino acid sequences were detected from the plasma of the remaining 8 subjects. In the groups of isolates from 5 subjects with p24 peptide-specific CD4+ T cell responses, dN/dS was greater inside than outside CD4+ T cell target regions only among the isolates from UH168 (Table IV). By contrast, isolates from UH156, UH185, UH200, and UH202 showed no increased dN/dS inside CD4+ T cell target regions, with dN/dS of zero in the CD4+ T cell target regions of UH185 and UH200.
Table IV.
Variation of plasma HIV-1 p24 amino acid sequences inside and outside of CD4+ T cell target regions.
| Subject | CD4+ T cell target regionsa | Untargeted regions | ||
|---|---|---|---|---|
| Proportionvariable aa (%)b | Average dN/dSc | Proportion variable aa (%) | Average dN/dS | |
| UH156 | 1/90 (1.1%) | 0.065 | 4/141 (2.8%) | 0.097 |
| UH168 | 4/60 (6.7%) | 0.147 | 2/171 (1.2%) | 0.096 |
| UH178 | – | – | 6/231 (2.6%) | 0.135 |
| UH185 | 0/20 (0.0%) | 0.000 | 5/211 (2.4%) | 0.063 |
| UH200 | 0/158 (0.0%) | 0.000 | 2/73 (2.7%) | 0.073 |
| UH202 | 1/108 (0.9%) | 0.183 | 1/123 (0.8%) | 0.279 |
| UH208 | – | – | 3/231 (1.3%) | 0.107 |
| UH210 | – | – | 2/231 (0.9%) | 0.033 |
| UH212 | – | – | 0/231 (0.0%) | 0.000 |
Residue positions spanned by peptides that stimulated CD4+ T cell responses in expansion IFN-γ ELISpot assays.
Proportion of residue positions within given region occupied by 2 or more different amino acids (aa) among subject’s group of p24 sequences.
Corrected ratio of non-synonymous to synonymous nucleotide variations among sequences coding for given region. Determined according to the method of Nei and Gojobori as described in Methods.
Differences between HXB.2 strain and autologous HIV-1 p24 amino acid sequences in subjects with HIV-1 RNA levels >100,000 copies/mL
We asked whether the subjects with the highest viral loads might be infected with HIV-1 strains that differed widely from the HXB.2 strain, and might thus appear to lack p24-specific CD4+ T cell responses because autologous virus-specific responses could not cross-recognize HXB.2 peptides. Accordingly, the median p24 amino acid sequence non-identity between HXB.2 and the plasma p24 clones from each subject was calculated. Table V shows that differences between autologous and HXB.2 p24 sequences ranged from 0.9% (in UH210) to 10.2% (in UH156). Percentage difference from HXB.2 was not significantly greater in the 4 subjects lacking p24-specific CD4+ T cell reactivity than in the 5 responders (p = 0.556, Mann-Whitney), and greater percentage difference from HXB.2 did not predict either weaker or more narrowly-directed CD4+ T cell reactivity (Spearman R = 0.3805, P = 0.360, magnitude; R = 0.174, P = 0.634, breadth). Therefore, differences between autologous and HXB.2 p24 amino acid sequences did not appear to explain the limited p24-specific CD4+ T cell reactivity in subjects with viral loads >100,000 copies/mL.
Table V.
Percentage sequence difference between autologous plasma HIV-1 p24 clones and the HXB.2 sequence, and relationship to magnitude and breadth of HXB.2 strain p24-specific CD4+ T cell reactivity.
| Subject | Median difference from HXB.2a | p24-specific CD4+ T cell responses |
|
|---|---|---|---|
| Magnitude | # targets | ||
| UH156 | 23.5 (10.2%) | 1778 | 5 (4) |
| UH168 | 8 (3.5%) | 823 | 4 (3) |
| UH178 | 7.5 (3.2%) | – | 0 |
| UH185 | 3 (1.3%) | 3448 | 1 |
| UH200 | 5.5 (2.4%) | 8704 | 10 (7) |
| UH202 | 10 (4.3%) | NAb | 6 (5) |
| UH208 | 5 (2.2%) | – | 0 |
| UH210 | 2 (0.9%) | – | 0 |
| UH212 | 12 (5.2%) | – | 0 |
Number (percentage) of 231 amino acid residues not identical to the equivalent positions in the HXB.2 strain sequence of Gag p24. Value shown is the median from the 5-10 molecular clones from each subject’s plasma.
NA, not attempted.
It remained possible that the subjects with no detectable HXB.2 strain p24 peptide-specific CD4+ T cell responses were infected with HIV-1 strains that differed from HXB.2 by only a few critical amino acid residues. To test this, we searched the HIV Molecular Immunology Database for epitopes predicted to be presented by the MHC class II molecules expressed by these subjects. Subjects UH178 and UH210 both expressed HLA-DRβ1*1301, which has previously been shown in natural infection to restrict CD4+ T cell recognition of epitopes within HXB.2 strain peptides 251-265, 260-269, and 265-282 (Malhotra et al., 2001, Kaufmann et al., 2004). CD4+ T cell recognition of peptides overlapping these epitopes was detected from UH200, a subject with viral load <100,000 copies/mL who expressed HLA-DRβ1*1302 (see Tables II and III). By contrast, neither UH178 nor UH210 showed CD4+ T cell recognition of these epitopes even though at least 1 autologous p24 molecular clone sequenced from each subject exactly matched the corresponding HXB.2 sequence at each epitope (Table VI). Outside the predicted epitopes, autologous p24 sequences from subject UH210 consistently differed from the HXB.2 sequence only by a single leucine for isoleucine substitution; sequences from UH178 consistently differed by the HXB.2 sequence by 5 largely conservative substitutions (Table VI).
