

Abstract
The glycoprotein envelope of alphaviruses consists of two proteins, E1 and E2. E1 is responsible for fusion and E2 is responsible for receptor binding. An atomic structure is available for E1, but one for E2 has not been reported. In this study, transposon linker insertion mutagenesis was used to probe the function of different domains of E2. A library of mutants, containing 19 amino acid insertions in the E2 glycoprotein sequence of the prototype alphavirus, Sindbis virus (SINV), was generated. Fifty seven independent E2 insertions were characterized, of which more than half (67%) gave rise to viable virus. The wild type-like mutants identify regions that accommodate insertions without perturbing virus production and can be used to insert targeting moieties to direct SINV to specific receptors. The defective and lethal mutants give insight into regions of E2 important for protein stability, transport to the cell membrane, E1-E2 contacts, and receptor binding.
Keywords: Sindbis virus, transposon mutagenesis, receptor binding, glycoprotein
Introduction
Alphaviruses are enveloped positive-strand RNA viruses belonging to the family Togaviridae. They are arthropod-borne viruses that cause a range of diseases from the relatively mild fever, rash and arthritis caused by the prototype virus Sindbis (SINV) to the more severe encephalitis caused by the human and veterinary pathogens, Eastern, Western and Venezuelan equine encephalitis viruses. The alphaviruses have been found in all continents throughout the world (La Linn, 2001; Strauss and Strauss, 1994).
The mature virion is composed of three major structural proteins, the capsid protein and two integral membrane glycoproteins E1 and E2. A small 6K protein is found in sub-stoichiometric amounts in the particle (Gaedigk-Nitschko and Schlesinger, 1990; Lusa et al., 1991). The 11,703 nucleotide (nt) viral genome of SINV is encapsidated by 240 copies of the capsid protein in the cytoplasm of infected cells. The E1 and E2 glycoproteins are translated from a subgenomic RNA and cotranslationally inserted into the endoplasmic reticulum where they are glycosylated and then transported through the Golgi apparatus to the plasma membrane (Melancon and Garoff, 1987; Wirth et al., 1977). The nucleocapsid core then acquires a lipid envelope, embedded with E1 and E2 glycoproteins, and buds out of the cell. In the mature virion, the glycoproteins are arranged into 80 spike-like structures, each spike being composed of a trimer of E2/E1 heterodimers (von Bonsdorff and Harrison, 1975, 1978; Cheng et al., 1995; Mancini et al., 2000; Mukhopadhyay et al., 2006; Paredes et al., 1993; Pletnev et al., 2001; Zhang et al., 2002).
E2 is a 423 amino acid (~50 kDa) Type I transmembrane protein. It consists of an N-terminal (1–364, SINV numbering) hydrophilic region, followed by a 26 residue membrane-spanning region (365–390) and a 33 residue (391–423) cytoplasmic endo domain. SINV E2 is glycosylated with two asparagine (N)-linked carbohydrate chains at N-196 and N-318 (Mayne et al., 1985). In addition, certain cysteine residues in the cytoplasmic domain and membrane-spanning region of E2 have been implicated as sites for palmitoylation (Schmidt et al., 1979). During the course of the SINV life cycle the E2 glycoprotein is responsible for cell receptor binding while E1 is involved in the subsequent fusion process with the host cell membrane.
Although alphaviruses have long served as a model system for the study of virus structure and assembly, an atomic resolution structure for E2 is still lacking. In the absence of structural data for E2, a random transposon linker insertion mutagenesis system was used to probe the functions of the different domains of the glycoprotein. A 19 amino acid insertion was introduced into E2 using a Tn5 transposon-based linker insertion mutagenesis system resulting in the generation of a library consisting of 57 independent E2 insertion mutations. The location of each of the insertions was determined by sequencing the E2 region of the cDNA and virus was generated by RNA transcription and transfection. The insertion mutants were characterized as to their level of E2 expression, E2 transport to the plasma membrane and the affect of the insertion on virus viability. The effects of the insertions on virus-receptor interactions were probed by virus neutralization assays. The results indicate that a majority of the insertions gave rise to viable virus. Some insertions had no impact on virus growth as determined by plaque assays and one-step growth assays. These sites may be used for inserting targeting ligands in order to target SINV to specific cell receptors in certain gene therapy applications. The results of this mutational analysis describe domains of E2 critical for the structural integrity of the protein, critical for transport to the plasma membrane, and critical for virus-host interactions.
Results
Generation of an E2 insertion library
To probe the structure of E2 and identify the regions of the protein integral to its function, a random insertion library was generated by transposon linker insertion mutagenesis. In order to rapidly generate a large number of independent insertions in E2, the Tn5 transposase-based EZ::TN In Frame Linker Insertion Kit (Epicentre, Madison WI) was used. Tn5 transposon insertions into target DNA are random, and the transposase will transpose any DNA sequence contained between its 19 bp Mosaic End (ME) Tn5 transposase recognition sequences (Goryshin and Reznikoff, 1998). The EZ::TN <NotI/KAN-3> transposon is an 1167 bp fragment of DNA which contains a kanamycin resistance (KanR) marker between the Tn5 transposase ME sequences. Removal of the KanR gene from the transposon by NotI digestion leaves behind a 57 nt insertion which is open in all three reading frames ensuring that all insertions will be productive (Table 1).
Table 1.
Amino acid sequences of the three possible open reading frames of the transposon insertion sequence
| ORFa | Amino Acid Sequenceb | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | L | S | L | V | H | I | L | R | P | Q | D | V | Y | K | R | Q |
| 2 | C | L | L | Y | T | S | C | G | R | K | M | C | T | R | D | |
| 3 | V | S | C | T | H | L | A | A | A | R | C | V | Q | E | T | |
ORF: open reading frame.
Depending on the ORF there are 16 (ORF1) or 15 (ORF2 and 3) invariant amino acids. The remaining 3 or 4 amino acids are the result of duplication of host target sequences.
To maximize the number of transposition events occurring in the E2 target sequence, a fragment of pToto64 (SINV cDNA) (Owen and Kuhn, 1996) that encoded the first 407 amino acids of E2 (ectodomain and transmembrane domain) was subcloned into pJRtac99 (American Type Culture Collection) to generate pJRtac99-E2. This vector was subsequently used in an in vitro transposon reaction that resulted in the random insertion of the transposon into pJRtac99-E2 to generate pJRtac99-E2TnKan (Fig. 1). Plasmids containing the transposon were selected by plating on media containing both kanamycin and chloramphenicol. Approximately 1400 colonies were recovered in this manner. The frequency of transposition into the plasmid was calculated to be about 14%, which is within the expected range (0.5–20%). In order to identify plasmids with insertions specifically in the SINV cDNA fragment, pJRtac99-E2TnKan was purified from individual KanR/CamR colonies (~200 colonies), restriction digested to release the SINV cDNA, and the fragments were analyzed on an agarose gel. SINV cDNA fragments with inserts were ~1100 bp larger, and 104 plasmids out of 200 (52%) had a transposon insertion in the cDNA sequence. These SINV cDNA fragments were gel purified for ligation back into pToto64, to generate pToto64-TnKan. NotI digestion of pToto64-TnKan eliminated the KanR gene and regenerated the SINV cDNA, pToto64–Tn, which now contained a 57 nt insertion in E2 (Fig. 1). In order to determine the location of the insertions, Toto64-Tn plasmids were sequenced with primers flanking the E2 coding sequence. 104 independent clones were sequenced and 57 (55%) had transposon insertions in E2.
Figure 1.

Generation of an E2 transposon insertion library. A schematic outlining the strategy employed to generate the library of E2 transposon mutants.
