

Abstract
Objective
To describe atypical phenotypes associated with the retinoschisis (X-linked, juvenile) 1 mutation (RS1).
Methods
Seven patients with multiple fine white dots at the macula and reduced visual acuity were evaluated. Six patients underwent pattern and full-field electroretinography (ERG). On-off ERG, optical coherence tomography, and fundus autofluorescence imaging were performed in some patients. Mutational screening of RS1 was prompted by the ERG findings.
Results
Fine white dots resembling drusenlike deposits and sometimes associated with retinal pigment epithelial abnormalities were present in the maculae. An electronegative bright-flash ERG configuration was present in all patients tested, and abnormal pattern ERG findings confirmed macular dysfunction. A parafoveal ring of high-density autofluorescence was present in 3 eyes; 1 patient showed high-density foci concordant with the white dots. Optical coherence tomography did not show foveal schisis in 3 of 4 eyes. All patients carried mutations in RS1, including 1 with a novel 206T→C mutation in exon 4.
Conclusions
Multiple fine white dots at the macula may be the initial fundus feature in RS1 mutation. Electrophysiologic findings suggest dysfunction after phototransduction and enable focused mutational screening. Autofluorescence imaging results suggest early retinal pigment epithelium involvement; a parafoveal ring of high-density autofluorescence has not previously been described in this disorder.
X-linked retinoschisis (XLRS) is a common cause of macular dysfunction in young men,1,2 with an incidence of 1 in 5000 to 1 in 25 000.3 Individuals with XLRS are typically first identified as school-aged boys who fail vision screening examinations (60%), have strabismus (30%), or have vitreous hemorrhage.1,2,4 Initially, the characteristic “spoke-wheel” appearance may be seen at the fovea, caused by cystic changes,3 but this is commonly replaced with nonspecific macular atrophy in middle age.5 There is allelic heterogeneity at the retinoschisis (X-linked, juvenile) 1 locus (RS1), with different mutations causing a wide spectrum of phenotypes.
The gene mutated in XLRS, RS1, has 6 exons that encode the 224–amino acid protein retinoschisin, which is processed by means of N-terminal cleavage of a 23–amino acid hydrophobic leader sequence into a mature protein of 201 amino acid residues (23 kDa) and is assembled into a disulfide-linked octamer.6 In many cases, this mutant protein cannot fold properly and is retained intracellularly.4 The predicted protein sequence contains a highly conserved discoidin domain, which is implicated in cell-cell adhesion and phospholipid binding. This function is consistent with the observed splitting of the fiber layer of Henle in the retina of patients with XLRS,7,8 suggesting that retinoschisin is essential during retinal development.9 The electronegative electroretinographic (ERG) configuration that is typically seen in patients with XLRS describes a waveform with a b-wave smaller than the a-wave and suggests that the primary functional defect occurs after phototransduction or is inner retinal.
This study describes 7 patients with genetically confirmed mutations in RS1 that manifest atypical signs in the form of multiple white spots at the macula. The genotype and functional (ERG) phenotype are described.
Methods
The tenets of the Declaration of Helsinki were followed. Seven individuals with reduced visual acuity and macular white dots were ascertained. Electroretinography, performed in 6 patients, suggested the possibility of XLRS and the need for genetic screening. In the remaining patient, screening was prompted by the presence of typical foveal lesions in an affected nephew. Findings from fundus photographs, fundus autofluorescence, and optical coherence tomography (OCT) were reviewed. Autofluorescence images were obtained by illuminating the fundus with argon laser light (488 nm) and viewing the resultant fluorescence through a bandpass filter with a short wavelength cut off at 495 nm.10,11
Full-field ERGs were performed using extended testing protocols incorporating the International Society for Clinical Electrophysiology of Vision (ISCEV) standard12 to assess generalized retinal function. A stimulus 0.6 log unit greater than the ISCEV standard flash was also used, better to demonstrate the a-wave, as suggested in the recent revision of the ISCEV standard for ERG.12 Long-duration on-off ERGs, performed in 4 patients, were used to assess postreceptoral cone on and off pathways, predominantly arising in relation to depolarizing and hyperpolarizing bipolar cell function.13,14 The duration of the amber stimulus (620 nm) was 150 or 200 milliseconds. Stimulus luminance was 560 candela (cd)/m2 with a green background (530 nm) of 160 cd/m2, suitable to suppress rod function.15 Pupils were dilated before full-field ERG testing using tropicamide (1%) and phenylephrine hydrochloride (2.5%). The ISCEV standard pattern ERGs were performed16 before mydriasis, and the P50 component was used to assess macular function.17
All 7 patients were screened for RS1 mutations. DNA was extracted from blood. Extracted genomic DNA was amplified for sequencing in 50 μL of polymerase chain reaction solution using standard methods.18
Results
Clinical and genetic features of all 7 patients are summarized in Table 1; fundus photographs are shown in Figures 1, 2, 3, and 4. Six of 7 patients initially sought care in early childhood for reduced visual acuity. Three patients retained a visual acuity of 20/80 or better in at least 1 eye, including 2 older patients, aged 49 years (patient 3) and 46 years (patient 5). One patient had a 20-year history of floaters in the right eye after blunt trauma but recently sought care for worsening night vision (patient 3). All 7 patients had an abnormal fundus appearance at the macula characterized by multiple fine white spots that resembled drusenlike deposits at the level of the retinal pigment epithelium (RPE), and these were bilateral in all patients (Figures 1, 2, 3, and 4). Pigmentation abnormalities in the RPE were seen bilaterally in 7 patients. Autofluorescence imaging confirmed atrophic changes in patients 2 (Figure 2C and D) and 3 (Figure 3C and D). No patient showed typical foveal schisis, although this had been documented in 1 patient 7 years previously (patient 5). Schitic lesions were present inferiorly in both eyes of patient 7 and in the left eye of patient 1 (Figure 1H). Mutations in intron 1 (1 patient) and exons 1 (1 patient), 4 (4 patients), and 6 (1 patient) of the RS1 gene (Xp22.2-Xp22.1) established the diagnosis.