Table VI.
Differences between autologous and HXB.2 reference strain p24 amino acid sequences in HLA-DRβ1*1301-expressing subjects lacking p24-specific CD4+ T cell reactivity.
| Subject | Predicted HLA-DRβ1*1301-restricted epitopes a |
Other non-HXB.2 positionsc | |||
|---|---|---|---|---|---|
| Position | Sequence | Autologous variants | Autologous %HXB.2b | ||
| UH178 | 251-265 | TNNPPIPVGEIYKRW |
N252
S (6/8) I256 T (7/8) |
12.5% | I138 L, V215 L, E312 D, A340 G, G357 S |
| 260-269 | EIYKRWIILG | None | 100% | ||
| 265-282 | WIILGLNKIVRMYSPTSI | None | 100% | ||
| UH210 | 251-265 | TNNPPIPVGEIYKRW | None | 100% | I138 L |
| 260-269 | EIYKRWIILG | None | 100% | ||
| 265-282 | WIILGLNKIVRMYSPTSI | None | 100% | ||
HXB.2 strain epitopes that bound tightly to HLA-DRβ1*13 molecules and were targeted by CD4+ T cells from HIV-1-infected subjects expressing these HLA molecules in previous studies (251-265 and 260-269, (Malhotra et al., 2001); 265-282, (Kaufmann et al., 2004)).
Percentage of the molecular clones from the given subject encoding amino acid sequences identical to the HXB.2 sequence at these positions.
HIV-1 Gag p24 residue positions outside predicted HLA-DRβ1*1301-restricted epitopes at which all autologous p24 molecular clones sequenced differed from the HXB.2 strain sequence. First letters refer to the HXB.2 sequence, numbers refer to Gag residue position numbers, and second letters refer to the subjects’ autologous sequences.
Discussion
A key finding of this study is that peripheral blood CD4+ T cells from several treatment-naive subjects with advanced HIV-1 infection targeted reference HXB.2 strain amino acid sequences also present in autologous plasma HIV-1. In previous studies, CD4+ T cell recognition of autologous HIV-1 epitopes was observed during early infection. Two groups have reported on individual CD4+ T cell clones targeting autologous Env or Gag epitopes from subjects starting antiretroviral therapy during acute disease (Malhotra et al., 2003, Norris et al., 2004). Harcourt et al. found Gag-specific CD4+ T cells targeting provirus-encoded epitopes from 2 subjects with normal CD4 counts (Harcourt et al., 1998). Finally, a study from our group demonstrated CD4+ T cell targeting of autologous plasma HIV-1 Gag sequences in subjects with CD4 counts as low as 430 cells/μL (Koeppe et al., 2006). In subjects with later stage infection, however, reference strain HIV-1 epitopes targeted by CD4+ T cells have not previously been compared against autologous HIV-1 amino acid sequences. The present study included a majority of subjects meeting CD4 count criteria for AIDS, 2 of whom had multiple CD4+ T cell responses against Gag p24 epitopes that matched the corresponding epitopes from autologous virus.
An important interpretation of this finding is that the CD4+ T cell responses from these subjects targeting reference strain HIV-1 antigens were not simply residual responses against epitopes no longer present in vivo. Because HIV-1 protein-coding sequences during chronic, progressive infection may rapidly diverge from those of the original transmitted strain (Delwart et al., 1997, Liu et al., 1997, Shankarappa et al., 1999), CD4+ T cells primed by early strains may not recognize emerging variants during the chronic phase. Given the preferential infection of HIV-1-specific CD4+ T cells with HIV-1, continued recognition of emerging variants may cause HIV-1-specific CD4+ T cells to be deleted in vivo. We had therefore hypothesized that CD4+ T cell responses against reference strain HIV-1 antigens during advanced disease might represent those HIV-1-specific CD4+ T cells originally primed by epitopes that had rapidly mutated early in the disease course. Our results are not consistent with this hypothesis, and instead suggest that the failure of HIV-1-specific CD4+ T cells detected during chronic infection to mediate HIV-1-specific immune protection does not result entirely from an inability to recognize autologous viral epitopes. Instead, as the CD4+ T cell responses in this study were detected only after in vitro expansion using optimized culture conditions, defects in their magnitude or effector functions may have limited their in vivo protective effects.