Distribution of transposon insertions in E2
Transposon insertions were found throughout the E2 sequence (Figs. 2A and 2B). However, certain sites were represented more than once among the 57 independent clones characterized, and as a result, only 42 of the 57 (74%) transposon insertion sites were unique. Sites with multiple transposition events may represent hotspots for Tn5 transposase-based mutagenesis (Fig. 2C). In contrast, several regions of the E2 sequence (amino acids 36–85, 120–152 and 170–220) did not have any insertions (Fig. 2C). Aminoacid residues 170–220 are especially interesting as it has been shown to contain numerous SINV neutralizing epitopes (reviewed in Strauss and Strauss, 1994). However, none of the transposon insertions were located within this region. The closest insertion was lethal for the virus and occurred at amino acid 221. Since insertions within residues 170–220 may interfere with virus-receptor interactions and provide insight into the specific residues involved in receptor binding, we decided to screen specifically for insertions in this region. A high-throughput approach was employed where CamR/KanR bacterial colonies were picked using an automated colony picker into 96-well plates containing LB broth, and the resulting cultures were used for colony PCR in order to amplify the 170–220 region. Transposon insertions could be easily recognized by their size on an agarose gel (1100 bp larger). Three 96-well plates were screened in this manner (288 colonies), but we were unable to identify any insertions in this region. Together with the initial colonies screened to generate the E2 transposon insertion library, over 400 colonies have been screened and none of them contained insertions in the 170–220 region. This region may have nucleotide sequences or secondary structures that inhibit Tn5 transposase-based insertions.
Figure 2.
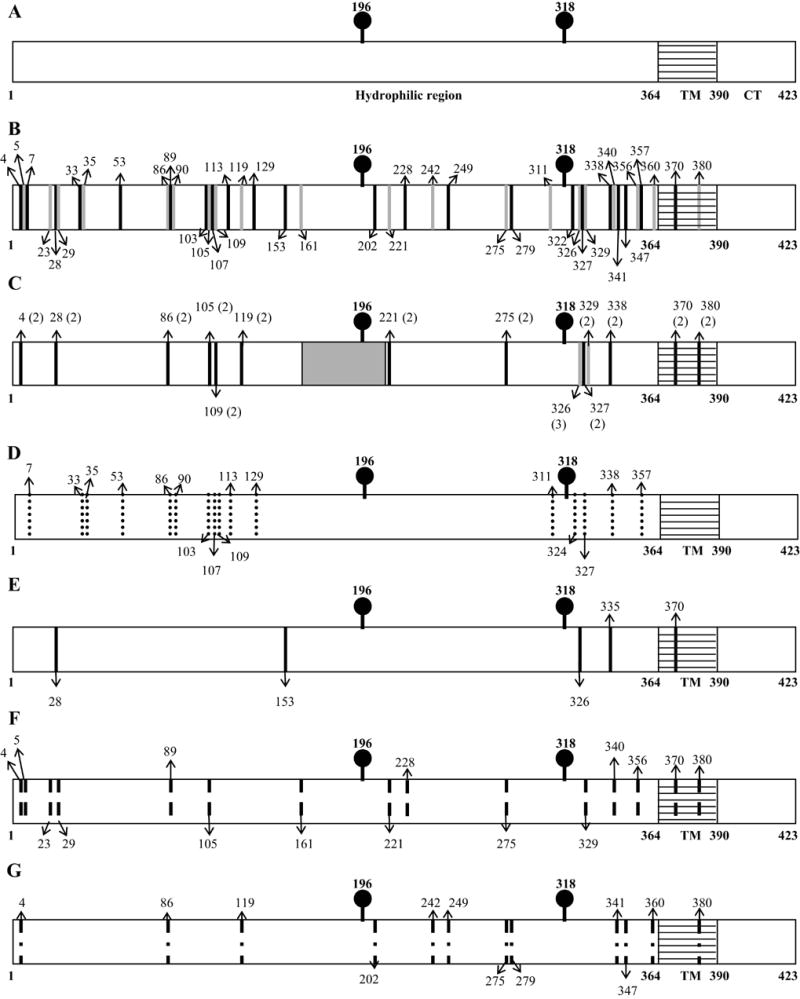
Distribution of transposon insertions in E2. A: A schematic of the SINV E2 protein. The hatched area represents the transmembrane (TM) region. The glycosylation sites at 196 and 318 are represented by a stick and ball. CT: cytoplasmic tail. B: location of all identified transposon insertions. The insertion sites are represented as bars with the residue after which the insertion occurred indicated by arrows at the top or bottom. C: location of possible hotspots for transposition (bars). The number of independent clones with insertions at that particular location is show in parentheses. The gray shaded area represents the putative receptor binding region where no transposon insertions were recovered. D: distribution of transposon insertions resulting in a lethal phenotype; E: large plaque phenotype (wild type); F: medium plaque phenotype and G: small plaque phenotype.
Majority of the insertions in E2 do not abolish protein expression
To assess the affect of the insertions on E2 protein expression, BHK-15 cells were transfected with Toto64-Tn RNA, and cytoplasmic extracts were analyzed by Western blot. Cells were harvested 12 hours post-transfection and probed with anti-E2 (cytoplasmic domain) and anti-capsid protein antibodies. Table 2 lists all the transposon insertion mutants along with the relative level of E2 and capsid protein expression for each mutant. The expression of the capsid protein served as an internal control to demonstrate the replication and translational efficiency of the cDNA clone. All the cDNA clones tested expressed levels of CP similar to wild-type SINV (Table 2). IFAs were also done on BHK cells electroporated with the mutant constructs which further confirmed the Western blot results (data not shown).
Table 2.
Library of transposon insertions in SINV E2
| Constructa | Location of Insertion | ORFb | Plaque Phenotype c | CP Productiond | E2 Productiond | Geometric Meane |
|---|---|---|---|---|---|---|
| E2-4:1 | 4 | 1 | MP | ++++ | +++ | 19 |
| E2-4:2 | 4 | 2 | SP | ++++ | + | 5 |
| E2-5 | 5 | 3 | MP | ++++ | ++++ | 35 |
| E2-7 | 7 | 3 | x | ++++ | +++ | 8 |
| E2-23 | 23 | 3 | MP | ++++ | +++ | 14 |
| E2-28:1 | 28 | 2 | LP | ++++ | ++++ | 39 |
| E2-28:2 | 28 | 2 | LP | ++++ | ++++ | 40 |
| E2-29 | 29 | 3 | LP | ++++ | ++++ | 32 |
| E2-33 | 33 | 2 | x | +++ | + | 4 |
| E2-35 | 35 | 3 | x | +++ | - | ND |
| E2-53 | 53 | 1 | x | +++ | + | 4 |
| E2-86:1 | 86 | 2 | SP | +++ | +++ | 12 |
| E2-86:2 | 86 | 3 | x | +++ | +++ | 7 |
| E2-89 | 89 | 2 | MP | ++++ | ++++ | 37 |
| E2-90 | 90 | 3 | x | ++++ | ++++ | 8 |
| E2-103 | 103 | 3 | x | ++++ | - | 3 |
| E2-105:1 | 105 | 3 | MP | ++++ | +++ | 35 |
| E2-105:2 | 105 | 3 | MP | ++++ | ++++ | 38 |
| E2-107 | 107 | 2 | x | ++++ | +++ | 4 |
| E2-109:1 | 109 | 3 | x | ++++ | +++ | 6 |
| E2-109:2 | 109 | 3 | x | ++++ | +++ | 8 |
| E2-113 | 113 | 3 | x | ++++ | ++++ | 7 |
| E2-119:1 | 119 | 2 | SP | ++++ | ++++ | 22 |
| E2-119:2 | 119 | 3 | SP | ++++ | ++++ | 17 |
| E2-153 | 153 | 1 | LP | ++++ | ++++ | 37 |
| E2-161 | 161 | 3 | MP | ++++ | ++++ | 18 |
| E2-202* | 202 | 2 | SP | ++++ | ++++ | 25 |
| E2-221:1 | 221 | 3 | MP | ++++ | ++++ | 20 |
| E2-221:2 | 221 | 3 | MP | ++++ | ++++ | 33 |
| E2-228 | 228 | 2 | MP | ++++ | +++ | 12 |
| E2-242 | 242 | 3 | SP | ++++ | ++++ | 15 |
| E2-249 | 249 | 2 | SP | ++++ | ++++ | 24 |
| E2-275:1 | 275 | 3 | MP | ++++ | ++++ | 37 |
| E2-275:2 | 275 | 1 | SP | ++++ | ++++ | 18 |
| E2-279 | 279 | 1 | SP | ++++ | ++++ | 11 |
| E2-311 | 311 | 1 | x | +++ | +++ | 5 |
| E2-324 | 324 | 3 | x | +++ | - | 3 |
| E2-326:1 | 326 | 2 | LP | ++++ | ++++ | ND |
| E2-326:2 | 326 | 2 | LP | ++++ | ++++ | ND |
| E2-326:3 | 326 | 2 | LP | ++++ | ++++ | ND |
| E2-327:1 | 327 | 3 | x | ++++ | ++++ | 4 |
| E2-327:2 | 327 | 3 | x | +++ | +++ | 5 |
| E2-329:1 | 329 | 2 | MP | ++++ | ++++ | 13 |
| E2-329:2 | 329 | 2 | MP | ++++ | ++++ | 15 |
| E2-335 | 335 | 2 | LP | ++++ | ++++ | ND |
| E2-338:1 | 338 | 1 | x | ++++ | ++++ | 7 |
| E2-338:2 | 338 | 1 | x | ++++ | ++++ | 7 |
| E2-340 | 340 | 2 | MP | ++++ | ++++ | 20 |
| E2-341 | 341 | 2 | SP | ++++ | ++++ | 33 |
| E2-347 | 347 | 1 | SP | ++++ | +++ | 10 |
| E2-356 | 356 | 3 | MP | ++++ | ++++ | 21 |
| E2-357 | 357 | 2 | x | ++++ | ++++ | 8 |
| E2-360 | 360 | 1 | SP | ++++ | ++++ | 39 |
| E2-370:1 | 370 | 1 | LP | ++++ | ++++ | ND |
| E2-370:2 | 370 | 2 | MP | ++++ | +++ | 37 |
| E2-380:1 | 380 | 1 | MP | ++++ | +++ | 10 |
| E2-380:2 | 380 | 2 | SP | ++++ | +++ | 6 |
| Toto64 (wild type) | - | - | LP | ++++ | ++++ | 35 |
The number after the dash (−) represents the location of the insertion. Multiple insertions at the same location were distinguished by a number following a colon (:).
