

Abstract
Alamethicin is a 19-residue hydrophobic peptide, which is extended by a C-terminal phenylalaninol but lacks residues that might anchor the ends of the peptide at the lipid-water interface. Voltage-dependent ion channels formed by alamethicin depend strongly in their characteristics on chain length of the host lipid membranes. EPR spectroscopy is used to investigate the dependence on lipid chain length of the incorporation of spin-labeled alamethicin in phosphatidylcholine bilayer membranes. The spin-label amino acid TOAC is substituted at residue positions n = 1, 8, or 16 in the sequence of alamethicin F50/5 [TOACn, Glu(OMe)7,18,19]. Polarity-dependent isotropic hyperfine couplings of the three TOAC derivatives indicate that alamethicin assumes approximately the same location, relative to the membrane midplane, in fluid diCNPtdCho bilayers with chain lengths ranging from N = 10–18. Residue TOAC8 is situated closest to the bilayer midplane, whereas TOAC16 is located farther from the midplane in the hydrophobic core of the opposing lipid leaflet, and TOAC1 remains in the lipid polar headgroup region. Orientational order parameters indicate that the tilt of alamethicin relative to the membrane normal is relatively small, even at high temperatures in the fluid phase, and increases rather slowly with decreasing chain length (from 13° to 23° for N = 18 and 10, respectively, at 75°C). This is insufficient for alamethicin to achieve hydrophobic matching. Alamethicin differs in its mode of incorporation from other helical peptides for which transmembrane orientation has been determined as a function of lipid chain length.
INTRODUCTION
Alamethicin is a predominantly α-helical, 19-amino acid residue peptide from Trichoderma viride, which has N-terminal and C-terminal residues that are blocked by an acetyl and a phenylalaninol group, respectively (1,2). This hydrophobic peptide is able to induce voltage-activated ion conduction across lipid membranes, the channel characteristics of which depend strongly on lipid chain length (3). The slope of the I/V curves and the effective number of monomers per alamethicin channel increase steeply with increasing chain length of the lipids that make up the host bilayer membrane. Spectroscopic studies reveal that the peptide is monomeric in fluid phospholipid membranes in the absence of a membrane potential (4,5), and electrophysiological experiments suggest that consecutive channel conductance levels are generated by incorporation of alamethicin monomers into existing pore aggregates (6).
Studies on the effects of hydrophobic matching between transmembrane peptides/proteins and the membrane lipids have revealed that, whereas single bitopic helices tend to tilt or oligomerize in response to a positive mismatch, larger integral proteins tend to respond by inducing distortions in conformation of the surrounding lipids (7,8). A negative mismatch, on the other hand, would tend to induce lipid distortions in both cases. In addition, it has been found that the indole side chains of interfacial tryptophan residues are able to adapt their orientation such as to compensate, at least partially, for hydrophobic mismatch with the adjacent lipids (9,10).
Alamethicin, although hydrophobic, is devoid of tryptophan residues and the length of the peptide, even for a straight helix, is on the border of being sufficient to span the hydrophobic thickness of a typical membrane bilayer ((4,11) and http://mosberglab.phar.umich.edu/projects/proj11.php). The orientation and/or aggregation state of alamethicin may therefore depend strongly on the chain length of the host lipid. Recently, we have shown that EPR of TOAC spin labels incorporated in the peptide backbone can be used to determine the orientation and mode of incorporation of alamethicin analogs in lipid membranes (5). Here we use this method to investigate the dependence on lipid chain length by using diacyl phosphatidylcholines with chain lengths from C10 to C18, the fluid-state bilayers of which encompass the likely hydrophobic thicknesses of natural membranes (12).
MATERIALS AND METHODS
Materials
Synthetic phosphatidylcholines with symmetrical saturated chains (diC10PtdCho, diC12PtdCho, diC14PtdCho, diC16PtdCho, and diC18PtdCho) were obtained from Avanti Polar Lipids (Alabaster, AL). Spin-labeled analogs of alamethicin F50/5 [TOACn, Glu(OMe)7,18,19], with n = 1, 8, and 16, were synthesized according to Peggion et al. (13,14). The complete amino-acid sequences of the three analogs are
Ac-TOAC-Pro-Aib-Ala-Aib-Ala-Glu(OMe)-Aib-Val-Aib-Gly-Leu-Aib-Pro-Val-Aib-Aib-Glu(OMe)-Glu(OMe)-Phol [TOAC1,Glu(OMe)7,18,19]
Ac-Aib-Pro-Aib-Ala-Aib-Ala-Glu(OMe)-TOAC-Val-Aib-Gly-Leu-Aib-Pro-Val-Aib-Aib-Glu(OMe)-Glu(OMe)-Phol [TOAC8,Glu(OMe)7,18,19]
Ac-Aib-Pro-Aib-Ala-Aib-Ala-Glu(OMe)-Aib-Val-Aib-Gly-Leu-Aib-Pro-Val-TOAC-Aib-Glu(OMe)-Glu(OMe)-Phol [TOAC16,Glu(OMe)7,18,19].
In each case, TOAC is substituted for an Aib residue, which is a conservative replacement because both are Cα,α-disubstituted amino acids that favor helix formation, as is confirmed by x-ray diffraction on the [TOAC16, Glu(OMe)7,18,19] alamethicin analog (15). Circular dichroism in MeOH additionally shows that all three analogs are folded in similar, highly helical conformations (16). Electrophysiological experiments demonstrate ion channel activity for these synthetic alamethicin analogs in membranes (15).
Sample preparation
Diacyl phosphatidylcholine, diCNPtdCho (∼1 mg), and 1 mol % of TOAC spin-labeled alamethicin (in MeOH) were codissolved in CH2Cl2, and the solution then evaporated with dry nitrogen. After keeping under vacuum overnight, the dry mixture was hydrated in 50 μl of 10 mM Hepes, 10 mM NaCl, 10 mM EDTA, pH 7.8 buffer, with vortex mixing at a temperature above the chain-melting transition of the phosphatidylcholine. The lipid dispersion was then transferred to a 1-mm-diameter glass capillary and pelleted in a benchtop centrifuge. Excess supernatant was removed and the capillaries were flame sealed.