Table 1. Phenotypic Manifestations of RS1 Mutations.
| Patient No./Age, y | VA, OD/OS | RS1 Mutation | Macular Appearance | Clinical History |
|---|---|---|---|---|
| 1/12 | 20/40 20/60 | 305G→A in exon 4 | Fine white parafoveal dots in both eyes | Previously diagnosed as having Stargardt disease. Normal vessels and discs. White dots correspond with high-density AF. OCT showed normal foveae and peripheral schisis in the left eye. |
| 2/47 | 20/200 20/200 | 206T→C in exon 4 (novel) | Fine white parafoveal dots and RPE atrophy in both eyes | Previously diagnosed as having Stargardt disease. No visible peripheral schisis. Normal vessels and discs. AF showed low-density AF over macular atrophy surrounded by a ring of high density. |
| 3/49 | 20/40 20/30 | C579dupC in exon 6 | Granular appearance and fine white dots in both eyes | Low-density area of AF at maculae, larger in the right eye, surrounded by a ring of high density in the right eye. Right eye trauma 20 y ago. Inferior leaf breaks bilaterally, worse in the left eye. Inferior shallow schisis in both eyes. |
| 4/46 | 20/100 20/200 | IVS1 + 2T→C in intron 1 | Fine white parafoveal dots in both eyes; atrophic scar in the left eye | No visible foveal schisis but a temporal schitic lesion extending beyond the superior arcade in the left eye. |
| 5/46 | 20/80 20/200 | 214G→A in exon 4 | Fine white dots and RPE granularity in both maculae | Initially diagnosed as having Goldmann-Favre syndrome on the basis of macular schisis. White dots and pigment clumping seen 7 y ago; VA: 20/100 OD and 20/100 OS. |
| 6/54 | 20/120 20/200 | 325G→T in exon 4 | Fine white dots and RPE pigmentation abnormalities in both maculae | Initially diagnosed as having “macular degeneration.” Normal vessels and discs. VA: 20/200 OU 3 y after initial examination. |
| 7/50 | 20/200 20/200 | Deletion of the entire exon 1 | White dots and RPE pigmentation abnormalities in both eyes | Previously diagnosed as having “macular degeneration.” Inferior schitic lesions in both eyes. Normal vessels and discs. Nephew (aged 32 y) has typical foveal schisis. |
Abbreviations: AF, autofluorescence; OCT, optical coherence tomography; RPE, retinal pigment epithelium; RS1, retinoschisis (X-linked, juvenile) 1; VA, visual acuity.
Figure 1.

Various imaging findings in patient 1. A, and B, Fundus photographs showed fine intraretinal white lesions along the venules and arterioles of the perifoveolar vascular network. Retinal pigment epithelium disturbance associated with mildly thickened retina is seen over the superior part of the macula of the left eye. C, Red-free photograph of the right eye. D, Red-free photograph of the left eye. E and F, Confocal scanning laser ophthalmoscopic images revealed increased autofluorescence signal associated with the white dots. G, Foveal schisis was not detected by means of optical coherence tomography in the right macula. H, Parafoveal and peripheral microcystic changes were seen via optical coherence tomography in the left eye. I, Full-field and pattern electroretinograms (ERGs). Rows 1 and 2 show recordings from the right and left eyes, respectively, of the patient; row 3, normal recordings. LU indicates log units. Ordinal axes are shown in microvolts, allowing measurement of ERG amplitudes.