The mechanisms by which specific CD4+ T cell responses against actively replicating HIV-1 strains might persist into advanced HIV-1 disease are unclear. It is possible that some CD4+ T cells recognizing autologous viral epitopes in vitro nevertheless fail to respond to these sequences in vivo. In particular, the use of costimulatory molecules and high peptide concentrations in the ELISpot assay may allow in vitro activation of HIV-specific CD4+ T cells with insufficient functional avidity to react against APCs presenting autologous HIV-1 antigens in vivo. However, it is also possible that some HIV-1-specific CD4+ T cells respond to antigen in vivo without being infected. HIV-1-specific CD4+ T cells might resist HIV-1 infection in vivo by expressing insufficient levels of chemokine receptors or other genes required for HIV-1 entry, reverse transcription, or integration. Alternatively, these cells might express coreceptor-blocking chemokines, as has been observed previously (Abdelwahab et al., 2003, Saha et al., 1998). Finally, it remains possible that the HIV-1-specific CD4+ T cells we detected in this study were infected with HIV-1, and that they persist in vivo at the cost of continuous, rapid turnover. Further investigation will be required to determine the exact mechanisms by which CD4+ T cells recognizing autologous HIV-1 antigens persist in vivo during chronic HIV-1 infection.
It was interesting that many CD4+ T cell responses in this study recognized HXB.2 strain p24 amino acid sequences differing by 1 or more residues from corresponding autologous sequences. Although this study was limited by not directly testing CD4+ T cell responses to autologous epitope peptides, we speculate that some of these responses could still have represented CD4+ T cells capable of targeting the viral epitopes present in vivo. Because target epitopes were mapped using 20-residue peptides, it is possible that differences between these peptides and autologous sequences fell outside the regions required for CD4+ T cell recognition. Furthermore, because of the promiscuity of MHC class II-peptide binding (Southwood et al., 1998), the conservative sequence differences we observed might not disrupt CD4+ T cell recognition even if these differences occurred at critical positions. Nevertheless, it remains possible that some of the HXB.2 strain-specific CD4+ T cell responses in this study would not have cross-recognized the autologous variant epitopes we detected, but instead reflected priming by autologous variants no longer present in vivo. Analysis of earlier plasma samples, or of “archived” proviral sequences in PBMC DNA, might have revealed such variants. Further studies will be required to address this possibility.
The non-synonymous mutation rates inside CD4+ T cell target regions were lower than the rates outside these regions among autologous sequences from 4 of 5 subjects, and thus failed to support direct CD4+ T cell-mediated selective pressure on target epitopes in vivo. This could mean that HIV-1-specific CD4+ T cells do not select for mutations at their target epitopes during chronic, progressive HIV-1 infection. As discussed above, it is possible that the cells detected in this study had insufficient functional avidity to respond to their target epitopes in vivo. It is also possible that the cells we detected had no direct antiviral effector activity even after activation in vivo, and thus contributed to selective pressure only at epitopes targeted by specific B cells and CTL receiving their “help.” On the other hand, we may have failed to detect HIV-1-specific CD4+ T cell-mediated selective pressure due largely to the limitations of our experimental design. The use of reference strain HIV-1 Gag peptides to detect T cell responses may have biased toward recognition of conserved epitopes. Also, cross-sectional dN/dS analysis detects genomic areas in which amino acid variation benefits the virus at the time of analysis, but does not detect areas where immune escape has already become fixed. Furthermore, mutations within the p24 antigen may be particularly deleterious to the virus, as has been observed in a study of p24-specific CD8+ T cell-mediated selection (Martinez-Picado et al., 2006). Therefore, non-synonymous genetic variation is probably more prevalent elsewhere in the genome. Longitudinal studies of CD4+ T cell responses against regions that better tolerate mutation will aid in understanding how mutational escape from these responses might influence HIV-1 pathogenesis.
Finally, we examined whether subjects with plasma HIV-1 RNA levels >100,000 copies/mL harbored a predominance of HIV-1 strains with p24 amino acid sequences differing widely from the HXB.2 sequence. We hypothesized that the inverse association we and others have documented between such high viral loads and HIV-1-specific CD4+ T cell IFN-γ production in vitro (Boritz, Palmer, and Wilson, 2004, Oxenius et al., 2002) might reflect an inability of CD4+ T cells targeting autologous viral epitopes in highly viremic subjects to cross-recognize corresponding HXB.2 epitopes. Although we did not test recognition of autologous variant epitopes in this study, our results do not favor this explanation. Most amino acid sequence differences between HXB.2 and autologous sequences in subjects with the highest viral loads were conservative, and in subject UH210, there was only a single position within the p24 sequence at which all autologous clones sequenced differed from HXB.2. Furthermore, at the three HXB.2 epitope positions predicted to be HLA-DRβ1*1301-restricted CD4+ T cell targets (Malhotra et al., 2001; Kaufmann et al., 2004), the HXB.2 sequence exactly matched at least one autologous sequence from both highly viremic, DRβ1*1301-expressing subjects. We therefore speculate that the most highly viremic subjects lacked functional HIV-1-specific CD4+ T cell responses even against autologous p24 epitopes, perhaps reflecting ablation of virus-specific T cells by persistently high viremia (Fuller and Zajac, 2003), T cell receptor antagonism of these cells by low frequency epitope variants in vivo (Frasca et al., 1999, McElrath and Kent, 1997), or other mechanisms. Nevertheless, we cannot exclude the possibility that CD4+ T cells targeting the few autologous viral variant sequences were present in these highly viremic subjects. Further studies addressing this possibility will have to test the variant-specific responses of these subjects, as has been done using HIV-1-specific CD8+ T cell epitopes (Altfeld et al., 2003).