EPR spectroscopy
EPR spectra were recorded on a Varian (Palo Alto, CA) Century-Line 9-GHz spectrometer with 100-kHz field modulation (50 kHz for second-harmonic detection). Sample capillaries were accommodated in standard quartz EPR tubes that contained light silicone oil for thermal stability. Temperature was regulated by thermostated nitrogen gas flow through a quartz Dewar and was measured with a fine-wire thermocouple situated in the silicone oil at the top of the microwave cavity. Samples of ∼5-mm height were centered in the rectangular TE102 resonator to minimize microwave- and modulation-field inhomogeneities (17). The microwave H1-field at the sample was measured as described in the latter reference. Conventional EPR spectra were recorded in the in-phase first-harmonic absorption mode (V1-display) and ST-EPR spectra in the out-of-phase second-harmonic absorption mode (V2′-display) under standard conditions for microwave and modulation field amplitudes (18).
Conventional EPR spectra were analyzed in terms of the outer and inner hyperfine splittings, 2Amax and 2Amin, respectively. In the motional narrowing regime, at high temperature, Amax is equal to the parallel element, A//, of the partially motionally averaged, axial hyperfine tensor. The perpendicular element, A⊥, is obtained from the separation, 2Amin, of the inner extrema, according to Schorn and Marsh (19):
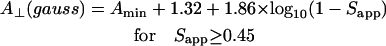 |
(1) |
and
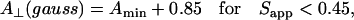 |
(2) |
where 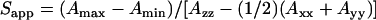 for a spin-label hyperfine tensor with Cartesian elements (Axx,Ayy,Azz). The environmental polarity was characterized by means of the isotropic 14N-hyperfine coupling, ao (20), which is given by
for a spin-label hyperfine tensor with Cartesian elements (Axx,Ayy,Azz). The environmental polarity was characterized by means of the isotropic 14N-hyperfine coupling, ao (20), which is given by
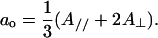 |
(3) |
Near constancy, with respect to temperature, of the value of ao obtained from Eq. 3 was used as a criterion to identify the fast motional regime (5,21,22).
The orientational order parameter of the spin-label z axis, in the fast motional regime, is given by
 |
(4) |
where the final factor on the right is a polarity correction to the hyperfine tensor elements. Values taken for the hyperfine tensor elements are (Axx,Ayy,Azz) = (6.0 G, 7.3 G, 34.5 G) from 2,2,6,6-tetramethylpiperidine-1-oxy in a toluene glass (23). For uniaxial motional averaging, the EPR order parameter of TOAC is given by the addition theorem for spherical harmonics:
 |
(5) |
where γ is the angle that the principal rotational diffusion axis, R, of the TOAC-labeled alamethicin molecule makes with the membrane normal, N, and θz is the inclination of the spin-label z axis to R (Fig. 1). P2(x) = (3x2 − 1)/2 is a second-order Legendre polynomial, and the angular brackets indicate a time average over the rotational motion.
FIGURE 1.

Orientation of TOAC-labeled alamethicin in a lipid membrane. The principal molecular diffusion axis, R, is inclined at instantaneous angle γ to the membrane normal N. The nitroxide z axis is oriented at constant angle θz to R. The experimental order parameter, Szz, of the nitroxide z axis is given by Eq. 5 where, for axial symmetry, 〈P2(cosγ)〉 is the order parameter of the alamethicin diffusion axis.
Saturation transfer EPR spectra were analyzed in terms of the diagnostic line-height ratios, L″/L, C′/C, and H″/H, defined in the low-field, central, and high-field regions of the spectra, respectively (24), and by the normalized integrated intensity, IST (25). Effective rotational correlation times, 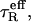 were obtained from ST-EPR line-height ratios, R, by using calibrations with spin-labeled hemoglobin in solutions of known viscosity from Horváth and Marsh (25,26).
were obtained from ST-EPR line-height ratios, R, by using calibrations with spin-labeled hemoglobin in solutions of known viscosity from Horváth and Marsh (25,26).
RESULTS
Conventional spin-label EPR spectra
Fig. 2 shows the EPR spectra of the three different TOAC1, TOAC8, and TOAC16 analogs of [Glu(OMe)7,18,19] alamethicin in fluid-phase bilayer membranes of phosphatidylcholines with different chain lengths. At this relatively high temperature (75°C) in the fluid phase, all spectra are approximately in the fast motional regime, with the possible exception of those of the TOAC16 analog. This conclusion is substantiated by the constancy with temperature of the effective isotropic hyperfine coupling (see later and (5)). Spin-spin broadening is absent from the EPR spectra, which indicates that the spin-labeled alamethicins are dispersed as monomers in the lipid membrane. The spectra from all three TOAC derivatives display axial motional averaging, as indicated by the well-defined outer and inner hyperfine splittings, 2Amax and 2Amin, respectively (27).
FIGURE 2.

Conventional EPR spectra (V1-display) of [Glu(OMe)7,18,19] alamethicin analogs with TOAC substituted for: residue 1, TOAC1; residue 8, TOAC8; or residue 16, TOAC16 in phosphatidylcholine bilayers of chain lengths C10–C18, as indicated, at 75°C. Total scan width = 100 G.
Fig. 3 shows the temperature dependences of the outer hyperfine splitting, 2Amax, for alamethicin with the different positions of TOAC labeling in phosphatidylcholine membranes with different lipid chain lengths, N. The chain-melting transition of diCNPtdCho bilayers takes place at temperatures of −6°C, −2°C, 24°C, 41°C, and 55°C for N = 10, 12, 14, 16, and 18, respectively (12). For the most part, there are small discontinuities in the temperature dependence of Amax at these chain-melting transitions. However, the values of Amax at temperatures above the lipid transition are still indicative of a high degree of order, or limited amplitude of angular motion, of the spin-labeled alamethicin. There are, nonetheless, clear differences between the temperature dependences for the three label positions. These differences are similar in the five host lipids and arise from the different intramolecular orientations, θz, of the TOAC spin-label group. The values of Amax decrease steadily in response to the increased extent of lipid chain motion with increasing temperature, to comparable extents for each lipid host. At a given temperature in the fluid phase, the values of Amax increase with increasing lipid chain length.
FIGURE 3.

Temperature dependences of the outer hyperfine splittings, 2Amax, for [Glu(OMe)7,18,19] alamethicin TOAC1 (squares), TOAC8 (circles), and TOAC16 (triangles) analogs in phosphatidylcholine bilayers of chain lengths C10–C18, as indicated.