ORF=open reading frame. See Table 1 for sequence.
LP = large plaque, MP = medium plaque, SP = small plaque and x = lethal
‘++++’=100% of wild-type protein expression; ‘+++’= 75% of wild-type protein expression; ‘++’= 50% of wild-type protein expression and ‘+’= 25% of wild-type protein expression.
The results are an average of three experiments.
E2-202 was created by cloning the transposon insertion sequence into this position.
Of the 57 independent cDNA clones tested, only 3 did not produce any detectable E2 as determined by Western blot analysis (Table 2). These results indicate that a majority of the insertions did not abolish E2 protein expression, although some did show reduced levels of the protein. The insertions that did abolish E2 expression were located at E2-35, E2-103 and E2-324. Transposon insertions in the vicinity of residue 35 also resulted in dramatically reduced E2 expression. Insertions at amino acid positions 33 and 53 were lethal and E2 expression was less than 25% of wild type (Table 2). Therefore, the region between amino acids 33–53 appears to be important for proper protein folding.
Although no E2 expression was detected for E2-103 and E2-324, other insertions adjacent to these residues had levels of E2 expression 75%–100% of wild type (Table 2). Therefore, the lack of protein expression in these mutants may be a function of the particular ORF expressed.
Majority of insertions in E2 result in viable virus
The effect of the insertions on the virus life cycle was determined by the ability of the mutants to form plaques on BHK monolayers. In vitro transcribed RNAs from the E2 transposon insertion mutations were transfected into BHK cells, and the resulting viruses were isolated and characterized by plaque assay. The plaque sizes were compared to that of wild-type SINV at 48 hours post-infection. Wild-type SINV forms plaques that are 3.5–4 mm in diameter 48 hours post-infection, and this was classified as a large-plaque phenotype (LP). A virus with a medium-plaque phenotype (MP) forms plaques 2.5–3.5 mm in diameter and, the plaques of a virus with a small-plaque phenotype (SP) are 2–2.5 mm in diameter, all relative to the size of wild type plaques. Table 2 lists all the transposon insertion mutants characterized and their plaque phenotypes. The results indicate that a majority of the insertions are viable, and some even exhibit wild-type levels of growth. Of the 57 independent clones tested, only 18 (33%) were lethal (NP). These 18 insertions represent 15 unique sites in E2 (Fig. 2D). The insertions that result in a lethal phenotype are primarily clustered towards the N-terminus and the C-terminus of E2. The lethal insertions are found between residues 33–113 at the N-terminus and residues 311–357 at the C-terminus.
Of the 40 independent clones that gave rise to viable virus, 9 (6 unique sites) had wild type-like plaque phenotypes, 17 (14 unique sites) had medium-plaque phenotypes and 12 (11 unique sites) had small-plaque phenotypes (Figs. 2E, F and G). There were several cases where two independent insertions at the same location resulted in different phenotypes. In all cases this could be attributed to different reading frames being used. For example an insertion at amino acid 4 resulted in a MP phenotype when ORF1 (Table 1) was expressed but a SP phenotype when ORF2 was expressed (Table 2 and Figs. 2F and 2G). ORF2 encodes three cysteine residues which may disrupt the pattern of disulfide bond formation within E2. This result indicates that the individual amino acids that are been expressed have an impact on how well the insertion is tolerated at a given location. Nineteen percent (19%) of the inserts expressed ORF1, 37% expressed ORF2 and 44% expressed ORF3. Therefore, there was a slight preference for ORF3 and ORF2 compared to ORF1. A tally of the phenotypes exhibited by each ORF showed that 64% of mutants expressing ORF1, 76% of mutants expressing ORF2 and 57% of mutants expressing ORF3, were viable. These results indicate that, although the expression of different ORFs at the same location can result in different phenotypes, no ORF was dramatically more disruptive than any other.
The plaque phenotype and growth properties of the transposon mutants were further verified by one-step growth analysis. Figure 3 represents the one-step growth curves of a representative cross-section of the transposon insertion library. All the transposon mutants with a large plaque phenotype had similar growth kinetics to wild type SINV (Fig. 3A). Figure 3B shows that a majority of the mutants with a medium plaque phenotype exhibited growth kinetics approximately two logs lower than that of wild type SINV. However, E2-105 and E2-221 grew to titers only one log lower than the parental virus. All the transposon mutants with a small plaque phenotype had final titers at least three logs lower than the parental virus (Fig. 3C). Thus, plaque phenotypes correlated well with the replication properties measured in single cycle growth curves.
Figure 3.

One-step growth curves of selected transposon mutants. The growth kinetics of the transposon mutants was compared to wild type SINV in BHK cells. A: transposon mutants with large plaque phenotype. B: transposon mutants with medium plaque phenotype. C: transposon mutants with small plaque phenotype. Media was replaced every 30 minutes for the first two hours, and then every hour for 12 hours post-infection. Supernatant was collected at 4, 6, 8, 10 and 12 hours post-infection and released virus was assayed by titration on BHK cells at 37ºC. The results represent the average of two experiments.
Insertions in E2 that result in reduced E2 transport to the plasma membrane
The amount of E2 at the cell surface was quantified using FACS analysis, in order to determine if any of the insertions affected E2 transport to the plasma membrane. For insertions that resulted in viable virus, BHK cells were infected at an MOI of 1, whereas, in the case of the insertions that were lethal, ~10 μg of in vitro transcribed RNA was electroporated into the cells. Cells were harvested 12 hours post-infection and stained with an anti-E2 polyclonal antibody, followed by a fluorescein-conjugated goat anti-rabbit secondary antibody prior to FACS analysis. Figure 4A shows that when BHK cells infected with wild-type SINV (unfilled curve) were stained with a polyclonal E2 antibody there was a shift in the number of strongly fluorescent cells compared to mock infected BHK cells (filled curve). The geometric mean represents the mean value of the fluorescent signal under the curve. Therefore, the fluorescent signal of the cells can be quantified as the geometric mean (Fig. 4A), and this value can be used as a measure of the amount of E2 in the plasma membrane. BHK cells infected with a mutant expressing less E2 at the cell surface would have a lower geometric mean compared to wild-type SINV-infected BHK cells (compare Figs. 4A and 4B).
Figure 4.