Overall, our results emphasize the potential of the peripheral CD4+ T cell pool to target multiple HIV-1 epitopes even during advanced HIV-1 disease, and establish for the first time that the reference HXB.2 strain may accurately represent some CD4+ T cell target epitopes present in later disease stages. This raises the possibility that HIV-1-specific CD4+ T cells recognizing autologous viral sequences in vitro might resist preferential depletion by HIV-1 in vivo, either by remaining quiescent or by failing to support productive infection despite antigenic stimulation. However, subjects with the highest levels of in vivo HIV-1 replication often lack measurable CD4+ T cell responses against the HXB.2 strain even though HXB.2 sequences accurately represent important autologous p24 epitopes. It will be important to determine whether such subjects lose HIV-1-specific CD4+ T cell responses during their disease course, or whether they fail to raise such responses from the outset of infection. In the latter case, therapeutic immunization and other immune-based strategies may help generate CD4+ T cell memory against predicted epitopes, with potential beneficial effects on viral replication and disease progression.
Materials and Methods
Study subjects
HIV-1-infected subjects were recruited from the Adult Infectious Diseases Group Practice at the University of Colorado Hospital. Nine subjects who had never received antiretroviral drugs previously (i.e., were “treatment-naïve”) but who met clinical criteria for treatment initiation (recorded CD4 count value <350 cells/μL or plasma HIV-1 RNA >100,000 copies/mL) were selected for this study. Subjects with suspected acute or recent infection were excluded. All subjects gave informed consent to participate in this study, and the study was approved by the University of Colorado Health Sciences Center Institutional Review Board.
Human leukocyte antigen (HLA) typing
Molecular HLA typing was carried out by the University of Colorado Health Sciences Center Histocompatibility Laboratory. High-resolution HLA-DRB1 typing was performed using the sequence-based typing method with commercial kits from Perkin-Elmer (Boston, MA).
Cell culture
The standard culture medium for cell expansion cultures and IFN-γ ELISpot assays was RPMI 1640 (Invitrogen, Carlsbad, CA) + 10% heat-inactivated human AB serum (Gemini Bio-Products, Woodland, CA) + 1× penicillin/streptomycin/glutamine (Invitrogen). All cultures were incubated at 37°C in a humidified 5% CO2 atmosphere.
Expanded IFN-γ ELISpot assays
HIV-1 p24-specific, IFN-γ-producing CD4+ T cells were expanded from peripheral blood and detected as previously described (Boritz, Palmer, and Wilson, 2004). Peripheral blood mononuclear cells (PBMC) were harvested by density gradient centrifugation and depleted of CD8+ cells using CD8 microbeads (Miltenyi Biotec, Auburn, CA). CD8-depleted PBMC (1-3 × 107) were then suspended at 5 × 106 cells/mL in standard culture medium and stimulated with 5 μg/mL recombinant HIV-1 Gag p24 antigen (Protein Sciences, Meriden, CT) in 6-well, polystyrene plates. Cells were fed with 0.5× volume (1-3 mL) of standard culture medium after 2 days of incubation. After 7 days, cells were gently scraped from wells, depleted again of residual CD8+ cells using CD8 microbeads, and resuspended in standard culture medium. Cells were then plated at 4.5 × 104/well in 96-well, nitrocellulose-backed plates (Millipore, Bedford, MA) that had been coated with anti-IFN-γ monoclonal antibody (1-D1K, mouse IgG1; Mabtech, Nacka, Sweden), washed × 3, and blocked with standard culture medium. Stimuli used in these assays included pooled, 20- and 15-amino acid p24 peptides overlapping one another by 10 or 11 amino acids and spanning residues 131-363 of the HXB.2 strain of HIV-1 Gag (1μg/mL each peptide; provided by the NIH AIDS Research and Reference Reagent Program, Rockville, MD); individual p24 peptides from the pool at 5 μg/L each; 1 μg/mL phytohemagglutinin (PHA; Murex Diagnostics, Dartford, U.K.) as a positive control; and dimethyl sulfoxide (DMSO) at 0.2% (v/v) as a control for the solvent used in the peptide preparations. Monoclonal CD28 antibody (eBiosciences, San Diego, CA) was included at a final concentration of 1 μg/mL. Plates were then incubated and developed as previously described, and spots were counted using a dissecting microscope (Boritz, Palmer, and Wilson, 2004). The net p24 peptide-specific, IFN-γ spot-forming cells (SFCs) per million CD4+ T cells was calculated by normalizing the number of peptide-induced SFCs/well to the CD3+CD4+ lymphocyte count measured by flow cytometry in a parallel expansion culture. The formula used for this calculation was as follows: (median p24-induced spot forming cells (SFCs)/well - median medium-induced SFCs/well) × (106/4.5 × 104) × (1/proportion viable cells with CD3+CD4+ lymphocyte phenotype). Net p24 peptide-induced responses of 4 SFCs/well greater than the median net p24 peptide pool-induced response from the negative control group were considered positive.