Isotropic hyperfine couplings
Fig. 4 gives the temperature dependence of the effective isotropic hyperfine couplings,  defined by Eq. 3, for alamethicin with the three positions of TOAC labeling in diCNPtdCho membranes of different lipid chain lengths, N. With the exception of the diC10PtdCho host lipid, the values of ao for the TOAC1 and TOAC8 analogs are practically constant at temperatures of 50°C–55°C or higher. This finding means that the EPR spectra are in the fast motional regime at these temperatures. Consequently, the values of
defined by Eq. 3, for alamethicin with the three positions of TOAC labeling in diCNPtdCho membranes of different lipid chain lengths, N. With the exception of the diC10PtdCho host lipid, the values of ao for the TOAC1 and TOAC8 analogs are practically constant at temperatures of 50°C–55°C or higher. This finding means that the EPR spectra are in the fast motional regime at these temperatures. Consequently, the values of  derived from Eq. 3 over this temperature range are a true reflection of the polarity of the environment in which the spin label is situated (28,29), uncontaminated by artifactual contributions from slow motion. Similarly for the TOAC16 derivative in membranes of diC16PtdCho and diC18PtdCho, the values of ao tend to a constant limiting value, characteristic of fast motion, but at higher temperatures (above ∼70°C). Only for the TOAC16 derivative in diC12PtdCho and diC14PtdCho, and all derivatives in diC10PtdCho, is the temperature dependence of
derived from Eq. 3 over this temperature range are a true reflection of the polarity of the environment in which the spin label is situated (28,29), uncontaminated by artifactual contributions from slow motion. Similarly for the TOAC16 derivative in membranes of diC16PtdCho and diC18PtdCho, the values of ao tend to a constant limiting value, characteristic of fast motion, but at higher temperatures (above ∼70°C). Only for the TOAC16 derivative in diC12PtdCho and diC14PtdCho, and all derivatives in diC10PtdCho, is the temperature dependence of 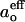 apparently anomalous throughout the entire range of measurement.
apparently anomalous throughout the entire range of measurement.
FIGURE 4.

Temperature dependences of the effective isotropic hyperfine couplings, 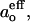 for [Glu(OMe)7,18,19] alamethicin TOAC1 (squares), TOAC8 (circles), and TOAC16 (triangles) analogs in phosphatidylcholine bilayers of chain lengths C10–C18, as indicated.
for [Glu(OMe)7,18,19] alamethicin TOAC1 (squares), TOAC8 (circles), and TOAC16 (triangles) analogs in phosphatidylcholine bilayers of chain lengths C10–C18, as indicated.
The left-hand panel of Fig. 5 shows the dependence on lipid chain length, N, of the isotropic hyperfine coupling increments, Δao, for the different positions of alamethicin TOAC labeling. Because these values are obtained from measurements at high temperature, in the region where ao is approximately constant (Fig. 4), they should depend only on the polarity of the spin-label environment. To correct for intrinsic differences between the three TOAC positions, the values are referred to the isotropic splittings of the respective analogs in methanol. From isotropic spectra at 20°C, the hyperfine splitting constants in MeOH are ao = 15.24 ± 0.03, 15.37 ± 0.02, and 15.11 ± 0.04 G for TOAC1, TOAC8, and TOAC16 alamethicin, respectively.
FIGURE 5.

Left-hand panel: chain-length dependence (N) of the effective isotropic hyperfine couplings, ao, for [Glu(OMe)7,18,19] alamethicin TOAC1 (squares), TOAC8 (circles), and TOAC16 (triangles) analogs in fluid diCNPtdCho bilayers. Right-hand panel: positional dependence (n) of the isotropic hyperfine couplings for nitroxides at position C-n in the sn-2 chain of n-PCSL phosphatidylcholine spin probes in diC14PtdCho (circles) or diC16PtdCho (squares) fluid bilayers (20). Values of ao are given relative to those in methanol: Δao = ao(PtdCho) − ao(MeOH). Horizontal dotted lines are the values of Δao in MeOH and in EtOH, as indicated.
The uncertainty ranges in Δao that are given by the vertical bars in Fig. 5 correspond to the standard deviation over the temperature range chosen for evaluation. For lipid chain lengths N < 16 with the TOAC16 analog, the uncertainty is relatively large because the effective value of ao that is derived from Eq. 3 continues to decrease with increasing temperature up to the highest temperatures of measurement. This result could indicate either that the spectra of this derivative are still not entirely in the fast motional regime at the highest temperatures or that the position of the spin label is changing with temperature. As found already for TOAC analogs of [Glu(OMe)7,18,19] alamethicin in diC14PtdCho bilayers (5), the lower values of Δao for TOAC8 and TOAC16 reveal that these residues are situated deeper in the hydrophobic core of the membrane than is the TOAC1 residue, at least at the high temperatures for which ao can be measured reliably. The corrected values also show that TOAC8 is located in a region of lower polarity than TOAC1 and TOAC16, indicating that the latter two residues are situated in opposite leaflets of the bilayer. For lipid chain lengths N > 10, the values of ao decrease with increasing chain length, corresponding to locations of decreasing polarity for all three residue positions.
Comparable data for Δao of spin-labeled phospholipid chains from Marsh (20) are also included in Fig. 5 (right panel). These will be used in the later Discussion.
Orientational order parameters
Fig. 6 shows the dependence on lipid chain length, N, of the order parameters, Szz, for the three TOAC analogs of alamethicin in diCNPtdCho bilayer membranes at 75°C and at 80°C in the fluid phase. With the possible exception of the TOAC16 analog (see above), the EPR spectra of [TOACn, Glu(OMe)7,18,19] alamethicins should be entirely in the fast motional regime at these elevated temperatures (cf. Fig. 4). The values of the order parameters given in Fig. 6 therefore should be reasonably representative of the time-average angular amplitude of motion of the spin-label z axis, relative to the director for the uniaxial rotational motion.
FIGURE 6.

Chain-length dependences (N) of the effective order parameters, Szz, for [Glu(OMe)7,18,19] alamethicin TOAC1 (squares), TOAC8 (circles), and TOAC16 (triangles) analogs in diCNPtdCho bilayers at 75°C (solid symbols) and 80°C (open symbols).