Figure 2.
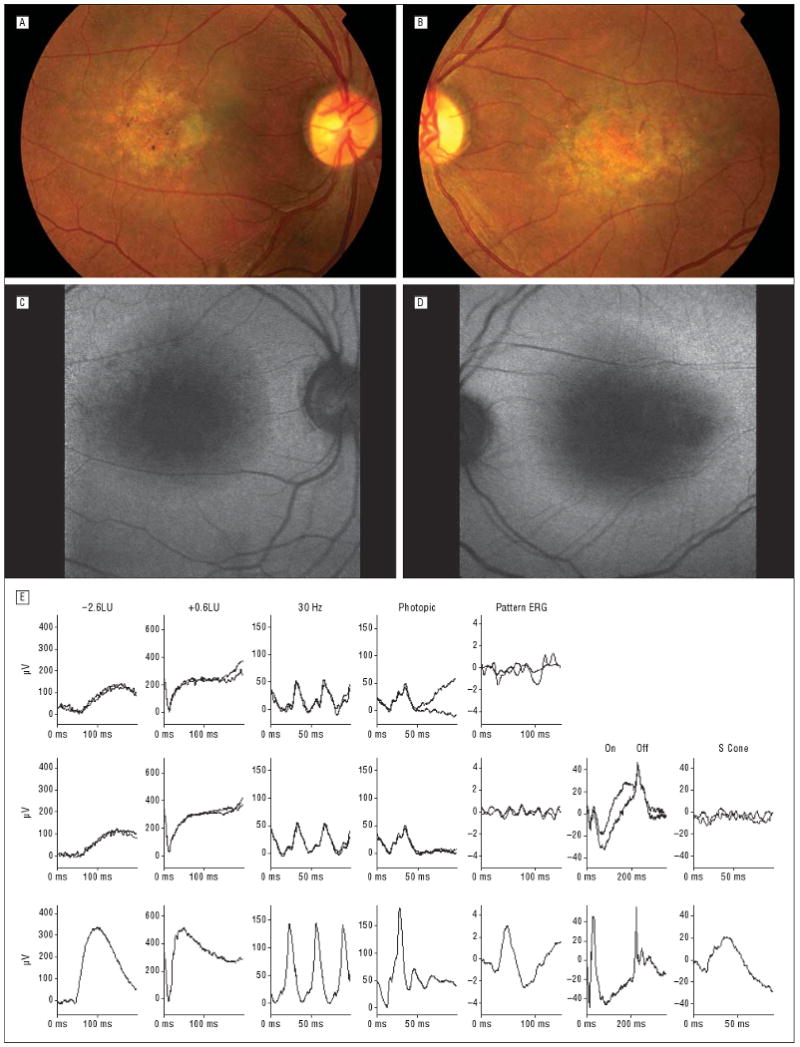
Fundus photographs (A and B) and fundus autofluorescence (C and D) in patient 2. Fine intraretinal white dots were seen in and adjacent to the central atrophic retinal pigment epithelium. Autofluorescence showed low-density signal over macular atrophy surrounded by a ring of high density. The observed ring of hyperfluorescence was consistent with retinal pigment epithelium disturbance outside of atrophic areas. E, Full-field and pattern electroretinograms (ERGs). Rows 1 and 2 show recordings from the right and left eyes, respectively, of the patient; row 3, normal recordings. LU indicates log units. Ordinal axes are shown in microvolts, allowing measurement of ERG amplitudes.
Figure 3.

Fundus photographs (A and B) and autofluorescence (C and D) in patient 3 showing multifocal fine intraretinal white dots in the foveal regions. Low-density autofluorescence at the maculae, larger in the right eye (C), was surrounded by a ring of high density, suggesting a retinal pigment epithelium disturbance outside of atrophic areas. E, Full-field and pattern electroretinograms (ERGs). Rows 1 and 2 show recordings from the right and left eyes, respectively, of the patient; row 3, normal recordings. LU indicates log units. Ordinal axes are shown in microvolts, allowing measurement of ERG amplitudes.
Figure 4.

Fundus photographs in patients 4 (A and B), 5 (C and D), 6 (E and F), and 7 (G and H) showing numerous white dots at the macula in all cases.
Pattern and full-field ERG findings in all 6 patients tested are summarized in Table 2. Pattern ERG P50 components were abnormal in all 6 patients. Two patients showed bilaterally undetectable pattern ERGs, with a further 2 having an undetectable pattern ERG in 1 eye. Full-field ERGs indicated generalized retinal dysfunction affecting rod and cone systems, and in all cases maximal ERG configurations were electronegative, consistent with a locus of dysfunction after phototransduction at the level of the inner retina. Three illustrative cases are described in detail.