Amplification of HIV-1 nucleotide sequences by reverse transcription PCR (RT-PCR)
Plasma from HIV-1-infected subjects was obtained at the time of expansion IFN-γ ELISpot assays by centrifugation of EDTA-treated blood. Plasma was then stored frozen at -20°C. After thawing, HIV-1 RNA was extracted using the QiaAmp Viral RNA Isolation Kit (Qiagen Inc., Valencia, CA) and dissolved in RNAse-free H20. Viral p24 cDNA was prepared using Omniscript reverse transcriptase (Qiagen) and the p24 outside reverse 2 primer (5’-ATC TTC CCT AAA AAA TTA GCC TGT C-3’; all primers synthesized by Integrated DNA Technologies, Inc., Coralville, IA). Viral cDNA was then amplified using the Expand High Fidelity PCR System (Roche Diagnostics GmbH, Penzberg, Germany) according to manufacturer’s instructions. To avoid founder effects resulting from chance amplification of different cDNA quasispecies in the PCR, each subject’s cDNA was amplified in four separate sets of reactions, and the products were pooled after the second PCR step. The first reaction used the primers p24 outside forward (5’-AGT ATG GGC AAG CAG GGA GC-3’) and p24 outside reverse 2; the second reaction used the primers p24 inside forward 1 (5’-AGA GAT AAA AGA CAC CCA GGA AGC-3’) and p24 inside reverse (5’-TTC CAC ATT TCC AAC AGC CC-3’). The second reaction produced a fragment including the entire p24 capsid sequence and a small amount of upstream flanking matrix and downstream flanking nucleocapsid sequences from the gag polyprotein gene. PCR products were then ligated into the pCR 2.1-TOPO vector using the TOPO TA Cloning kit (Invitrogen) and transformed into chemically-competent One Shot E. coli. Transformants were selected on LB-Ampicillin plates and plasmid DNA was prepared using the QiaPrep Spin Miniprep Kit (Qiagen).
Nucleotide and amino acid sequence analysis
The nucleotide sequences of individual molecular clones were determined in both sense and antisense directions with ABI Prism kits (Applied Biosystems, Foster City, CA) containing AmpliTaq DNA Polymerase. The reaction products were run on an ABI Prism 3730 DNA fluorescent capillary automated sequencer. Sequences were analyzed with Sequencing Analysis version 3.3 (Applied Biosystems); sense and antisense sequences from each clone were aligned with Sequencher 3.1 (Gene Codes Corporation, Ann Arbor, MI) and agreement of the two sequences through the gag p24 coding region was verified. An unreadable pair of sequences from a single molecular clone in subject UH212 was discarded. Phylogenetic sequence analysis was performed by the neighbor-joining method and subjected to 1,000 bootstrap replicates using the PHYLIP v3.65 software package (Felsenstein, 1989). Adjusted ratios of non-synonymous to synonymous mutational frequencies (dN/dS) among molecular p24 clones from individual study subjects were calculated according to the method of Nei and Gojobori (Nei and Gojobori, 1986) using the Synonymous Nonsynonymous Analysis Program (Korber, 2001) on the HIV Sequence Database (http://hiv-web.lanl.gov/content/hiv-db/mainpage.html).
HIV-1 p24 epitopes predicted to be CD4+ T cell targets in subjects lacking in vitro HIV-1-specific CD4+ T cell responses were sought using the HIV Molecular Immunology Database at http://hiv-web.lanl.gov/content/immunology/helper_search. A search was performed in 04/06 using the following search terms: protein = “p24,” subtype = “all,” immunogen = “HIV-1 infection,” and species = “human.” Each epitope from this search was used for further analysis under the following conditions: 1) the epitope was predicted by a previous study to bind an HLA molecule expressed by one or more of the above subjects, and 2) this HLA molecule was found to restrict CD4+ T cell recognition of the epitope by memory CD4+ T cells from other HIV-1-infected subjects in a previous study.
All nucleotide sequences obtained in this study were submitted to GenBank (http://www.ncbi.nlm.nih.gov). Accession numbers are DQ493657-DQ493719.
Supplementary Material
Acknowledgments
We thank the study subjects for their generous participation and the staff of the University of Colorado Hospital Infectious Diseases Group Practice for help with subject enrollment and phlebotomy. We thank Brent Palmer, Russell Young, Philip Kuo, and Moira Hagen for stimulating discussions and helpful guidance. Finally, E.B. acknowledges his debt to Steven Degar, whose example inspired this work and whose teaching made it possible.
Reagents for this study were provided by the DAIDS Vaccine and Prevention Research Program and the National Institutes of Health AIDS Reagent Program.