Surface expression of E2. BHK cells were infected with wild-type SINV (A) or an E2 transposon insertion mutant (E2-53) (B) at a multiplicity of infection of 1. Cells were harvested 12 hours post-infection and stained with a polyclonal α-E2 antibody and a fluorescein-conjugated secondary antibody. Cells were analyzed using a FACSCalibure flow cytometer. The grey filled-in curve represents the signal from uninfected BHK cells. The shift in the signal upon virus infection is shown by the clear curve and represents the expression of E2 at the cell surface. The number above the curve is the geometric mean for the region of the curve delineated by the line and is a measure of the amount of E2 at the plasma membrane. The results represent data from a single experiment. However, the experiment has been repeated three times, with consistent results.
Figure 4B represents the E2 surface expression for an insertion mutant at E2-53 which exhibits low levels of E2 protein production (Table 2). This shows that when the level of E2 synthesis is low (less than 25% of wild type in this case) or undetectable, the cells exhibit a level of fluorescent signal comparable to uninfected BHK cells. Therefore, there is no detectable E2 in the plasma membrane for the insertions at E2-33, E2-53 and E2-103 due to reduced levels of E2 expression. However, there are several mutations that have wild-type levels of E2 expression in the cytoplasm, but low levels of E2 at the cell surface. There are 4 insertions within residues 107–113 (E2-107, E2-109:1, E2-109:2 and E2-113) that express wild-type levels of E2, but have 3–6 fold lower E2 signal in the plasma membrane and exhibit a lethal phenotype (Table 2). Furthermore, two insertions at residue 119 (E2-119:1 and E2-119:2) resulted in small plaque phenotypes (Table 2). These mutations also exhibited wild-type levels of E2 expression but had a 2-fold reduction of the E2 signal at the cell surface. Therefore, although sufficient levels of E2 are being expressed, it is not making it to the plasma membrane, suggesting that insertions in this region are disrupting E2 transport.
Several insertions towards the C-terminus of E2 (311–380) also dramatically reduced levels of E2 protein at the plasma membrane (Table 2). An insertion in the membrane-spanning region at 380 resulted in a 4- to 8-fold reduction in the E2 signal at the cell surface. Insertions in this region may destabilize the protein in the membrane and therefore, affect protein transport to the plasma membrane. An insertion at residue 370 (also in the transmembrane), however, did not have a detrimental effect on E2 transport as E2 expression at the cell surface was similar to wild type.
A transposon-insertion at E2-360 results in wild-type levels of E2 at the plasma membrane, yet it has a small-plaque phenotype (Table 2). Insertions at E2-340, E2-341 and E2-356 also resulted in medium plaque or small plaque phenotypes, but had wild-type levels of E2 at the cell surface. Therefore, insertions in this region do not effect protein expression or transport but have defects in other aspects of the life cycle.
Effect of transposon insertions on the efficacy of neutralizing monoclonal antibodies that map to E2
Most of the MAbs that neutralize SINV have been mapped to E2. Insertions in E2 may alter the conformation of these surface-exposed sites such that these neutralizing MAbs no longer bind E2. By correlating the site of the insertion with the neutralizing MAb inhibited by the mutation, the putative MAb binding site can be mapped on E2. Virus neutralization assays were performed with two SINV neutralizing MAbs (202 and 209) that bind to different antigenic sites on E2 (Davis et al., 1987; Meyer and Johnston, 1993; Strauss et al., 1991). Neutralizing MAb 202 binds to the E2ab antigenic site of E2, which comprises amino acid residues 181–216, whereas neutralizing MAb 209 binds to the E2c antigenic site (Ubol and Griffin, 1991; Wang et al., 1991). Escape mutants to E2c have been mapped to residues 62, 96 and 159 (Pence et al., 1990). MAb 202 and 209 each neutralize wild-type SINV effectively as shown in Fig. 5A and 5B. However, an insertion at residue 119 resulted in decreased efficacy of neutralization by each antibody (Fig. 5A and 5B). In fact, as presented in Table 3, eight mutants were identified that are fully or partially resistant to neutralization, indicating that some of these mutants may have impaired ability to bind cellular receptors. Insertions at E2-4, E2-119, E2-202 and E2-380 of E2 were able to prevent neutralization by MAb 202, whereas insertions at residues E2-4, E2-23, E2-119, E2-161 and E2-202 were able to prevent neutralization by MAb 209. Some of these insertion sites may represent residues to which the MAbs bind directly, whereas some represent sites that merely alter the conformation of the protein such that the MAbs can no longer bind. In either case, this information would be useful in planning a strategy to ablate the natural tropism of SINV.
Figure 5.
Neutralization of E2 transposon insertion mutations by MAb 202 and MAb 209. 100 PFU of Toto64 or E2-119 were each incubated with different concentrations of MAb 202 (A) or MAb 209 (B) for 30 minutes. The titer of the resulting virus was determined by standard plaque assay on BHK cells at 37ºC. The results are the average of three experiments. The error bars indicate the standard deviation.
Table 3.
Neutralization of E2 insertion mutants by MAb 202 and MAb 209
| Virus | Plaquea | % Neutralizationb | |
|---|---|---|---|
| Phenotype | MAb 202 | MAb 209 | |
| E2-4:1 | MP | 9% | 7% |
| E2-23 | MP | ND | 1% |
| E2-119:1 | SP | 17% | 43% |
| E2-161 | MP | 96% | 35% |
| E2-202 | SP | 5% | 45% |
| E2-347 | SP | 94% | 88% |
| E2-380:1 | MP | 2% | 73% |
| Toto64 (wild type) | LP | 95% | 92% |
LP = large plaque; MP = medium plaque; SP = small plaque
See Materials and Methods for experimental details
Insertions in E2 that may produce noninfectious particles
An ELISA was used in order to determine if any of the insertion mutants with a lethal phenotype produced noninfectious virions. Ten-fold concentrated culture supernatant from BHK cells electroporated with the mutant constructs was assayed for the presence of virions using a polyclonal antibody against E2. Table 4 presents the ELISA data in terms of the optical density (OD at 450 nm) for each of the constructs tested as well as in terms of a percentage of the wild-type (Toto64) signal. The results indicate that a lethal insertion at E2-7 has an OD450 value of 0.427 which is 86% of the wild-type signal. This strongly suggests that this mutation may be producing noninfectious particles. BHK cells electroporated with the RNA genome of this construct were analyzed by transmission electron microscopy (TEM)12-hours post-electroporation. The TEM images demonstrated the presence of budding virions at the surface of BHK cells electroporated with the E2-7 construct (data not shown). In order to confirm that noninfectious virions were produced, an attempt was made to isolate these particles. BHK cells were electroporated with the in vitro transcribed RNA of the mutant construct and then a virion fraction was purified from the media using standard techniques. We were unable to visualize any virus particles by negative-stain EM despite the observations from the TEM. Thus, the virions may not have survived the harsh purification steps, or the number of virions may be below the detection limit for negative-stain EM analysis. The other lethal insertions tested had OD450 values closer to mock levels. E2-33 resulted in 48% of the wild-type E2 signal. However, immunoblot analysis has revealed that E2-33 has low levels of E2 expression (Table 2), and this disruption of E2 expression may explain the lethal nature of the insertion.
Table 4.
ELISA to detect production of non-infectious particles
| Virus | OD 450a | % of wild-type signal | Plaque Phenotypeb |
|---|---|---|---|
| E2-7 | 0.427 (0.02) | 86% | x |
| E2-33 | 0.241 (0.02) | 48% | x |
| E2-86:2 | 0.119 (0.01) | 24% | x |
| E2-90 | 0.165 (0.02) | 33% | x |
| E2-119:1 | 0.481 (0.02) | 97% | SP |
| E2-311 | 0.125 (0.03) | 25% | x |
| E2-338:1 | 0.248 (0.01) | 50% | x |
| Toto64 (wild type) | 0.498 (0.05) | 100% | LP |
| Mock | 0.121 (0.01) | 24% | - |
The results represent the average of three experiments. The standard deviation is presented in parentheses.