Table 2. Summary of Electrophysiologic Findings*.
| Patient No. | Rod ERG Amplitude | Maximum a-Wave Amplitude | Maximal ERG b/a Ratio | 30-Hz Timing | 30-Hz Amplitude | Photopic ERG a-Wave Amplitude | Photopic ERG b-Wave Amplitude | Photopic ERG b/a Ratio | Pattern ERG P50 Amplitude | On-Off ERGs |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | N | N | 0.98 | A | N | N (high amp) | N | 1.7 | A | NP |
| A | N | 0.96 | A | N | N (high amp) | N | 1.7 | A | NP | |
| 2 | A | N | 0.89 | A | N | N | A | 2.0 | U | Reduced b-wave, delayed d-wave, both eyes |
| A | N | 0.90 | A | A (mild) | N | A | 1.8 | U | ||
| 3 | A | N | 0.90 | A | N | N | A | 1.7 | U | Reduced b-wave, delayed d-wave, both eyes |
| A | N | 0.82 | A | N | N | A | 2.3 | U | ||
| 4 | A | N | 0.55 | A | A (mild) | N | A | 2.0 | U | Reduced b- and d-waves, both eyes |
| A | N | 0.60 | A | A (mild) | N | A | 2.0 | A+ | ||
| 5 | A | N | 0.95 | A | N | N (high amp) | N | 2.3 | A | t |
| A | N | 0.98 | A | N | N (high amp) | N | 2.2 | A | t | |
| 6 | t | N | 0.79 | A | A (mild) | N | A | 3.0 | U | Reduced b- and d-waves, right eye. NP, left eye. |
| t | N | 0.71 | A | A (mild) | N | A | 1.8 | A+ |
Abbreviations: A, abnormal; A+, very abnormal; ERG, electroretinography; N, normal; NP, not performed; t, technically unsatisfactory (eye movement) U, undetectable.
Right eye findings are shown above left eye findings except where stated.
Patient 1
A white boy was found to have decreased vision on a routine examination by his pediatrician at 3½ years of age. Shortly afterward he was noted to have “speckling at the back of both eyes.” There was no other relevant medical history.
Visual acuity at age 12 years was 20/40 OD and 20/60 OS. Fundus examination revealed fine white drusenlike deposits around the foveae (Figure 1A-D). No peripheral cystic changes were found at initial examination and up to 18 years of age. The white dots were more apparent on red-free photographs (Figure 1C and D). The diagnosis of Stargardt disease was initially entertained until the results of full-field ERG studies suggested otherwise.
Pattern ERG P50 components were subnormal bilaterally, in keeping with bilateral macular dysfunction (Figure 1I). Full-field ERGs showed borderline reduction in the rod-specific ERG; maximal ERG a-waves were normal but waveforms were mildly electronegative. Photopic 30-Hz flicker ERGs were mildly delayed, without significant amplitude reduction; transient photopic ERGs showed a low b/a ratio. Overall, the findings were consistent with generalized retinal dysfunction affecting rod and cone systems, with a locus of dysfunction after phototransduction.
During the next 6 years, his vision remained stable at 20/40 OD and 20/80 OS. Fundus examination showed no gross changes. Autofluorescence imaging revealed localized high-density pinpoint lesions that corresponded to the white dots (Figure 1E and F). No foveal schisis was detectable in either eye using OCT (Figure 1G and H), but the left eye showed cystic juxtafoveal changes (Figure 1H). Genetic analysis showed a 305G→A mutation in exon 4 of the RS1 gene. This mutation introduces an altered amino acid (R102Q) into the retinoschisin.
Patient 2
A 47-year-old Chinese man complained of slowly progressive worsening of vision in both eyes since age 8 years. Stargardt disease had been suspected. There was no known family history of ophthalmic problems. Visual acuity was 20/200 OU. Fundus examination revealed multiple fine white spots around the foveae, with some RPE pigmentation changes (Figure 2A and B). No peripheral schisis was detected. Optic nerve heads and retinal vessels were normal. Autofluorescence revealed low-density macular lesions consistent with atrophy surrounded by a ring of high density bilaterally (Figure 2C and D). Electroretinographic findings are shown in Figure 2E. The on-off ERGs were consistent with involvement of cone on and off bipolar pathways. Pattern ERGs were undetectable, indicating severe macular dysfunction bilaterally. Genetic analysis revealed a novel 206T→C mutation in exon 4 of the RS1 gene.