Fig. 7 (solid lines and symbols) shows the dependence on lipid chain length, N, of the order parameter, 〈P2(cosγ)〉, for the diffusion axis of alamethicin (see Fig. 1) in diCNPtdCho bilayer membranes at 75°C and 80°C in the fluid phase. These values were obtained by least-squares fitting of Eq. 5 to the measurements of Szz for the three TOAC alamethicin analogs in the five different lipid hosts at 75°C and 80°C, with the restriction that the orientation of the TOAC z axis, θz, does not change with temperature or lipid host. The corresponding values of P2(cosθz) for the three TOAC analogs are given by the horizontal dotted lines and open symbols in Fig. 7. Values of the latter are in the same relative order as those obtained previously by fitting a more extensive temperature dependence (60°C–85°C) of Szz for the three TOAC derivatives in a single lipid host (diC14PtdCho): P2(cosθz) = 0.63, 0.74, and 0.82 for TOAC1, TOAC8, and TOAC16 analogs, respectively (5), although the absolute values are somewhat reduced. It is seen from Fig. 7 that the orientational order parameter of alamethicin increases with increasing chain length of the lipid host at fixed temperature within the fluid phase. Also, as expected, the order parameters decrease with increasing temperature in each host lipid.
FIGURE 7.

Solid lines: chain-length dependence (N) of the order parameters, 〈P2(cosγ)〉, for the alamethicin diffusion axis in diCNPtdCho bilayers at 75°C (solid squares) and 80°C (solid circles). Dotted lines: orientation, P2(cosθz), of the nitroxide z axis for TOAC1 (open squares), TOAC8 (open circles), and TOAC16 (open triangles) in [Glu(OMe)7,18,19] alamethicin analogs. Values are determined from nonlinear least squares fitting of Eq. 5 to the chain-length dependence of Szz measured at 75°C and 80°C, with constant θz for each TOAC position (see text and Fig. 1).
Saturation transfer spin-label EPR spectra
Fig. 8 shows the saturation transfer EPR spectra of the TOAC1, TOAC8, and TOAC16 analogs of [Glu(OMe)7,18,19] alamethicin in bilayer membranes of diCNPtdCho with different chain lengths, N. Spectra for diC14, diC16, and diC18PtdCho are shown mostly at temperatures both above (45°C, or 60°C for diC18) and below (5°C) the respective bilayer chain-melting transitions. For diC10 and diC12PtdCho, both low and high temperatures (5°C and 45°C) correspond to the fluid phase of these bilayers. (In a few cases, where the ST-intensity was too low at the higher temperature, the second spectrum is at a somewhat lower temperature.)
FIGURE 8.

Saturation transfer EPR spectra (V2′-display) of [Glu(OMe)7,18,19] alamethicin analogs with TOAC substituted for: residue 1, TOAC1; residue 8, TOAC8; and residue 16, TOAC16 in phosphatidylcholine bilayers of chain lengths C10–C18 at the temperatures indicated. Total scan width = 160 G.
In the gel-phase, all the ST-EPR spectra (for N = 14–18) have appreciable intensity in the diagnostic regions at intermediate positions in the low-, high-, and center-field manifolds of the 14N-hyperfine structure that are sensitive to slow rotational motion. These nonvanishing ST-EPR line heights reflect the extremely slow rotational diffusion of the gel-phase lipids, with effective correlation times beyond the microsecond regime (30,31). At the fixed temperature of 5°C, the ST-EPR spectra indicate increasingly slow motion with increasing chain length, because the bilayers lie deeper in the gel phase as the lipid chain length increases. Above the chain-melting transition, the spectral line shape changes dramatically because of preferential reduction in the line heights at the intermediate diagnostic spectral positions relative to the stationary turning points for each of the three hyperfine manifolds at low-, central-, and high-field, respectively (24). For all chain lengths of the lipid host, the spectra are characteristic of very fast motion, no longer in the microsecond regime, and reflect the rapid lipid chain motions in the fluid membrane phase (30,32). Note that the V2′-EPR line shapes in diC10 and diC12PtdCho are rather similar at 5°C and 45°C because the bilayers are in the fluid phase at both temperatures.
The ST-EPR spectra in Fig. 8 are scaled to line height, rather than to the absolute intensity, which decreases with increasing temperature. Fig. 9 gives the temperature dependence of the normalized ST-intensity, IST, for the TOAC8 alamethicin analog in diCNPtdCho membranes. The ST-intensities are appreciable in the gel phase (IST ∼10−4−10−3) although they display a considerable temperature dependence that demonstrates mobility of the peptide on the microsecond timescale. At the chain-melting transition, the ST-intensities drop to almost zero (IST ∼10−5) for all chain lengths of the lipid host (or throughout the entire temperature range for diC10 and diC12PtdCho). These latter values of IST correspond to effective rotational correlation times of <2.9 × 10−8 s in the fluid phase (26). Such rapid rotational reorientation indicates that the peptide is not aggregated in fluid phosphatidylcholine membranes, even at the extreme chain lengths of N = 10 or 18. This conclusion is in agreement with the lack of spin-spin broadening in the conventional EPR spectra from fluid-phase bilayers.
FIGURE 9.

Temperature dependences of the integrated intensity, IST, from the ST-EPR spectra of the TOAC8 [Glu(OMe)7,18,19] alamethicin analog in diCNPtdCho bilayers with N = 12 (squares), 14 (circles), 16 (triangles), and 18 (diamonds). Data points for N = 10 superimpose on those for N = 12.
DISCUSSION
The central results obtained from this study are the dependences of the isotropic hyperfine coupling, ao, and of the order parameter, 〈P2(cosγ)〉, of the alamethicin diffusion axis on the chain length of the bilayer membrane lipid host (Figs. 5 and 7). Data on overall rotational diffusion from ST-EPR suggest that alamethicin is present predominantly as a monomer in the fluid-phase membranes at all lipid chain lengths studied.
Vertical positioning from ao
The location of the different TOAC residues in the bilayer membrane can be determined by comparing the isotropic hyperfine couplings with those determined for phosphatidylcholines (n-PCSL) that are spin labeled at different positions in the sn-2 lipid chain (20). For comparison with the TOAC-values that are given in the left-hand panel of Fig. 5, the incremental isotropic hyperfine couplings of the n-PCSL spin labels in fluid diC14PtdCho and diC16PtdCho bilayers are shown in the right-hand panel of the figure. These vary from a high plateau value of Δao = 0.05–0.1 G for n = 4−6 toward the polar-apolar interface to a low plateau value of Δao = −0.45 to −0.55 G for n = 10−16 toward the middle of the membrane. These values, like those for TOACn, are expressed relative to ao in methanol, i.e., Δao = ao(PC) − ao(MeOH), to correct for the intrinsic difference between the isotropic couplings of doxyl nitroxides (in n-PCSL) and TOAC nitroxides.