This work was supported by NIH grant P01AI48238 and was facilitated by the infrastructure and resources provided by the Colorado Center for AIDS Research (AI054907).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelwahab SF, Cocchi F, Bagley KC, Kamin-Lewis R, Gallo RC, DeVico A, Lewis GK. HIV-1-suppressive factors are secreted by CD4+ T cells during primary immune responses. Proc Natl Acad Sci U S A. 2003;100(25):15006–10. doi: 10.1073/pnas.2035075100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altfeld M, Addo MM, Shankarappa R, Lee PK, Allen TM, Yu XG, Rathod A, Harlow J, O’Sullivan K, Johnston MN, Goulder PJ, Mullins JI, Rosenberg ES, Brander C, Korber B, Walker BD. Enhanced detection of human immunodeficiency virus type 1-specific T-cell responses to highly variable regions by using peptides based on autologous virus sequences. J Virol. 2003;77(13):7330–40. doi: 10.1128/JVI.77.13.7330-7340.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouch DH, Kunstman J, Kuroda MJ, Schmitz JE, Santra S, Peyerl FW, Krivulka GR, Beaudry K, Lifton MA, Gorgone DA, Montefiori DC, Lewis MG, Wolinsky SM, Letvin NL. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature. 2002;415(6869):335–9. doi: 10.1038/415335a. [DOI] [PubMed] [Google Scholar]
- Birk M, Vahlne A, Sonnerborg A, Sallberg M. Nonsynonymous mutations within the human immunodeficiency virus type 1 p17 gene are clustered to sequences binding to the host human leukocyte antigen class I molecules. AIDS Res Hum Retroviruses. 1998;14(3):241–8. doi: 10.1089/aid.1998.14.241. [DOI] [PubMed] [Google Scholar]
- Boaz MJ, Waters A, Murad S, Easterbrook PJ, Vyakarnam A. Presence of HIV-1 Gag-specific IFN-gamma+IL-2+ and CD28+IL-2+ CD4 T cell responses is associated with nonprogression in HIV-1 infection. J Immunol. 2002;169(11):6376–85. doi: 10.4049/jimmunol.169.11.6376. [DOI] [PubMed] [Google Scholar]
- Boritz E, Palmer BE, Wilson CC. Human immunodeficiency virus type 1 (HIV-1)-specific CD4+ T cells that proliferate in vitro detected in samples from most viremic subjects and inversely associated with plasma HIV-1 levels. J Virol. 2004;78(22):12638–46. doi: 10.1128/JVI.78.22.12638-12646.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, McNevin J, Malhotra U, McElrath MJ. Evolution of CD8+ T cell immunity and viral escape following acute HIV-1 infection. J Immunol. 2003;171(7):3837–46. doi: 10.4049/jimmunol.171.7.3837. [DOI] [PubMed] [Google Scholar]
- Ciurea A, Hunziker L, Martinic MM, Oxenius A, Hengartner H, Zinkernagel RM. CD4+ T-cell-epitope escape mutant virus selected in vivo. Nat Med. 2001;7(7):795–800. doi: 10.1038/89915. [DOI] [PubMed] [Google Scholar]
- Coffin JM. Genetic diversity and evolution of retroviruses. Curr Top Microbiol Immunol. 1992;176:143–64. doi: 10.1007/978-3-642-77011-1_10. [DOI] [PubMed] [Google Scholar]
- Delwart EL, Pan H, Sheppard HW, Wolpert D, Neumann AU, Korber B, Mullins JI. Slower evolution of human immunodeficiency virus type 1 quasispecies during progression to AIDS. J Virol. 1997;71(10):7498–508. doi: 10.1128/jvi.71.10.7498-7508.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S, Grossman Z, Dybul M, Oxenius A, Price DA, Connors M, Koup RA. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417(6884):95–8. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–6. [Google Scholar]
- Frasca L, Del Porto P, Tuosto L, Marinari B, Scotta C, Carbonari M, Nicosia A, Piccolella E. Hypervariable region 1 variants act as TCR antagonists for hepatitis C virus-specific CD4+ T cells. J Immunol. 1999;163(2):650–8. [PubMed] [Google Scholar]
- Frost SD, Wrin T, Smith DM, Kosakovsky Pond SL, Liu Y, Paxinos E, Chappey C, Galovich J, Beauchaine J, Petropoulos CJ, Little SJ, Richman DD. Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proc Natl Acad Sci U S A. 2005;102(51):18514–9. doi: 10.1073/pnas.0504658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MJ, Zajac AJ. Ablation of CD8 and CD4 T cell responses by high viral loads. J Immunol. 2003;170(1):477–86. doi: 10.4049/jimmunol.170.1.477. [DOI] [PubMed] [Google Scholar]
- Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med. 1997;3(2):212–7. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- Harari A, Petitpierre S, Vallelian F, Pantaleo G. Skewed representation of functionally distinct populations of virus-specific CD4 T cells in HIV-1-infected subjects with progressive disease: changes after antiretroviral therapy. Blood. 2004;103(3):966–72. doi: 10.1182/blood-2003-04-1203. [DOI] [PubMed] [Google Scholar]
- Harari A, Rizzardi GP, Ellefsen K, Ciuffreda D, Champagne P, Bart PA, Kaufmann D, Telenti A, Sahli R, Tambussi G, Kaiser L, Lazzarin A, Perrin L, Pantaleo G. Analysis of HIV-1- and CMV-specific memory CD4 T-cell responses during primary and chronic infection. Blood. 2002;100(4):1381–7. doi: 10.1182/blood-2001-11-0080. [DOI] [PubMed] [Google Scholar]