Patient 3
A 49-year-old hyperopic white man complained of progressive night blindness. He had a 20-year history of floaters in the right eye after blunt trauma. He had hyperopic correction. Changes in the right vitreous gel and a preretinal inferior vitreous band were reported. Both eyes showed inferior shallow schisis, and there was a hole in the left retinal periphery that was subsequently treated with photocoagulation. He had no family history of any ophthalmic condition. Four years later the patient reported intermittent distortion of vision in the right eye. Visual acuity was 20/40 OD and 20/30 OS. The right macula showed stippling with multiple white spots; the left retina showed a subtle granularity. There was no evidence of foveal schisis.
Six years after initial examination, the visual acuity had deteriorated to 20/200 OD and 20/40 OS. There was bilateral granularity of the macula (Figure 3A), worse in the right eye than the left (Figure 3B). Inferior leaf breaks were present and were more noticeable in the left fundus. In the right eye, low-density autofluorescence over the central macula was surrounded by a parafoveal ring of high density. There was also a low-density patch in the arcades, superior and slightly temporal to the optic disc. In the left eye, there were patches of low density over areas superior to and nasal to the fovea (Figure 3C and D).
Mildly electronegative maximal ERG findings were consistent with generalized inner retinal dysfunction. Photopic ERG findings were abnormal, and on-off ERG findings were consistent with involvement of cone on and off bipolar pathways. There was pattern ERG evidence of severe macular dysfunction (Table 2 and Figure 3E). Genetic analysis revealed a C579dupC mutation in exon 6 of the RS1 gene.
Comment
All 7 patients had an abnormal fundus appearance at the macula characterized by multiple fine white dots at the level of the RPE that resembled drusenlike deposits (Figures 1, 2, 3, and 4). The findings were bilateral in all patients and were often associated with pigmentation abnormalities of the RPE. No patient showed typical foveal schisis, although this had been reported in patient 5 seven years previously. Most patients were first seen by us at 45 years or older, and it is possible that macular schisis may have been present before the white dots appeared. The absence of foveal schisis was not always associated with confluent atrophy, and visual acuity was relatively preserved in some of the older patients (Table 1). Two patients had schitic lesions away from the central macula. Findings from ERG were consistent with generalized inner retinal dysfunction of both rod and cone systems (Table 2 and Figures 1, 2, and 3) and suggested the unsuspected diagnosis of XLRS in most patients.
Foveal or parafoveal white dots or flecks are commonly associated with a variety of disorders, including Stargardt-fundus flavimaculatus (initially considered in 2 of the patients described in the present study), retinitis punctata albescens, Goldmann-Favre syndrome, Wagner vitreoretinal dystrophy, yellow dot dystrophy, and other forms of macular dystrophy.19 However, none of those disorders typically manifest an electronegative ERG configuration. Fundus albipunctatus often shows a myriad of symmetrical round white flecks, with the greatest concentration in the midperiphery, but those patients always have night blindness, not a feature of the present cohort. The drusenlike multiple white spots described in the present patients were sometimes associated with atrophic changes at the macula, a common finding in older patients with XLRS, but could be the only fundus abnormality. The spots were not typical of those associated with age-related maculopathy. A similar matrix of extensive white flecks has been described in 2 individuals with juvenile retinoschisis20 and more recently in 2 additional patients with confirmed RS1 defects, including a 214G→A defect.21 Those flecks were associated with typical star-shaped macular lesions (foveal schisis) and did not resemble the drusenlike white dots described in the present study, even in a patient carrying an identical mutation (patient 5). Macular white dots have not previously been described in individuals carrying the previously reported RS1 alleles: 305G→A, 214G→A, 579dupC, 325G→T, IVS1 + 2T→C, and exon 1 deletion.4 The 206T→C mutation in patient 2 is a novel mutation.
It is difficult to assess the incidence of this phenotype. The primary aims of this retrospective study were to report a previously unsuspected phenotype of XLRS and to highlight the role of electrophysiologic assessment in identifying these patients. Patients with characteristic fundus features and a positive family history may have been diagnosed without using molecular genetic screening, and not all patients were referred for electrodiagnostic testing. For diagnostic purposes, equivocal or unusual cases were more likely to be screened genetically, giving a skewed impression of phenotype in this genetically confirmed cohort. All but 1 of the mutations in the present study have been reported by the Retinoschisis Consortium.18 The presence of white dots is unlikely to be age or genotype specific because patients with identical mutations have not manifest the same fundus appearance.