LP = large plaque; SP = small plaque; x = lethal
Discussion
Transposon linker insertion mutagenesis as a method to produce an insertion library
In the past decade much effort has focused on the development of alphaviruses as vectors for gene expression and as vaccines or therapeutic agents (Chikkanna-Gowda , 2005; Schlesinger and Dubensky, 1999; Xiong et al., 1989). The virus has been structurally characterized, having T=4 icosahedral symmetry, and the atomic structures of the capsid (amino acids 106–264) and E1 (ectodomain) proteins have been determined (Choi et al., 1991; Lee et al., 1996; Lescar et al., 2001; Zhang et al., 2002). However, the structure of the receptor-binding protein, E2, has been elusive. Attempts to express and crystallize have so far failed to produce diffracting crystals of E2 (unpublished data, R.J. Kuhn and M.G. Rossmann). Knowledge of the different functional domains of E2 would be invaluable in the design of a targeted alphavirus gene therapy vector. In order to functionally characterize the different regions of E2 in the absence of a three-dimensional structure, an E2 insertion library was generated. A previous random linker insertion mutagenesis study, where the 15 amino acid Rift Valley fever virus (RVFV) 4D4 epitope was inserted into E2, demonstrated that it was possible to incorporate heterologous peptides into E2 (London et al., 1992). That study lead to the characterization of two insertions sites (at E2-3 and E2-244) that were able to accommodate the RVFV 4D4 epitope. Our study extends this work by creating a transposon-based linker insertion library in E2 and characterizing both permissive and non-permissive sites for the insertion of the 19-residue transposon insertion.
A transposon based mutagenesis strategy was adopted, because it afforded a rapid way of randomly inserting a linker into E2 and generating many independent clones. The EZ::TN In Frame Linker Insertion Kit was chosen because the inserted linker is open in all three reading frames, thereby ensuring that all insertions are productive. From an initial pool of 200 KanR/CamR colonies, we were able to identify 56 that had insertions specifically in the E2 region of the SINV cDNA. The calculated percentage of insertions in the E2 sequence is 28%, which is close to the theoretical value of 34%, based on a completely random distribution of insertions. Although the mutagenesis system is in theory random, there are 15 locations represented two or more times among the 56 independent clones; thus, 27 % of the transposon insertions are repeats. The maximum number of times a particular location was represented in the library was three (E-326) (Table 2). Therefore, the transposon insertions are not random, and as a result there are some regions with a high incidence of insertions and some with no insertions. This is exemplified by the 170–220 region of E2. Assuming a completely random distribution of transposon insertions, the probability that there will not be an insertion in the 170–220 region among the 400 colonies screened, is very low (~10−16). It has previously been reported that Tn5-based transposases have a low efficiency of transposition into GC rich sequences (Herron et al., 2004). An analysis of the 170–220 region reveals that it has a high GC content compared to the average GC content of E2, while an analysis of the nucleotide composition in regions with a high incidence of insertions (such as the region from 103–113) reveals that they are relatively AT rich. Thus, this may explain the lack of transposon insertions in this region.
Two independent insertions at the same residue may express different ORFs and thus, exhibit different phenotypes based on the nature of the sequence. Two independent insertions at E2-86 (E2-86:1 and E2-86:2) had the most dramatic impact. E2-86:1 expressed ORF2 and gave rise to a viable mutant (SP) and E2-86:2 expressed ORF3 which resulted in a lethal phenotype (Table 2). This indicates that insertion mutations, although they may indicate regions which can or cannot tolerate insertions, are not necessarily a good indicator of how well a heterologous sequence will be tolerated at the same location.
Functional characterization of E2 based on transposon linker insertion mutagenesis
The ability of E2 to readily tolerate insertions in spite of its multiple roles in the SINV life cycle is quite remarkable. These roles include its requirement along with E1 to drive virus assembly and budding, its role in receptor binding, and the conformational changes necessary in the fusion process. Despite these important functional roles, E2 is the least conserved of the alphavirus structural proteins (Strauss and Strauss, 1994). An analysis of E2 protein expression in the transposon mutants by Western immunoblot revealed that only three insertions completely abolished E2 expression, (insertions at amino acids E2-35, E2-103 and E2-324). Other insertions surrounding amino acid 35 (E2-33 and E2-53) also resulted in dramatically reduced E2 expression (Table 2). This suggests that the region encompassing amino acids 33–53 is important for proper protein folding and stability. The fact that different ORFs resulted in the same phenotype, indicates that it is the insertions in general that are destabilizing the protein and not any one sequence in particular. An analysis of the secondary structure prediction for E2 suggests that this region comprises a helix, loop and an extended β-sheet structure (data not shown) (PredictProtein, Rost et al., 2003).
Lethal insertions were located between residues 33–113 at the N-terminus and 311–357, close to the membrane-spanning region of E2. Some of the lethal phenotypes can be explained by protein folding problems as explained above. However, a group of 4 insertions between residues 107–119 had close to wild-type levels of E2 expression, but had a lethal phenotype. FACS analysis revealed that there was very little E2 at the cell surface. The E2 signal for these mutants was 3- to 6-fold reduced compared to that of wild type (Table 2). Thus, insertions in the 107–119 region impair proper E2 transport to the plasma membrane. However, an insertion at E2-105 exhibited wild-type levels of E2 at the cell surface, while an insertion at 103 abolished E2 protein expression. A previous linker insertion mutagenesis study using the RVFV 4D4 epitope (London et al., 1992) identified an insertion at residue 88 that was lethal. In that study, the authors were unable to detect any protein synthesis. We have recovered an insertion at E2-90 that has wild-type levels of protein expression, but impaired transport.
The insertions that produced viable mutants were distributed throughout the E2 glycoprotein, with a high density being found between residues 1–30 and 329–381. The replication phenotypes of the transposon mutants were independently verified by plaque size and one-step growth analyses (Fig. 3). The growth kinetics and plaque phenotypes were found to be consistent for a representative cross section of the transposon insertion library (see Fig. 3A, 3B and 3C). Three of the insertions that gave rise to viruses with large plaque phenotypes, were located towards the N-terminal half of E2, at amino acid position 28, 29 and 153. E2-28 and E2-29 are located at the beginning of a predicted helix, whereas E2-153 is located in a predicted random coil (data not shown). These insertions are of interest, because they identify potential sites for the insertion of targeting ligands. The insertions at position 28 and 29 are most probably surface-accessible based on the location of the N-terminus of E2 and residue 45 (Mukhopadhyay et al., 2006; Paredes et al., 1998). Based on the position of residue 160 as previously reported, the insertion at 153 would also be surface accessible (Mukhopadhyay et al., 2006). Western blot analysis and FACS analysis have shown that these mutants exhibit wild-type levels of E2 expression and transport to the cell surface (Table 2). One-step growth analysis reveals that the growth kinetics of these transposon insertion mutants are similar to wild-type SINV (Fig. 3A).
The other three mutants with large plaque phenotypes are located at residues 326, 338 and 370 and, therefore, unlikely to be surface-accessible. The site at 370 is in the middle of the transmembrane region of the glycoprotein. In order for the 26 amino acid TM region to tolerate a 19 amino acid insertion, the E2 protein needs to shift the extra amino acids to either side of the membrane. Due to space constraints within the virion, and the need for the cytoplasmic domain of E2 to interact with the capsid protein, it is more likely that the bulk of these extra amino acids would end up on the ectodomain of the virus (Hernandez et al., 2005). In fact, the predicted location of the transmembrane helix by several secondary structure prediction algorithms supports this conclusion (data not shown) (PredictProtein, Rost et al., 2003).
The insertion at residue 202 is in a highly antigenic region of E2. Many escape mutants to neutralizing MAbs of alphaviruses have mapped to the amino acid residues between 170 and 220, implicating them in receptor binding (Davis et al., 1987; Meyer and Johnston, 1993; Smith et al., 1995; Strauss et al., 1991). Therefore, the small plaque phenotype of the mutant at 202 may be explained by disruptions in receptor binding as a result of the insertion. To determine if any of the insertions may have affected cell receptor binding, we assayed the ability of SINV neutralizing MAbs to neutralize the insertion mutants. We chose mutants that had medium or small plaque phenotypes and expressed wild-type levels of E2. The neutralization assays show that some of the insertions did prevent virus neutralization by MAb 202 and 209 (Fig. 5 and Table 3). The insertions that were able to escape or partially escape the neutralizing effects of the MAbs may also have impaired ability to interact with cellular receptors and this may partly explain their medium or small plaque phenotypes. These insertions could be of great value in the design of a SINV gene therapy vector. Insertions into this region may simultaneously allow directed targeting of the virus as well as partial ablation of native receptor-binding ability. Previously Dubuisson and Rice (1993) demonstrated that a small insertion (15 amino acids) between E2 amino acids 69 and 74 dramatically reduced receptor binding. However, insertion of large targeting ligands at this location adversely affected virus assembly and resulted in a reduction in viral titers (Ohno et al., 1997; Sawai, 1998). The insertions mentioned above represent alternate sites for the incorporation of targeting ligands in to E2.