- Harcourt GC, Garrard S, Davenport MP, Edwards A, Phillips RE. HIV-1 variation diminishes CD4 T lymphocyte recognition. J Exp Med. 1998;188(10):1785–93. doi: 10.1084/jem.188.10.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyasere C, Tilton JC, Johnson AJ, Younes S, Yassine-Diab B, Sekaly RP, Kwok WW, Migueles SA, Laborico AC, Shupert WL, Hallahan CW, Davey RT, Jr, Dybul M, Vogel S, Metcalf J, Connors M. Diminished proliferation of human immunodeficiency virus-specific CD4+ T cells is associated with diminished interleukin-2 (IL-2) production and is recovered by exogenous IL-2. J Virol. 2003;77(20):10900–9. doi: 10.1128/JVI.77.20.10900-10909.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson BD, Yang OO, Hultin L, Hausner MA, Hultin P, Matud J, Kunstman K, Killian S, Altman J, Kommander K, Korber B, Giorgi J, Wolinsky S. Epitope escape mutation and decay of human immunodeficiency virus type 1-specific CTL responses. J Immunol. 2003;171(10):5372–9. doi: 10.4049/jimmunol.171.10.5372. [DOI] [PubMed] [Google Scholar]
- Kaufmann DE, Bailey PM, Sidney J, Wagner B, Norris PJ, Johnston MN, Cosimi LA, Addo MM, Lichterfeld M, Altfeld M, Frahm N, Brander C, Sette A, Walker BD, Rosenberg ES. Comprehensive analysis of human immunodeficiency virus type 1-specific CD4 responses reveals marked immunodominance of gag and nef and the presence of broadly recognized peptides. J Virol. 2004;78(9):4463–77. doi: 10.1128/JVI.78.9.4463-4477.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher AD, Long C, Holmes EC, Allen RL, Wilson J, Conlon C, Workman C, Shaunak S, Olson K, Goulder P, Brander C, Ogg G, Sullivan JS, Dyer W, Jones I, McMichael AJ, Rowland-Jones S, Phillips RE. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J Exp Med. 2001;193(3):375–86. doi: 10.1084/jem.193.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppe JR, Campbell TB, Rapaport EL, Wilson CC. HIV-1-Specific CD4+ T-Cell Responses Are Not Associated With Significant Viral Epitope Variation in Persons With Persistent Plasma Viremia. J Acquir Immune Defic Syndr. 2006;41(2):140–8. doi: 10.1097/01.qai.0000195608.32885.38. [DOI] [PubMed] [Google Scholar]
- Korber B. HIV Signature and Sequence Variation Analysis. In: Rodrigo AG, Learn GH, editors. Computational Analysis of HIV Molecular Sequences. Kluwer Academic Publishers; Dordrecht, Netherlands: 2001. pp. 55–72. [Google Scholar]
- Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJ. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004;10(3):282–9. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- Lichterfeld M, Kaufmann DE, Yu XG, Mui SK, Addo MM, Johnston MN, Cohen D, Robbins GK, Pae E, Alter G, Wurcel A, Stone D, Rosenberg ES, Walker BD, Altfeld M. Loss of HIV-1-specific CD8+ T cell proliferation after acute HIV-1 infection and restoration by vaccine-induced HIV-1-specific CD4+ T cells. J Exp Med. 2004;200(6):701–12. doi: 10.1084/jem.20041270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SL, Schacker T, Musey L, Shriner D, McElrath MJ, Corey L, Mullins JI. Divergent patterns of progression to AIDS after infection from the same source: human immunodeficiency virus type 1 evolution and antiviral responses. J Virol. 1997;71(6):4284–95. doi: 10.1128/jvi.71.6.4284-4295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra U, Holte S, Dutta S, Berrey MM, Delpit E, Koelle DM, Sette A, Corey L, McElrath MJ. Role for HLA class II molecules in HIV-1 suppression and cellular immunity following antiretroviral treatment. J Clin Invest. 2001;107(4):505–17. doi: 10.1172/JCI11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra U, Holte S, Zhu T, Delpit E, Huntsberry C, Sette A, Shankarappa R, Maenza J, Corey L, McElrath MJ. Early induction and maintenance of Env-specific T-helper cells following human immunodeficiency virus type 1 infection. J Virol. 2003;77(4):2663–74. doi: 10.1128/JVI.77.4.2663-2674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeybourne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins JI, Brander C, Walker BD, Stuart DI, Kiepiela P, Goulder PJ. Fitness Cost of Escape Mutations in p24 Gag in Association with Control of Human Immunodeficiency Virus Type 1. J Virol. 2006;80(7):3617–23. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElrath MJ, Kent SJ. Antagonism of vaccine-induced HIV-1-specific CD4+ T cells by primary HIV-1 infection: potential mechanism of vaccine failure. AIDS Res Hum Retroviruses. 1997;13(3):205–10. [PubMed] [Google Scholar]
- McNeil AC, Shupert WL, Iyasere CA, Hallahan CW, Mican JA, Davey RT, Jr, Connors M. High-level HIV-1 viremia suppresses viral antigen-specific CD4(+) T cell proliferation. Proc Natl Acad Sci U S A. 2001;98(24):13878–83. doi: 10.1073/pnas.251539598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, Van Baarle D, Kostense S, Miedema F, McLaughlin M, Ehler L, Metcalf J, Liu S, Connors M. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol. 2002;3(11):1061–8. doi: 10.1038/ni845. [DOI] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3(5):418–26. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Norris PJ, Moffett HF, Brander C, Allen TM, O’Sullivan KM, Cosimi LA, Kaufmann DE, Walker BD, Rosenberg ES. Fine specificity and cross-clade reactivity of HIV type 1 Gag-specific CD4+ T cells. AIDS Res Hum Retroviruses. 2004;20(3):315–25. doi: 10.1089/088922204322996554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenius A, Price DA, Dawson SJ, Gunthard HF, Fischer M, Perrin L, Ramirez E, Fagard C, Hirschel B, Scullard G, Weber JN, McLean AR, Phillips RE, Swiss, H. I. V. c. s Residual HIV-specific CD4 and CD8 T cell frequencies after prolonged antiretroviral therapy reflect pretreatment plasma virus load. AIDS. 2002;16(17):2317–22. doi: 10.1097/00002030-200211220-00012. [DOI] [PubMed] [Google Scholar]