Autofluorescence imaging revealed abnormal foci of high density that corresponded with the white dots (Figure 1). The autofluorescence emission spectrum closely resembles that of lipofuscin, a byproduct of outer segment shedding. High-density autofluorescence lesions suggest localized accumulation of lipofuscin and disruption of RPE metabolism that may eventually be associated with photoreceptor death. Atrophy of the RPE manifests as absent autofluorescence. In 2 other patients (Figures 2 and 3), low-density central areas corresponded with macular atrophy in 3 of 4 eyes that were surrounded by a ring of high density not previously described in XLRS. These annuli of high density resemble those present in some patients with retinitis pigmentosa and normal visual acuity22-24 that have been shown to surround areas of normal autofluorescence and preserved photopic function. They may constrict with time.25 Rings surrounding areas of central atrophy have also been described in some cases of cone-rod dystrophy26,27 that show a gradient of sensitivity loss across the arc of hyperfluorescence.28 The autofluorescence rings in XLRS may represent an intermediate stage of increased photoreceptor-RPE metabolic load before cell loss and atrophy seen in many older individuals (>45 years) with XLRS.
The 6 patients tested electrophysiologically had findings in keeping with inner retinal dysfunction, typical of XLRS.5,29,30 Rod-specific ERGs of inner retinal origin are usually undetectable or markedly reduced.31 Scotopic bright-flash ERGs are typically electronegative, although in rare cases maximal b-wave amplitudes have been reported as normal.30,32 Photopic and 30-Hz flicker ERGs are usually subnormal and delayed. Pattern ERGs showed variable degrees of reduction in all 6 patients, in keeping with previous studies31,33 of macular dysfunction in this disorder. Full-field ERG abnormalities indicate generalized retinal dysfunction that is consistent with widespread expression of retinoschisin in the retina. In a 19-year-old normal human eye, retinoschisin was found abundantly in the inner segments of the photoreceptors and the outer nuclear layer; moderate levels were also detected in the inner nuclear and plexiform layers.34 Reduced retinoschisin immunoactivity was observed in the peripheral retina of a patient of the same age with XLRS.34
Retinoschisin may be essential in on-bipolar and associated cell-cell interactions. Loss of retinoschisin in the mouse results in disrupted synaptic interactions between photoreceptors and bipolar neurons in the outer plexiform layer.35,36 Abnormal centrifugal displacement of dendrites and synapses during foveal development may explain the radial macular lesions often seen in humans.37 The on-off ERG recordings of some patients with XLRS have shown a reduced b-wave–d-wave ratio, suggesting that cone on-bipolar signaling may be the primary defect.13 In other patients, the on and off responses are affected,31,38 consistent with abnormal signaling by the on and off pathways.31,38 It has been our experience that some patients with XLRS have relative off-pathway sparing and others have clear off-pathway involvement.31
In summary, multiple fine white macular dots may be a manifestation of XLRS unaccompanied by the typical foveal spoke-wheel lesions. Findings from autofluorescence studies suggest that the RPE is involved in the early stages of the disease. Electrophysiologic assessment plays an important role in the documentation of postreceptoral dysfunction and may enable focused DNA screening.
Acknowledgments
Funding/Support: This study is supported by a fellowship from the Burroughs-Wellcome Program in Biomedical Sciences, the Dennis Jahnigen Award from the American Geriatrics Society, the Hirschl Charitable Trust, the Joel Hoffmann Foundation, the Schneeweiss Stem Cell Fund, and the Association of University Professors in Ophthalmology–Research to Prevent Blindness and Eye Surgery Fund (Dr Tsang); by the Foundation Fighting Blindness (Drs Tsang, Bird, and Robson); and by grant EY004081 from the National Institutes of Health (Dr Tsang).
We thank Andrew R. Webster, MD, Sharon Jenkins, MSc, and the members of their laboratories for sharing ideas and resources and for critical reading of the manuscript and Stanley Chang, MD, for guidance and advice.
Footnotes
Financial Disclosure: None reported.