Several insertion mutants exhibited close to wild-type levels of E2 protein expression and sufficient transport to the plasma membrane, yet had lethal phenotypes. The levels of E2 transport to the cell membrane in these mutants were higher than some of the viable mutants. This suggests that lower levels of E2 may not be the only reason for the lethal phenotype of these mutants. Such mutants may have problems with assembly, fusion or receptor binding. If the mutants had problems with fusion or receptor binding, but not assembly, then one would expect to see the production of noninfectious virions. An ELISA–based detection system was used to determine if any of the mutants with lethal phenotypes were producing noninfectious particles. The results indicate that E2-7 could be producing noninfectious particles. However, attempts to purify the noninfectious virions were not successful (data not shown).
The linker insertion mutagenesis study by London et al. (1992) recovered 10 insertions near the N-terminus of E2 and 16 insertions near residue 240. The researchers determined that a majority of the insertions near the N-terminus produced the E2 glycoprotein and they further demonstrated that an insertion at E2-3 was viable and in fact exhibited wild-type like growth properties. This is consistent with our results, where the N-terminus was shown to tolerate 19 amino acid insertions (Fig. 2). London and colleagues also determined that an insertion at E2-244 was viable. Our results show that insertions at E2-242 and E2-249 are also viable in agreement with these previous results.
Figure 6 summarizes the functional regions of E2 based on the characterization of the transposon linker-insertion library. The N-terminus of E2 (amino acids 1–30) is amenable to insertion of the transposon and is involved in receptor binding. Residues 33–53 comprise an important secondary structural element as transposon insertions in this region eliminate E2 protein production. Amino acid residues 86–119 are also important for protein stability as insertions in this region are lethal, and moreover, this region is predicted to be part of an ordered structure (FoldIndex, Prilusky et al., 2005; data not shown). This conclusion is further supported by the results of London et al (1992) that demonstrated that an insertion near residue 88 was defective in protein production. Insertions at certain residues within 86–119 (amino acids 86, 90 and 107–113) have greatly reduced protein expression at the plasma membrane, suggesting a possible role in protein transport for this region. However, since this region lies within the lumen of the ER it is unlikely to directly interact with the transport machinery of the cell. Therefore it is likely that insertions in this region are impacting protein folding, thus triggering the ER quality control machinery to block the transport of the glycoprotein to the plasma membrane. The region between amino acids 120 and 161 is amenable to insertion of the transposon and is also implicated in receptor binding. The transposon insertion at E2-202 impaired receptor binding, which is consistent with its role as the receptor binding region of E2. Finally, insertions adjacent to and within the membrane-spanning region impaired the transport of E2 to the plasma membrane. Insertions in this region may disrupt E1-E2 and E2-E2 interactions thus affecting transport. Insertions in the membrane-spanning region may destabilize the protein within the membrane, thereby disrupting transport.
Figure 6.

Functional regions of SINV E2. The functional regions of E2 based on the characterization of the E2 transposon linker-insertion library have been mapped onto a schematic of SINV E2. The color scheme represents the plaque phenotype of the insertion mutants. Blue represents a wild type-like phenotype (large plaque), green represents a medium plaque phenotype, brown represents a small plaque phenotype and red indicates that the insertions were lethal.
The E2 transposon insertion library, in addition to revealing the function of different regions of E2, has also helped identify several regions within the protein which can tolerate insertions without adverse effects on virus growth. Some of these sites (E2-28, E2-29 and E2-153) are predicted to be surface accessible and represent potential sites for the insertion of targeting ligands. The ability to insert a targeting ligand without perturbing virus growth is significant as it will allow the generation of the high titers that are required for gene-therapy applications.
Materials and Methods
Viruses and cells
All viruses were grown at 37ºC in BHK-15 cells propagated in Eagle minimal essential medium (MEM) (Invitrogen) supplemented with 7.5% fetal bovine serum (FBS), unless otherwise noted. All E2 transposon insertion mutations were constructed from pToto64, a full-length cDNA clone of SINV that has been previously described (Owen and Kuhn, 1996). All cells were grown at 37ºC in the presence of 5% CO2.
Random transposon linker-insertion mutagenesis
A fragment of the SINV cDNA clone, pToto64 (the BstEII-BssHII fragment, nt 7472–9804), encoding the capsid protein, E3 and the first 407 amino acids of E2, was subcloned into pJRtac99 (CamR) (American Type Culture Collection) to generate pJRtac99-E2 (Fig. 1). This plasmid was then used in an in vitro transposon linker insertion reaction according to the manufacturer’s instructions (EZ::TN In-Frame Linker Insertion Kit, Epicentre Technologies, Madison, WI). This resulted in the insertion of the ~1100 base pair (bp) EZ::TN <NotI/Kan-3> transposon randomly into the plasmid to generate pJRtac99-E2/TnKan. Plasmids containing the transposon were selected by plating on media containing both kanamycin (50 μg/ml) and chloramphenicol (25 μg/ml). The resulting colonies were individually grown in 3 ml of Luria-Bertani (LB) medium supplemented with both chloramphenicol (50 μg/ml) and kanamycin (25 μg/ml). pJRtac99-E2/TnKan DNA was extracted from these cultures with the Qiaprep Spin Miniprep Kit (Qiagen) according to the manufacturer’s instructions. In order to identify and isolate transposition events that occurred within the SINV cDNA sequence, the resulting plasmids were digested with BstEII and BssHII. Restriction-digested DNA was analyzed by gel electrophoresis, and the BstEII-BssHII fragments containing transposon inserts were identified by their increase in size by 1100 bp. Fragments with transposon inserts were gel purified using the GFX Kit (Amersham Biosciences) for DNA purification according to the manufacturer’s instructions. The BstEII-BssHII fragments were then cloned into the SINV cDNA by ligation with a BstEII-BssHII digested pToto64 plasmid to generate pToto64-TnKan. The pToto64-TnKan plasmids were further restriction digested with NotI in order to eliminate the KanR gene and the resulting plasmid self-ligated to regenerate the full-length SINV cDNA, pToto64Tn, now containing a 19 amino acid insertion in the E2 coding sequence. The resulting pToto64Tn plasmids were sequenced with primers flanking the E2 coding sequence to determine the location of the transposon inserts.
High-throughput screen to identify insertions between E2 residues 170-220
KanR/CamR colonies were picked using the QPix2 (Genetix) automated colony picker and placed in 200 μl of LB broth (34 μg/ml Kan, 25 μg/ml Cam) in 96-well plates. Five 96-well plates were generated in this manner and incubated at 37°C for 5 hours. 5 μl of this culture was used to set up colony PCR reactions in a total volume of 25 μl in 96-well plates. The primers used specifically amplified the sequence between E2 residues 160–230. If there was a transposon insertion within this region the size of the PCR product would have been ~1300 bp vs. ~200 bp if there was no insertion. The PCR products were analyzed on 96-well precast 1% agarose gels (E-Gels 96, Invitrogen). The BioMek Fx (Beckman Coulter) was used to transfer the PCR products to the gels. In all, 3 x 96-well plate PCR reactions were screened.
Construction of an insertion at E2 residue 202
Since we were unable to isolate any insertions in the 170–220 region, the 15 amino acids of the transposon insertion sequence was inserted at E2 residue 202 by an overlap PCR technique. The insertion was created in pJRtac99-E2 using a complimentary pair of mutagenic oligonucleotide primers and two outside primers to generate pJRtac99-E2/202Tn. This plasmid was digested with BstEII and BssHII and the SINV cDNA fragment was inserted into a similarly digested pToto64 vector to generate pToto64-Tn202.