For all three TOAC positions, the effective isotropic hyperfine splitting constants decrease with increasing lipid chain length, from N = 14 onward. This result implies that alamethicin is anchored in the hydrophobic core of the membrane, rather than at some specific interfacial location of an anchoring residue that is fixed at one end of the peptide. In diC12PtdCho and diC14PtdCho, the TOAC1 residue of alamethicin is situated in a more polar location than the 4-C atom of the phosphatidylcholine sn-2 chain. However, the TOAC1 residue is by no means fully exposed to water in these lipids because the isotropic hyperfine coupling increment in that case would be much higher (Δao = 1.0−1.3 G, depending on the pH/charge state of the peptide) (33). For longer chain lengths, TOAC1 is located at the polar-apolar interface or in the plateau region of the 4-C atom. In lipids with chain lengths N = 10−14, the TOAC8 residue of alamethicin is located at a depth similar to that of the 8–9 C-atom of the lipid chains. In lipids with longer chain lengths, the TOAC8 residue is situated toward the center of the bilayer, in the region of the 10 C-atom of the lipid chains or beyond. The TOAC16 residue of alamethicin is located close to the 8 C-atom of the lipid chains in diC16PtdCho and diC18PtdCho, and in the region of the 7 C-atom or higher in diC14PtdCho. Clearly, TOAC16 resides in the opposite bilayer leaflet from that of TOAC8 and TOAC1, and is closer to the polar-apolar interface than is TOAC8, which lies closer to the bilayer midplane. Note that x-ray diffraction studies exclude the possibility of extensively bent or looped structures for the peptide backbone. In [TOAC16, Glu(OMe)7,18,19] alamethicin, replacement of Aib by TOAC does not significantly affect the backbone conformation (15). In particular, the overall fold of molecules A and B of the TOAC analog (15) is quite similar to that reported for the three independent molecules of alamethicin itself (1), although molecule A of the analog is bent slightly more sharply near Pro14 than is molecule B.
A positioning of alamethicin consistent with the isotropic hyperfine couplings in diC16PtdCho is obtained if the TOAC1 residue is located close to the polar-apolar interface of the bilayer. A similar location relative to the bilayer midplane is also consistent with the isotropic hyperfine couplings in diC14PtdCho. Fig. 10 shows the most recent x-ray refinements of the bilayer dimensions for diC12PtdCho and diC14PtdCho (34) and for diC16PtdCho (35) in the fluid phase at 50°C. The approximate position of the residues in alamethicin is indicated by the heavy dumbbells. For the purposes of illustration, a straight α-helix is assumed. If the position relative to the bilayer midplane is retained for all chain lengths, the N-terminal of alamethicin lies within the lipid polar group region for chain lengths N < 18. Similarly, the C-terminal Phol resides in the polar group region of the opposite leaflet for the same lipid chain lengths. Such a location of the C-terminal is essentially consistent with the proposals in Barranger-Mathys and Cafiso (4), although perhaps closer to that of the leucine analog, which is compared with native alamethicin in this latter reference. (Note that diC18PtdCho bilayers are still in the gel phase at 50°C, and the extrapolated values of dC and db must be reduced by 5% to reach 65°C in the fluid phase.) With the disposition indicated in Fig. 10, the TOAC8 and TOAC16 residues are situated within the hydrocarbon core of the membrane for all lipid chain lengths, consistent with the isotropic hyperfine splittings in Fig. 5.
FIGURE 10.

Chain-length dependences of the thickness of diCNPtdCho bilayers for N = 12, 14 (34), and 16 (35) at 50°C. Solid squares: thickness of the hydrocarbon core, dC; open squares: anhydrous bilayer thickness, db; open circles: steric thickness of the bilayer, dst = dC + 1.8 nm. Measurements of dC and db for N = 12 and 14 at 30°C are corrected to 50°C by using an expansion coefficient αd = (1/d)(∂d/∂T) = −0.0032K−1 (34). Sloping lines are linear regressions. The approximate position of [Glu(OMe)7,18,19] alamethicin in diC14PtdCho and diC16PtdCho bilayers, consistent with Fig. 5, is indicated by the heavy vertical line and horizontal dotted lines (Cα-atoms), which are located at ∼ +1.45, +0.4, and −0.8 nm for TOAC1, TOAC8, and TOAC16, respectively.
Peptide tilt from order parameters
The distance between the Cα atoms of residues Aib1 and Phol20 in the crystal structure of [TOAC16,Glu(OMe)7,18,19] alamethicin is 2.8 nm (15), close to the repeat distance for a straight α-helix of this length. The entire length of the molecule is ∼3.3−3.4 nm and contains no polar anchoring groups. Hydrophobic matching will therefore be achieved approximately with diC16PtdCho bilayers, for which the thickness of the hydrocarbon core is dC = 2.79 nm in the fluid phase at 50°C (35). This conclusion is essentially consistent with the deductions made above from isotropic hyperfine splittings that TOAC1 is situated only 0.15 nm outside the hydrocarbon core of diC16PtdCho bilayers (Fig. 10). On the other hand, the total length of alamethicin corresponds to the total anhydrous thickness of diC14PtdCho bilayers (db = 3.4 nm at 50°C) and is less than the estimated total steric extent of diC12PtdCho bilayers (dst = 3.76 nm).