- Palmer BE, Boritz E, Blyveis N, Wilson CC. Discordance between frequency of human immunodeficiency virus type 1 (HIV-1)-specific gamma interferon-producing CD4(+) T cells and HIV-1- specific lymphoproliferation in HIV-1-infected subjects with active viral replication. J Virol. 2002;76(12):5925–36. doi: 10.1128/JVI.76.12.5925-5936.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer BE, Boritz E, Wilson CC. Effects of Sustained HIV-1 Plasma Viremia on HIV-1 Gag-Specific CD4+ T Cell Maturation and Function. J Immunol. 2004;172(5):3337–3347. doi: 10.4049/jimmunol.172.5.3337. [DOI] [PubMed] [Google Scholar]
- Pitcher CJ, Quittner C, Peterson DM, Connors M, Koup RA, Maino VC, Picker LJ. HIV-1-specific CD4+ T cells are detectable in most individuals with active HIV-1 infection, but decline with prolonged viral suppression. Nat Med. 1999;5(5):518–25. doi: 10.1038/8400. [DOI] [PubMed] [Google Scholar]
- Saha K, Bentsman G, Chess L, Volsky DJ. Endogenous production of beta-chemokines by CD4+, but not CD8+, T-cell clones correlates with the clinical state of human immunodeficiency virus type 1 (HIV-1)-infected individuals and may be responsible for blocking infection with non-syncytium-inducing HIV-1 in vitro. J Virol. 1998;72(1):876–81. doi: 10.1128/jvi.72.1.876-881.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriba TJ, Zhang HT, Brown HL, Oxenius A, Tamm N, Fidler S, Fox J, Weber JN, Klenerman P, Day CL, Lucas M, Phillips RE. HIV-1-specific CD4+ T lymphocyte turnover and activation increase upon viral rebound. J Clin Invest. 2005;115(2):443–50. doi: 10.1172/JCI23084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth N, Kaufmann D, Lahey T, Rosenberg ES, Wucherpfennig KW. Expansion and contraction of HIV-specific CD4 T cells with short bursts of viremia, but physical loss of the majority of these cells with sustained viral replication. J Immunol. 2005;175(10):6948–58. doi: 10.4049/jimmunol.175.10.6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73(12):10489–502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudeyns H, Paolucci S, Chappey C, Daucher MB, Graziosi C, Vaccarezza M, Cohen OJ, Fauci AS, Pantaleo G. Selective pressure exerted by immunodominant HIV-1-specific cytotoxic T lymphocyte responses during primary infection drives genetic variation restricted to the cognate epitope. Eur J Immunol. 1999;29(11):3629–35. doi: 10.1002/(SICI)1521-4141(199911)29:11<3629::AID-IMMU3629>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S, Kubo RT, Chesnut RW, Grey HM, Sette A. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol. 1998;160(7):3363–73. [PubMed] [Google Scholar]
- Wang FX, Kimura T, Nishihara K, Yoshimura K, Koito A, Matsushita S. Emergence of autologous neutralization-resistant variants from preexisting human immunodeficiency virus (HIV) quasi species during virus rebound in HIV type 1-infected patients undergoing highly active antiretroviral therapy. J Infect Dis. 2002;185(5):608–17. doi: 10.1086/339015. [DOI] [PubMed] [Google Scholar]
- Wilson CC, Brown RC, Korber BT, Wilkes BM, Ruhl DJ, Sakamoto D, Kunstman K, Luzuriaga K, Hanson IC, Widmayer SM, Wiznia A, Clapp S, Ammann AJ, Koup RA, Wolinsky SM, Walker BD. Frequent detection of escape from cytotoxic T-lymphocyte recognition in perinatal human immunodeficiency virus (HIV) type 1 transmission: the ariel project for the prevention of transmission of HIV from mother to infant. J Virol. 1999;73(5):3975–85. doi: 10.1128/jvi.73.5.3975-3985.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JD, Imami N, Watkins A, Gill J, Hay P, Gazzard B, Westby M, Gotch FM. Loss of CD4+ T cell proliferative ability but not loss of human immunodeficiency virus type 1 specificity equates with progression to disease. J Infect Dis. 2000;182(3):792–8. doi: 10.1086/315764. [DOI] [PubMed] [Google Scholar]
- Yang OO, Sarkis PT, Ali A, Harlow JD, Brander C, Kalams SA, Walker BD. Determinants of HIV-1 mutational escape from cytotoxic T lymphocytes. J Exp Med. 2003;197(10):1365–75. doi: 10.1084/jem.20022138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes SA, Yassine-Diab B, Dumont AR, Boulassel MR, Grossman Z, Routy JP, Sekaly RP. HIV-1 viremia prevents the establishment of interleukin 2-producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J Exp Med. 2003;198(12):1909–1922. doi: 10.1084/jem.20031598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.