References
- 1.Deutman AF, Pickers AJL, Aan de Kerk AL. Dominantly inherited cystoid macular edema. Am J Ophthalmol. 1976;82:540–548. doi: 10.1016/0002-9394(76)90540-7. [DOI] [PubMed] [Google Scholar]
- 2.Traboulsi E, editor. Genetic Disease of the Eye. Oxford, England: Oxford University Press; 1998. Oxford Monographs on Medical Genetics; No. 36. [Google Scholar]
- 3.George ND, Yates JR, Moore AT. X linked retinoschisis. Br J Ophthalmol. 1995;79:697–702. doi: 10.1136/bjo.79.7.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pimenides D, George ND, Yates JR, et al. X-linked retinoschisis: clinical phenotype and RS1 genotype in 86 UK patients. J Med Genet. 2005;42:e35. doi: 10.1136/jmg.2004.029769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kellner U, Brummer S, Foerster MH, Wessing A. X-linked congenital retinoschisis. Graefes Arch Clin Exp Ophthalmol. 1990;228:432–437. doi: 10.1007/BF00927256. [DOI] [PubMed] [Google Scholar]
- 6.Wu WW, Wong JP, Kast J, Molday RS. RS1, a discoidin domain-containing retinal cell adhesion protein associated with X-linked retinoschisis, exists as a novel disulfide-linked octamer. J Biol Chem. 2005;280:10721–10730. doi: 10.1074/jbc.M413117200. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi T, Omoto S, Takeuchi T, Kozaki K, Ueoka Y, Kitahara K. Four Japanese male patients with juvenile retinoschisis: only three have mutations in the RS1 gene. Am J Ophthalmol. 2004;138:788–798. doi: 10.1016/j.ajo.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 8.Yanoff M, Kertesz Rahn E, Zimmerman LE. Histopathology of juvenile retinoschisis. Arch Ophthalmol. 1968;79:49–53. doi: 10.1001/archopht.1968.03850040051014. [DOI] [PubMed] [Google Scholar]
- 9.Arden GB, Gorin MB, Polkinghorne PJ, Jay M, Bird AC. Detection of the carrier state of X-linked retinoschisis. Am J Ophthalmol. 1988;105:590–595. doi: 10.1016/0002-9394(88)90049-9. [DOI] [PubMed] [Google Scholar]
- 10.Robson AG, Moreland JD, Pauleikhoff D, et al. Macular pigment density and distribution: comparison of fundus autofluorescence with minimum motion photometry. Vision Res. 2003;43:1765–1775. doi: 10.1016/s0042-6989(03)00280-3. [DOI] [PubMed] [Google Scholar]
- 11.von Ruckmann A, Fitzke FW, Bird AC. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol. 1995;79:407–412. doi: 10.1136/bjo.79.5.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marmor MF, Holder G, Seeliger M, Yamamoto S. International Society for Clinical Electrophysiology of Vision. Standard for clinical electroretinography (2004 update) Doc Ophthalmol. 2004;108:107–114. doi: 10.1023/b:doop.0000036793.44912.45. [DOI] [PubMed] [Google Scholar]
- 13.Alexander KR, Barnes CS, Fishman GA. High-frequency attenuation of the cone ERG and ON-response deficits in X-linked retinoschisis. Invest Ophthalmol Vis Sci. 2001;42:2094–2101. [PubMed] [Google Scholar]
- 14.Sieving PA. Photopic ON- and OFF-pathway abnormalities in retinal dystrophies. Trans Am Ophthalmol Soc. 1993;91:701–773. [PMC free article] [PubMed] [Google Scholar]
- 15.Robson AG, Richardson EC, Koh AH, et al. Unilateral electronegative ERG of non-vascular aetiology. Br J Ophthalmol. 2005;89:1620–1626. doi: 10.1136/bjo.2005.071357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bach M, Hawlina M, Holder GE, et al. International Society for Clinical Electrophysiology of Vision. Standard for pattern electroretinography. Doc Ophthalmol. 2000;101:11–18. doi: 10.1023/a:1002732114721. [DOI] [PubMed] [Google Scholar]
- 17.Holder GE. Pattern electroretinography (PERG) and an integrated approach to visual pathway diagnosis. Prog Retin Eye Res. 2001;20:531–561. doi: 10.1016/s1350-9462(00)00030-6. [DOI] [PubMed] [Google Scholar]
- 18.Retinoschisis Consortium. Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. Hum Mol Genet. 1998;7:1185–1192. doi: 10.1093/hmg/7.7.1185. [DOI] [PubMed] [Google Scholar]
- 19.Ho A, Brown G, McNamara J, Recchia F, Regillo C, Vander J. Retina. Barcelona, Spain: McGraw-Hill; 2003. [Google Scholar]
- 20.van Schooneveld MJ, Miyake Y. Fundus albipunctatus-like lesions in juvenile retinoschisis. Br J Ophthalmol. 1994;78:659–661. doi: 10.1136/bjo.78.8.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotta Y, Nakamura M, Okamoto Y, Nomura R, Terasaki H, Miyake Y. Different mutation of the XLRS1 gene causes juvenile retinoschisis with retinal white flecks. Br J Ophthalmol. 2001;85:238–239. doi: 10.1136/bjo.85.2.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robson AG, El-Amir A, Bailey C, et al. Pattern ERG correlates of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Invest Ophthalmol Vis Sci. 2003;44:3544–3550. doi: 10.1167/iovs.02-1278. [DOI] [PubMed] [Google Scholar]