In vitro transcription and transfection of E2 transposon mutants and rescue of mutant viruses
Following sequencing to determine the location of the transposon insertions, the full-length SINV derived cDNA clones were linearized with SacI and in vitro transcribed with SP6 RNA polymerase (Amersham Biosciences, Piscataway, NJ). For electroporation of BHK-15 cells, subconfluent monolayers of cells grown in T-75 culture flasks (~1.5 × 107 cells) were harvested by trypsinization and washed twice with phosphate-buffered saline (PBS) before final suspension in 400 μl of PBS. The resulting cells were combined with the in vitro transcribed RNA, placed in a 2-mm gap cuvette (BioRad, Hercules, CA), and electroporated (two pulses at settings of 1.5 kV, 25 μF and 200 ) using a GenePulser II electroporation apparatus (BioRad). Following a 5-min recovery at room temperature, cells were suspended in MEM supplemented with 10% FBS. The media was harvested 48 hours post-electroporation and assayed for the presence of infectious virus using a standard plaque assay. Viable E2 transposon insertions were plaque purified to generate viral stocks. Plaque phenotypes were determined 48 hours post-infection by comparing Toto64-Tn plaques to wild-type Toto64 plaques.
Western blot analysis
The expression of E2 in lysates of BHK-15 cells electroporated with Toto64-Tn RNA was analyzed by Western immunoblots using a polyclonal anti-E2 antibody specific for the cytoplasmic domain of E2 (a generous gift of M. Schlesinger). The expression of the capsid protein was also analyzed as an internal control with a polyclonal anti-capsid protein antibody. BHK-15 cells were harvested 24 hours post-electroporation and cytoplasmic extracts generated. Samples were run under denaturing conditions in 13% Bis-Tris acrylamide gels. Western immunoblots were performed using the ECL Western Blot Detection System (Amersham Biosciences).
Immunofluorescence Assay
Immunofluorescence assay (IFA) was performed on BHK cells grown on glass coverslips. Cells were washed with PBS and subsequently treated with methanol for 15 minutes at room temperature. Following fixation, cells were washed with PBS followed by a 5 min incubation in PBS with 10 mg/ml BSA. Methanol-treated cells were then subjected to adsorption with primary antibody (anti-E2 or anti-capsid protein) for 45 min at 37°C. Following primary antibody incubation, cells were washed with PBS and subsequently incubated for 45 minutes at 37°C with a fluorescein-conjugated goat anti-rabbit antibody in PBS (10 mg/ml BSA). Following secondary antibody adsorption, cells were washed with PBS and finally with distilled water. Coverslips were then placed on microscope slides, cell side down, in the presence of Fluorosave reagent (Gibco). Fluorescent images were visualized using an inverted epi-fluorescent microscope equipped with a digital CCD camera.
One-step growth analysis
BHK cells were infected with the indicated virus at a multiplicity of infection of 1. Media over the cells was replaced every 30 minutes for the first two hours and then every hour for 12 hours post-infection. Supernatant was collected at the indicated times and assayed for released virus by titration on BHK cell monolayers.
Flow Cytometry
For E2 transposon-insertion mutants that formed viable virus, BHK-15 cells (~2 x 106) were infected at a multiplicity of infection of 1. The cells were harvested 12 hours post-infection by treating with trypsin-EDTA for 30 seconds and then suspending the cells gently in MEM supplemented with 10% FBS. The cells were transferred to an eppendorf tube and washed twice with PBS supplemented with 1% FBS. The cells were incubated on ice for 1 hour with a 1/100 dilution of a polyclonal antibody against the ectodomain of E2 (a generous gift from J.H. Strauss). The cells were washed three time with PBS (1% FBS) and then incubated on ice in the dark for 30 minutes with a fluorescein-conjugated goat anti-rabbit secondary antibody. The cells were washed three times with PBS (1% FBS) and suspended in 500 μl of PBS. The cells were analyzed using a FACSCalibure flow cytometer (BD Biosciences, CA). Control staining was performed with uninfected cells or by omitting the primary anti-E2 antibody. Results are expressed as a plot of frequency versus log fluorescence or the geometric mean. For insertion mutants of E2 that were lethal, ~10 μg of in vitro transcribed RNA were electroporated into BHK-15 cells as described previously. Cells were harvested and stained as described above.
Virus Neutralization Assay
To examine the effect of transposon linker insertions in E2 on the ability of E2 neutralizing antibodies to neutralize SINV infections, 100 plaque-forming units (pfu) of the E2 insertion mutants were incubated with 10-fold dilutions of the neutralizing antibody and rabbit complement for 1 hour. The different virus-antibody complexes were adsorbed to cells for 1 hour at room temperature and then overlaid with 1% agarose in 1x MEM, supplemented with 1% FBS. Plaques were stained (4% Neutral Red in PBS) and counted 48 hours post-infection.
Transmission electron microscopy
BHK-15 cells were electroporated with Toto64-Tn or Toto64 in vitro transcribed RNA as described previously. The cells were fixed (2% glutaldehyde and reduced osmium) 12 hours post-electroporation and then embedded in 1.5% agarose. The samples were diced and dehydrated for transmission electron microscopy.
ELISA
BHK-15 cells were electroporated with Toto64-Tn or Toto64 in vitro transcribed RNA as described previously. The media was harvested 24 hours post-electroporation and concentrated with a 100,000 MWCO concentrator (Microcon). The concentrated media (50 μl) was applied to a 96-well Polysorp ELISA plate overnight at 4ºC. The samples were blocked with 5% BSA in PBS for 2 hours followed by three washes with PBS (0.1 % Tween 20). A 1/100 dilution of a polyclonal anti-E2 antibody was applied to the sample for 1.5 hours, followed by three washes with PBS (0.1% Tween 20). A 1/1000 dilution of a HRP-conjugated goat anti-rabbit secondary antibody was applied to the sample for 1 hour, followed by three PBS (0.1% Tween 20) washes. The TMB Peroxidase Detection system was used to develop the ELISA according to the manufacturer’s instructions. The optical density (OD) was determined at 450 nm. Uninfected cells were used as a negative control and cells electroporated with Toto64 in vitro transcribed RNA were used as a positive control.
Acknowledgments
We thank Dr. M. J. Schlesinger for providing the anti-E2 cytoplasmic domain antibodies. We also acknowledge Dr. J. H. Strauss for supplying polyclonal anti-E2 antibodies. The authors are grateful to Dr. R. Perera for helpful comments on the manuscript. This research was supported by Public Health Service Grants GM56279 and AI55672 from the National Institutes of Health, and funds from the Indiana Elks Charities Inc. provided by the Purdue University Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cheng RH, Kuhn RJ, Olson NH, Rossmann MG, Choi HK, Smith TJH-K, Baker TSH-K. Nucleocapsid and glycoprotein organization in an enveloped virus. Cell. 1995;80:621–630. doi: 10.1016/0092-8674(95)90516-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikkanna-Gowda CP, Sheahan BJ, Fleeton MN, Atkins GJ. Regression of mouse tumours and inhibition of metastases following administration of a Semliki Forest virus vector with enhanced expression of IL-12. Gene Ther. 2005;12(16):1253–63. doi: 10.1038/sj.gt.3302561. [DOI] [PubMed] [Google Scholar]
- Choi HK, Tong L, Minor W, Dumas P, Boege U, Rossmann MG, Wengler G. Structure of Sindbis virus core protein reveals a chymotrypsin-like serine proteinase and the organization of the virion. Nature. 1991;354:37–43. doi: 10.1038/354037a0. [DOI] [PubMed] [Google Scholar]
- Davis NL, Pence DF, Meyer WJ, Schmaljohn AL, Johnston RE. Alternative forms of a strain-specific neutralizing antigenic site on the Sindbis virus E2 glycoprotein. Virology. 1987;161(1):101–8. doi: 10.1016/0042-6822(87)90175-9. [DOI] [PubMed] [Google Scholar]