The order parameters, 〈P2(cosγ)〉, of the long axis of alamethicin are relatively large, even at the high temperatures for which they can be measured reliably (Fig. 7). Most significantly, the order parameters decrease rather slowly with decreasing chain length of the host lipid. The latter result is not consistent with a tight matching of the peptide tilt to the thickness of the hydrophobic core of the membrane. Fig. 11 shows model calculations for tightly coupled hydrophobic matching. The order parameter is calculated assuming either a fixed tilt γ
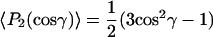 |
(6) |
or a random wobble within a cone of maximum amplitude γ
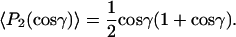 |
(7) |
The geometric condition for hydrophobic matching is given by
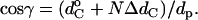 |
(8) |
where dp is the hydrophobic thickness of alamethicin and the parameters 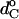 and ΔdC governing the thickness of the hydrophobic lipid core, dC, are obtained from the linear regressions in Fig. 10. Clearly, geometrical hydrophobic matching would require a far greater dependence of order parameter on lipid chain length than is observed. Attempts to fit Eqs. 7 and 8 (or equivalently Eqs. 6 and 8) to the experimental data give rise to effective thicknesses of the lipid bilayer that vary far less with chain length (and correspondingly far larger end contributions) than is found experimentally. The chain-length dependence of the hydrocarbon core thickness for phosphatidylcholines that is given in Fig. 10 is characterized by values of ΔdC/dp = 0.0741 ± 0.0005 (0.0610 ± 0.0004) and
and ΔdC governing the thickness of the hydrophobic lipid core, dC, are obtained from the linear regressions in Fig. 10. Clearly, geometrical hydrophobic matching would require a far greater dependence of order parameter on lipid chain length than is observed. Attempts to fit Eqs. 7 and 8 (or equivalently Eqs. 6 and 8) to the experimental data give rise to effective thicknesses of the lipid bilayer that vary far less with chain length (and correspondingly far larger end contributions) than is found experimentally. The chain-length dependence of the hydrocarbon core thickness for phosphatidylcholines that is given in Fig. 10 is characterized by values of ΔdC/dp = 0.0741 ± 0.0005 (0.0610 ± 0.0004) and 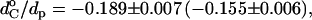 assuming that dp = 2.8 (3.4) nm. These parameters are very different from those required to describe the chain-length dependence of the order parameters in Fig. 11.
assuming that dp = 2.8 (3.4) nm. These parameters are very different from those required to describe the chain-length dependence of the order parameters in Fig. 11.
FIGURE 11.

Predicted dependence of the order parameter of the long axis of alamethicin on chain length of host diCNPtdCho bilayers according to Eq. 6 (solid lines) or Eq. 7 (dashed lines) and Eq. 8 for hydrophobic matching. Dimensions of the lipid hydrocarbon core, dC, are taken from Fig. 10, and the hydrophobic span of alamethicin is dp = 2.8 nm or 3.4 nm, as indicated. Squares (circles) are order parameter measurements at 75°C (85°C); dotted lines are nonlinear fits of Eqs. 7 and 8 with 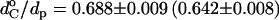 and ΔdC/dp = 0.0147 ± 0.0006 (0.0159 ± 0.0005).
and ΔdC/dp = 0.0147 ± 0.0006 (0.0159 ± 0.0005).
At 75°C, the effective angle of tilt deduced from Eq. 6 varies from γ = 13° for diC18PtdCho to γ = 23° for diC10PtdCho. For comparison, the geometrical criterion for hydrophobic matching would require that γ = 56° for diC10PtdCho, taking dp = 2.8 nm in Eq. 8. Clearly, alamethicin remains oriented relatively close to the membrane normal, even in thin phosphatidylcholine bilayers. The mode of incorporation is that indicated schematically in Fig. 10 and deduced from the isotropic hyperfine couplings. The peptide is disposed in a similar way relative to the bilayer midplane for all lipid chain lengths and remains relatively untilted.
Comparison with other data
Extensive experiments on hydrophobic matching have been performed with alanine-leucine model peptides that, unlike [Glu(OMe)7,18,19] alamethicin F50/5, have well-defined terminal anchoring residues: either tryptophans in the WALP peptides or lysines in the KALP peptides (36–39). Of these, GW2(AL)8LW2A (WALP23) and GK2(AL)8LK2A (KALP23) are likely to have a hydrophobic span similar to that of alamethicin, although they are of longer total length. Both peptides are found to have very small tilts, with only small systematic variations, in phosphatidylcholine bilayers of different thicknesses (39). At 40°C, WALP23 is found to have tilts of 5.2° and 8.1° in bilayers of diC14PtdCho and diC12PtdCho, respectively, and the corresponding tilt angles for KALP23 are 7.6° and 11.2°, respectively. Qualitatively similar results are obtained with a shorter peptide WALP19, which has the same anchoring sequences as WALP23 but two fewer AL dipeptide units (37). Similarly, the transmembrane segment of the EGF receptor, which like the more symmetrical KALP peptides is also anchored by charged groups, exhibits only small tilt that is insensitive to hydrophobic matching (40). For the WALP and KALP peptides, adaptation to hydrophobic membrane thickness is thought to be achieved by adjustment of the position of the side chains of the anchoring residues (8,10,41). Significantly, appreciable tilts of ∼30° are registered by longer peptides, WALP27 and WALP31, in diC14PtdCho (36). This degree of freedom by side-chain adjustment is not available, however, to the terminal residues of alamethicin (note that Phol does not possess the indole anchoring propensity of tryptophan). It is therefore likely that alamethicin is anchored rather differently from the WALP and KALP peptides solely by embedding in the hydrophobic region of lipid membranes, as is indicated in Fig. 10. This situation may well require distortion in the polar region of the membrane and also contribute to the potential of alamethicin to form pore assemblies under the influence of a transmembrane potential. It could also account for the observed dependence of the ion channel properties on lipid chain length (3) because propensity to form channels will increase with increasing distortion of the polar-group region.
The mode of membrane anchoring of alamethicin differs much more from certain other peptides, for instance the transmembrane domain of the Vpu protein from the HIV-1 virus (42), than it does from the WALP and KALP model peptides. The transmembrane helix of Vpu responds almost quantitatively to membrane thickness by tilting by up to 51° in the thinnest bilayers (diC10PtdCho). The Vpu helix construct has a strongly positive C-terminal domain but no obvious anchoring residues at the N-terminal. Relatively large tilts have also been obtained with the M2 transmembrane peptide from influenza A virus (43) that are more than sufficient to achieve hydrophobic matching with the host lipid. In the latter case, both charged residues at the N- and C-termini and a tryptophan may be involved in the transmembrane anchoring. Appreciable tilts are also achieved with the M13 coat protein (44) although the dependence on lipid chain length is insufficient alone to achieve simple hydrophobic matching. This latter peptide is also expected to be anchored by highly charged domains at the N- and C-termini.
Acknowledgments
We thank Frau B. Angerstein and Frau B. Freyberg for skillful technical assistance.