- 23.Robson AG, Egan CA, Luong VA, Bird AC, Holder GE, Fitzke FW. Comparison of fundus autofluorescence with photopic and scotopic fine-matrix mapping in patients with retinitis pigmentosa and normal visual acuity. Invest Ophthalmol Vis Sci. 2004;45:4119–4125. doi: 10.1167/iovs.04-0211. [DOI] [PubMed] [Google Scholar]
- 24.Wabbels B, Demmler A, Paunescu K, Wegscheider E, Preising MN, Lorenz B. Fundus autofluorescence in children and teenagers with hereditary retinal diseases. Graefes Arch Clin Exp Ophthalmol. 2006;244:36–45. doi: 10.1007/s00417-005-0043-2. [DOI] [PubMed] [Google Scholar]
- 25.Robson AG, Saihan Z, Jenkins SA, et al. Functional characterisation and serial imaging of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Br J Ophthalmol. 2006;90:472–479. doi: 10.1136/bjo.2005.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michaelides M, Holder GE, Hunt DM, Fitzke FW, Bird AC, Moore AT. A detailed study of the phenotype of an autosomal dominant cone-rod dystrophy (CORD7) associated with mutation in the gene for RIM1. Br J Ophthalmol. 2005;89:198–206. doi: 10.1136/bjo.2004.050773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ebenezer ND, Michaelides M, Jenkins SA, et al. Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest Ophthalmol Vis Sci. 2005;46:1891–1898. doi: 10.1167/iovs.04-1482. [DOI] [PubMed] [Google Scholar]
- 28.Robson A, Michaelides M, Webster A, et al. Comparison of pattern ERG, multifocal ERG and psychophysical correlates of fundus autofluorescence abnormalities in patients with cone-rod (RPGR, RIM1) or rod-cone dystrophy [ARVO abstract] Invest Ophthalmol Vis Sci. 2005;43:552. [Google Scholar]
- 29.Peachey NS, Fishman GA, Derlacki DJ, Brigell MG. Psychophysical and electroretinographic findings in X-linked juvenile retinoschisis. Arch Ophthalmol. 1987;105:513–516. doi: 10.1001/archopht.1987.01060040083038. [DOI] [PubMed] [Google Scholar]
- 30.Bradshaw K, George N, Moore A, Trump D. Mutations of the XLRS1 gene cause abnormalities of photoreceptor as well as inner retinal responses of the ERG. Doc Ophthalmol. 1999;98:153–173. doi: 10.1023/a:1002432919073. [DOI] [PubMed] [Google Scholar]
- 31.Stanga PE, Chong NH, Reck AC, Hardcastle AJ, Holder GE. Optical coherence tomography and electrophysiology in X-linked juvenile retinoschisis associated with a novel mutation in the XLRS1 gene. Retina. 2001;21:78–80. doi: 10.1097/00006982-200102000-00019. [DOI] [PubMed] [Google Scholar]
- 32.Sieving PA, Bingham EL, Kemp J, Richards J, Hiriyanna K. Juvenile X-linked retinoschisis from XLRS1 Arg213Trp mutation with preservation of the electroretinogram scotopic b-wave. Am J Ophthalmol. 1999;128:179–184. doi: 10.1016/s0002-9394(99)00144-0. [DOI] [PubMed] [Google Scholar]
- 33.Clarke MP, Mitchell KW, McDonnell S. Electroretinographic findings in macular dystrophy. Doc Ophthalmol. 1996;92:325–339. doi: 10.1007/BF02584086. [DOI] [PubMed] [Google Scholar]
- 34.Mooy CM, Van Den Born LI, Baarsma S, et al. Hereditary X-linked juvenile retinoschisis: a review of the role of Muller cells. Arch Ophthalmol. 2002;120:979–984. [PubMed] [Google Scholar]
- 35.Zeng Y, Takada Y, Kjellstrom S, et al. RS-1 gene delivery to an adult Rs1h knockout mouse model restores ERG b-wave with reversal of the electronegative waveform of X-linked retinoschisis. Invest Ophthalmol Vis Sci. 2004;45:3279–3285. doi: 10.1167/iovs.04-0576. [DOI] [PubMed] [Google Scholar]
- 36.Weber BH, Schrewe H, Molday LL, et al. Inactivation of the murine X-linked juvenile retinoschisis gene, Rs1h, suggests a role of retinoschisin in retinal cell layer organization and synaptic structure. Proc Natl Acad Sci U S A. 2002;99:6222–6227. doi: 10.1073/pnas.092528599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takada Y, Fariss RN, Tanikawa A, et al. A retinal neuronal developmental wave of retinoschisin expression begins in ganglion cells during layer formation. Invest Ophthalmol Vis Sci. 2004;45:3302–3312. doi: 10.1167/iovs.04-0156. [DOI] [PubMed] [Google Scholar]
- 38.Khan NW, Jamison JA, Kemp JA, Sieving PA. Analysis of photoreceptor function and inner retinal activity in juvenile X-linked retinoschisis. Vision Res. 2001;41:3931–3942. doi: 10.1016/s0042-6989(01)00188-2. [DOI] [PubMed] [Google Scholar]