- Dubuisson J, Rice CM. Sindbis virus attachment: isolation and characterization of mutants with impaired binding to vertebrate cells. J Virol. 1993;67:3363–3374. doi: 10.1128/jvi.67.6.3363-3374.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaedigk-Nitschko K, Schlesinger MJ. The Sindbis virus 6K protein can be detected in virions and is acylated with fatty acids. Virology. 1990;175:274–281. doi: 10.1016/0042-6822(90)90209-a. [DOI] [PubMed] [Google Scholar]
- Goryshin IY, Reznikoff WS. Tn5 in vitro transposition. J Biol Chem. 1998;273(13):7367–74. doi: 10.1074/jbc.273.13.7367. [DOI] [PubMed] [Google Scholar]
- Hernandez R, Ferreira D, Sinodis C, Litton K, Brown DT. Single amino acid insertions at the junction of the sindbis virus E2 transmembrane domain and endodomain disrupt virus envelopment and alter infectivity. J Virol. 2005;79(12):7682–97. doi: 10.1128/JVI.79.12.7682-7697.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron PR, Hughes G, Chandra G, Fielding S, Dyson PJ. Transposon Express, a software application to report the identity of insertions obtained by comprehensive transposon mutagenesis of sequenced genomes: analysis of the preference for in vitro Tn5 transposition into GC-rich DNA. Nucleic Acids Res. 2004;32(14):e113. doi: 10.1093/nar/gnh112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Linn M, Gardner J, Warrilow D, Darnell GA, McMahon CR, Field I, Hyatt AD, Slade RW, Suhrbier A. Arbovirus of marine mammals: a new alphavirus isolated from the elephant seal louse, Lepidophthirus macrorhini. J Virol. 2001;75(9):4103–9. doi: 10.1128/JVI.75.9.4103-4109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Owen KE, Choi HK, Lee H, Lu G, Wengler G, Brown DT, Rossmann MG, Kuhn RJ. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure. 1996;4:531–541. doi: 10.1016/s0969-2126(96)00059-7. [DOI] [PubMed] [Google Scholar]
- Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Rey FA. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell. 2001;105:137–148. doi: 10.1016/s0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- London SD, Schmauohn AL, Dalrymple JM, Rice CM. Infectious enveloped RNA virus antigenic chimeras. Proc Natl Acad Sci USA. 1992;89:207–211. doi: 10.1073/pnas.89.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusa S, Garoff H, Liljestrom P. Fate of the 6K membrane protein of Semliki Forest virus during virus assembly. Virology. 1991;185:843–846. doi: 10.1016/0042-6822(91)90556-q. [DOI] [PubMed] [Google Scholar]
- Mancini EJ, Clarke M, Gowen BE, Rutten T, Fuller SD. Cryo-electron microscopy reveals the functional organization of an enveloped virus, Semliki Forest virus. Mol Cell. 2000;5:255–266. doi: 10.1016/s1097-2765(00)80421-9. [DOI] [PubMed] [Google Scholar]
- Mayne JT, Bell JR, Strauss EG, Strauss JH. Pattern of glycosylation of Sindbis virus envelope proteins synthesized in hamster and chicken cells. Virology. 1985;142:121–133. doi: 10.1016/0042-6822(85)90427-1. [DOI] [PubMed] [Google Scholar]
- Melancon P, Garoff H. Processing of the Semliki Forest virus structural polyprotein: role of the capsid protease. J Virol. 1987;61:1301–1309. doi: 10.1128/jvi.61.5.1301-1309.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer WJ, Johnston RE. Structural rearrangement of infecting Sindbis virions at the cell surface: mapping of newly accessible epitopes. J Virol. 1993;67(9):5117–25. doi: 10.1128/jvi.67.9.5117-5125.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Zhang W, Gabler S, Chipman PR, Strauss EG, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. Structure. 2006;14:63–73. doi: 10.1016/j.str.2005.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Sawai K, Ijima Y, Levin B, Meruelo D. Cell-specific targeting of Sindbis virus vectors displaying IgG-binding domains of protein A. Nature Biotechnology. 1997;15:763–767. doi: 10.1038/nbt0897-763. [DOI] [PubMed] [Google Scholar]
- Owen KE, Kuhn RJ. Identification of a region in the Sindbis virus nucleocapsid protein that is involved in specificity of RNA encapsidation. J Virol. 1996;70(5):2757–63. doi: 10.1128/jvi.70.5.2757-2763.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes AM, Brown DT, Rothnagel R, Chiu W, Schoepp RJ, Johnston RE, Prasad BV. Three-dimensional structure of a membrane-containing virus. Proc Natl Acad Sci USA. 1993;90:9095–9099. doi: 10.1073/pnas.90.19.9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes AM, Heidner H, Thuman-Commike PV, Venkatarm P, Johnston RE, Chiu W. Structural localization of the E3 glycoprotein in attenuated Sindbis virus mutants. J Vriol. 1998;72:1534–1541. doi: 10.1128/jvi.72.2.1534-1541.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pence DF, Davis NL, Johnston RE. Antigenic and genetic characterization of Sindbis virus monoclonal antibody escape mutants which define a pathogenesis domain on glycoprotein E2. Virology. 1990;175(1):41–9. doi: 10.1016/0042-6822(90)90184-s. [DOI] [PubMed] [Google Scholar]
- Pletnev SV, Zhang W, Mukhopadhyay S, Fisher BR, Hernandez R, Brown DT, Baker TS, Rossmann MG, Kuhn RJ. Locations of carbohydrate sites on alphavirus glycoproteins show that E1 forms an icosahedral scaffold. Cell. 2001;105:127–136. doi: 10.1016/s0092-8674(01)00302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21(16):3435–8. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- Rost B, Yachdav G, Liu J. The PredictProtein Server. Nucleic Acids Research. 2003;32 (Web Server issue):W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai K, Meruelo D. Cell-Specific Transfection of Choriocarcinoma Cells by using Sindbis virus hCG expressing chimeric vector. Biochemical and Biophysical Research Communications. 1998;248:315–323. doi: 10.1006/bbrc.1998.8922. [DOI] [PubMed] [Google Scholar]
- Schlesinger S, Dubensky TW. Alphavirus vectors for gene expression and vaccines. Curr Opin Biotechnol. 1999;10(5):434–9. doi: 10.1016/s0958-1669(99)00006-3. [DOI] [PubMed] [Google Scholar]
- Schmidt MF, Bracha M, Schlesinger MJ. Evidence for covalent attachment of fatty acids to Sindbis virus glycoproteins. Proc Natl Acad Sci U S A. 1979;76(4):1687–91. doi: 10.1073/pnas.76.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TJ, Cheng RH, Olson NH, Peterson P, Chase E, Kuhn RJ, Baker TS. Putative receptor binding sites on alphaviruses as visualized by cryoelectron microscopy. Proc Natl Acad Sci USA. 1995;92(23):10648–52. doi: 10.1073/pnas.92.23.10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss EG, Stec DS, Schmaljohn AL, Strauss JH. Identification of antigenically important domains in the glycoproteins of Sindbis virus by analysis of antibody escape variants. J Virol. 1991;65(9):4654–64. doi: 10.1128/jvi.65.9.4654-4664.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev. 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubol S, Griffin D. Identification of a Putative Alphavirus Receptor on Mouse Neural Cells. J Vriol. 1991;65:6913–6921. doi: 10.1128/jvi.65.12.6913-6921.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bonsdorff CH, Harrison SC. Sindbis virus glycoproteins form a regular icosahedral surface lattice. J Virol. 1975;16:141–145. doi: 10.1128/jvi.16.1.141-145.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bonsdorff CH, Harrison SC. Hexagonal glycoprotein arrays from Sindbis virus membranes. J Virol. 1978;28:578–583. doi: 10.1128/jvi.28.2.578-583.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KS, Schmaljohn AL, Kuhn RJ, Strauss JH. Antiidiotypic antibodies as probes for the Sindbis virus receptor. Virology. 1991;181(2):694–702. doi: 10.1016/0042-6822(91)90903-o. [DOI] [PubMed] [Google Scholar]
- Wirth DF, Katz F, Small B, Lodish HF. How a single Sindbis virus mRNA directs the synthesis of one soluble protein and two integral membrane glycoproteins. Cell. 1977;10:253–263. doi: 10.1016/0092-8674(77)90219-7. [DOI] [PubMed] [Google Scholar]
- Xiong C, Levis R, Shen P, Schlesinger S, Rice CM, Huang HV. Sindbis virus: an efficient, broad host range vector for gene expression in animal cells. Science. 1989;243(4895):1188–91. doi: 10.1126/science.2922607. [DOI] [PubMed] [Google Scholar]
- Zhang W, Mukhopadhayay S, Pletnev SV, Baker TS, Kuhn RJ, Rossmann MG. Placement of the structural proteins in Sindbis virus. J Virol. 2002;76:11645–11658. doi: 10.1128/JVI.76.22.11645-11658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]