Abbreviations: Aib, α-aminoisobutyric acid; diC10PtdCho, 1,2-didecanoyl-sn-glycero-3-phosphocholine; diC12PtdCho, 1,2-dilauroyl-sn-glycero-3-phosphocholine; diC14PtdCho, 1,2-dimyristoyl-sn-glycero-3-phosphocholine; diC16PtdCho, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; diC18PtdCho, 1,2-distearoyl-sn-glycero-3-phosphocholine; EPR, electron paramagnetic resonance; n-PCSL, 1-acyl-2-[n-(4,4-dimethyl-oxazolidin-N-oxyl)]stearoyl-sn-glycero-3-phosphocholine; OEt, ethoxy; OMe, methoxy; Phol, phenylalaninol; ST-EPR, saturation transfer EPR; TOAC, 2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4-carboxylic acid; V1, first-harmonic absorption EPR spectrum detected in phase with respect to the static magnetic field modulation; V2′, second-harmonic absorption EPR spectrum detected 90° out-of-phase with respect to the static magnetic field modulation.
References
- 1.Fox, R. O. Jr., and F. M. Richards. 1982. A voltage-gated ion channel model inferred from the crystal structure of alamethicin at 1.5 Å resolution. Nature. 300:325–330. [DOI] [PubMed] [Google Scholar]
- 2.Nagaraj, R., and P. Balaram. 1981. Alamethicin, a transmembrane channel. Acc. Chem. Res. 14:356–362. [Google Scholar]
- 3.Hall, J. E., I. Vodyanoy, T. M. Balasubramanian, and G. R. Marshall. 1984. Alamethicin—a rich model for channel behavior. Biophys. J. 45:233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barranger-Mathys, M., and D. S. Cafiso. 1996. Membrane structure of voltage-gated channel forming peptides by site-directed spin-labeling. Biochemistry. 35:498–505. [DOI] [PubMed] [Google Scholar]
- 5.Marsh, D., M. Jost, C. Peggion, and C. Toniolo. 2007. TOAC spin labels in the backbone of alamethicin: EPR studies in lipid membranes. Biophys. J. 92:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boheim, G., and H.-A. Kolb. 1978. Analysis of the multi-pore system of alamethicin in a lipid membrane. I. Voltage-jump current-relaxation measurements. J. Membr. Biol. 38:99–150. [Google Scholar]
- 7.Lee, A. G. 2004. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 1666:62–87. [DOI] [PubMed] [Google Scholar]
- 8.Killian, J. A., and T. K. M. Nyholm. 2006. Peptides in lipid bilayers: the power of simple models. Curr. Opin. Struct. Biol. 16:473–479. [DOI] [PubMed] [Google Scholar]
- 9.de Planque, M. R. R., D. V. Greathouse, R. E. Koeppe II, H. Schäfer, D. Marsh, and J. A. Killian. 1998. Influence of lipid/peptide hydrophobic mismatch on the thickness of diacylphosphatidylcholine bilayers. A 2H NMR and ESR study using designed transmembrane α-helical peptides and gramicidin A. Biochemistry. 37:9333–9345. [DOI] [PubMed] [Google Scholar]
- 10.de Planque, M. R. R., B. B. Bonev, J. A. A. Demmers, D. V. Greathouse, R. E. Koeppe II, F. Separovic, A. Watts, and J. A. Killian. 2003. Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry. 42:5341–5348. [DOI] [PubMed] [Google Scholar]
- 11.Lomize, M. A., A. L. Lomize, I. Pogozheva, and H. I. Mosberg. 2006. OPM: orientations of proteins in membranes database. Bioinformatics. 22:623–625. [DOI] [PubMed] [Google Scholar]
- 12.Marsh, D. 1990. Handbook of Lipid Bilayers. CRC Press, Boca Raton, FL.
- 13.Peggion, C., M. Jost, C. Baldini, F. Formaggio, and C. Toniolo. 2007. Total syntheses in solution of TOAC-labelled alamethicin F50/5 analogues. Chem. Biodiv. In press. [DOI] [PubMed]
- 14.Peggion, C., I. Coin, and C. Toniolo. 2004. Total synthesis in solution of alamethicin F50/5 by an easily tunable segment condensation approach. Biopolymers. 76:485–493. [DOI] [PubMed] [Google Scholar]
- 15.Crisma, M., C. Peggion, C. Baldini, E. J. MacLean, N. Vedovato, G. Rispoli, and C. Toniolo. 2007. Crystal structure of a spin-labelled, channel-forming, alamethicin analogue. Angew. Chem. Int. Ed. Engl. 46:2047–2050. [DOI] [PubMed] [Google Scholar]
- 16.Peggion, C., M. Jost, W. M. de Borggraeve, M. Crisma, F. Formaggio, and C. Toniolo. 2007. Conformational analysis of TOAC-labelled alamethicin F50/5 analogues. Chem. Biodiv. In press. [DOI] [PubMed]
- 17.Fajer, P., and D. Marsh. 1982. Microwave and modulation field inhomogeneities and the effect of cavity Q in saturation transfer ESR spectra. Dependence on sample size. J. Magn. Reson. 49:212–224. [Google Scholar]
- 18.Hemminga, M. A., P. A. De Jager, D. Marsh, and P. Fajer. 1984. Standard conditions for the measurement of saturation transfer ESR spectra. J. Magn. Reson. 59:160–163. [Google Scholar]
- 19.Schorn, K., and D. Marsh. 1997. Extracting order parameters from powder EPR lineshapes for spin-labelled lipids in membranes. Spectrochim. Acta [A]. 53:2235–2240. [Google Scholar]
- 20.Marsh, D. 2001. Polarity and permeation profiles in lipid membranes. Proc. Natl. Acad. Sci. USA. 98:7777–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rama Krishna, Y. V. S., and D. Marsh. 1990. Spin label ESR and 31P-NMR studies of the cubic and inverted hexagonal phases of dimyristoylphosphatidylcholine/myristic acid (1:2, mol/mol) mixtures. Biochim. Biophys. Acta. 1024:89–94. [DOI] [PubMed] [Google Scholar]
- 22.Schorn, K., and D. Marsh. 1996. Lipid chain dynamics and molecular location of diacylglycerol in hydrated binary mixtures with phosphatidylcholine: spin label ESR studies. Biochemistry. 35:3831–3836. [DOI] [PubMed] [Google Scholar]
- 23.Ondar, M. A., O. Ya. Grinberg, A. A. Dubinskii, and Ya. S. Lebedev. 1985. Study of the effect of the medium on the magnetic-resonance parameters of nitroxyl radicals by high-resolution EPR spectroscopy. Sov. J. Chem. Phys. 3:781–792. [Google Scholar]
- 24.Thomas, D. D., L. R. Dalton, and J. S. Hyde. 1976. Rotational diffusion studied by passage saturation transfer electron paramagnetic resonance. J. Chem. Phys. 65:3006–3024. [Google Scholar]
- 25.Horváth, L. I., and D. Marsh. 1983. Analysis of multicomponent saturation transfer ESR spectra using the integral method: application to membrane systems. J. Magn. Reson. 54:363–373. [Google Scholar]
- 26.Horváth, L. I., and D. Marsh. 1988. Improved numerical evaluation of saturation transfer electron spin resonance spectra. J. Magn. Reson. 80:314–317. [Google Scholar]
- 27.Marsh, D. 1981. Electron spin resonance: spin labels. In Membrane Spectroscopy. Molecular Biology, Biochemistry and Biophysics, Vol. 31. E. Grell, editor. Springer, Berlin. 51–142. [DOI] [PubMed]
- 28.Marsh, D. 2002. Membrane water-penetration profiles from spin labels. Eur. Biophys. J. 31:559–562. [DOI] [PubMed] [Google Scholar]
- 29.Marsh, D. 2002. Polarity contributions to hyperfine splittings of hydrogen-bonded nitroxides—the microenvironment of spin labels. J. Magn. Reson. 157:114–118. [DOI] [PubMed] [Google Scholar]
- 30.Marsh, D. 1980. Molecular motion in phospholipid bilayers in the gel phase: long axis rotation. Biochemistry. 19:1632–1637. [DOI] [PubMed] [Google Scholar]
- 31.Fajer, P., A. Watts, and D. Marsh. 1992. Saturation transfer, continuous wave saturation, and saturation recovery electron spin resonance studies of chain-spin labeled phosphatidylcholines in the low temperature phases of dipalmitoyl phosphatidylcholine bilayers. Effects of rotational dynamics and spin-spin interactions. Biophys. J. 61:879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartucci, R., T. Páli, and D. Marsh. 1993. Lipid chain motion in an interdigitated gel phase: conventional and saturation transfer ESR of spin-labelled lipids in dipalmitoylphosphatidylcholine-glycerol dispersions. Biochemistry. 32:274–281. [DOI] [PubMed] [Google Scholar]
- 33.Schreier, S., S. R. Barbosa, F. Casallanovo, R. Vieira, E. M. Cilli, A. C. M. Paiva, and C. R. Nakaie. 2004. Conformational basis for the biological activity of TOAC-labeled angiotensin II and bradykinin: electron paramagnetic resonance, circular dichroism, and fluorescence studies. Biopolymers. 74:389–402. [DOI] [PubMed] [Google Scholar]
- 34.Kučerka, N., Y. Liu, N. Chu, H. I. Petrache, S. Tristram-Nagle, and J. F. Nagle. 2005. Structure of fully hydrated fluid phase DMPC and DLPC lipid bilayers using x-ray scattering from oriented multilamellar arrays and from unilamellar vesicles. Biophys. J. 88:2626–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kučerka, N., S. Tristram-Nagle, and J. F. Nagle. 2006. Closer look at structure of fully hydrated fluid phase DPPC bilayers. Biophys. J. 90:L83–L85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Planque, M. R. R., E. Goormaghtigh, D. V. Greathouse, R. E. Koeppe II, J. A. W. Kruijtzer, R. M. J. Liskamp, B. De Kruijff, and J. A. Killian. 2001. Sensitivity of single membrane-spanning α-helical peptides to hydrophobic mismatch with a lipid bilayer: effects on backbone structure, orientation, and extent of membrane incorporation. Biochemistry. 40:5000–5010. [DOI] [PubMed] [Google Scholar]
- 37.van der Wel, P. C. A., E. Strandberg, J. A. Killian, and R. E. Koeppe II. 2002. Geometry and intrinsic tilt of a tryptophan-anchored transmembrane α-helix determined by 2H NMR. Biophys. J. 83:1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strandberg, E., S. Özdirekcan, D. T. S. Rijkers, P. C. A. van der Wel, R. E. Koeppe, R. M. J. Liskamp, and J. A. Killian. 2004. Tilt angles of transmembrane model peptides in oriented and non-oriented lipid bilayers as determined by 2H-solid-state NMR. Biophys. J. 86:3709–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Özdirekcan, S., D. T. S. Rijkers, R. M. J. Liskamp, and J. A. Killian. 2005. Influence of flanking residues on tilt and rotation angles of transmembrane peptides in lipid bilayers. A solid-state 2H NMR study. Biochemistry. 44:1004–1012. [DOI] [PubMed] [Google Scholar]
- 40.Jones, D. H., K. R. Barber, E. W. VanDerLoo, and C. W. M. Grant. 1998. Epidermal growth factor receptor transmembrane domain: 2H NMR implications for orientation and motion in a bilayer environment. Biochemistry. 37:16780–16787. [DOI] [PubMed] [Google Scholar]
- 41.Strandberg, E., S. Morein, D. T. S. Rijkers, R. M. J. Liskamp, P. C. A. van der Wel, and J. A. Killian. 2002. Lipid dependence of membrane anchoring properties and snorkeling behavior of aromatic and charged residues in transmembrane peptides. Biochemistry. 41:7190–7198. [DOI] [PubMed] [Google Scholar]
- 42.Park, S. H., and S. J. Opella. 2005. Tilt angle of a trans-membrane helix is determined by hydrophobic mismatch. J. Mol. Biol. 350:310–318. [DOI] [PubMed] [Google Scholar]
- 43.Kovacs, F. A., J. K. Denney, Z. Song, J. R. Quine, and T. A. Cross. 2000. Helix tilt of the M2 transmembrane peptide from influenza A virus: an intrinsic property. J. Mol. Biol. 295:117–125. [DOI] [PubMed] [Google Scholar]
- 44.Koehorst, R. B. M., R. B. Spruijt, F. J. Vergeldt, and M. A. Hemminga. 2004. Lipid bilayer topology of the transmembrane α-helix of M13 major coat protein and bilayer polarity profile by site-directed fluorescence spectroscopy. Biophys. J. 87:1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]