

Abstract

Direct diazo transfer proceeds smoothly with α-aryl ketones. The derived α-aryl-α-diazo ketones cyclize efficiently with Rh catalysis to give the corresponding α-aryl cyclopentanones.
Introduction
a-Aryl cyclopentanones are a class of useful intermediates for the synthesis of natural products and for pharmaceutical applications.1 A number of effective methods have been developed for the synthesis of a-aryl cyclopentanones, including the Pd-catalyzed arylation of cyclopentanones,2a the Heck arylation of enol ethers,2b and epoxide rearrangement.2c,d It occurred to us that acyclic a-aryl ketones such as 1a (Scheme 1) could be prepared by convergent coupling. If diazo transfer and Rh-mediated intramolecular C-H insertion were efficient, we would have established a new route to α-aryl cyclopentanones.
SCHEME 1.

Results and Discussion
α-Diazo carbonyl compounds are ideal substrates for generating carbenes,3 by reaction of the diazo precursors with transition metal catalysts.4 Dirhodium catalysts, in particular, can direct highly chemo-, regio- and stereoselective reactions.5 Selectivity is determined not only by the nature of the catalyst, but also by the steric demand and electronic characteristics of the diazo precursors.6
The diazo center has two substituents. They can be both electron-withdrawing, one electron-withdrawing and one neutral or one electron-withdrawing and one electron-donating. With two electron-withdrawing substituents, the intermediate carbenoid formed is highly electrophilic, and so potentially not highly selective. With one electron-withdrawing and one electron-donating substituent, the intermediate carbenoid is stabilized, and so is more likely to be selective. The first examples of intermolecular C-H insertion with this class of carbenoids were reported by Davies in 1997.7,8
The extension to intramolecular C-H insertion, using an all sp3-hybridized tether, seemed to be straightforward, and in fact had been attempted (Scheme 2),9 but had been reported not to proceed. In contrast to this conclusion, we now report that intramolecular C-H insertion of α-aryl-α-diazo ketones is a useful and efficient process.
SCHEME 2.

Preparation of the α-aryl ketones
This approach to α-aryl cyclopentanones was particularly attractive because the requisite α-aryl ketones10 could be prepared by convergent coupling of Grignard reagents with epoxides (Table 1). PCC-catalyzed oxidation11 efficiently converted the alcohols so formed to the desired ketones. Epoxide 4c was prepared by coupling of allyl bromide with (4-bromophenyl)magnesium bromide followed by bromohydrin formation and exposure to base. Alternatively, aryl Grignard reagents could be used to open the commercially-available epoxides 4a, 4b and 4d. By these two complementary approaches, different substitution patterns on the arene and different C-H insertion sites were easily accessible.
TABLE 1.
Preparation of the α-Aryl Ketones
| entry | epoxide | Grignard | ketone | overall yield (%) |
|---|---|---|---|---|
| 1 |

|

|

|
40 |
| 2 |

|

|

|
71 |
| 3 |

|

|

|
60 |
| 4 |

|

|

|
89 |
| 5 | 4d |

|

|
62 |
| 6 | 4c |

|

|
76 |
Previously reported (Ref. 12).
Diazo transfer reaction
There were several procedures available for the diazo transfer reaction.9,13 Although mesyl azide13b with DBU worked well for ketone 1c (entry 1, Table 2), these conditions gave poor yields with ketone 1e (entry 2). 4-Nitrobenzenesulfonyl azide (PNBSA)14 gave a similar yield (entry 3). Since the only difference between 1c and 1e was the different substituent on the benzene ring, it was apparent that the electron-donating group on the para position of the aromatic ring made the α-position of the ketone less reactive. This was in contrast to diazo transfer on a range of arylacetic esters.15 To solve this problem, we screened several diazo transfer reagents along with different solvents (Table 2). We found that exactly 1.0 eq. of 2,4,6-triisopropylbenzenesulfonyl azide (TIBSA)13c in toluene gave the best yield. Additional TIBSA was not necessary and should be avoided since separation of the excess TIBSA was difficult. A simplified workup procedure was also developed, which ensured a high yield of the pure diazo ketones.
TABLE 2.
Optimization of the Diazo Transfer Reaction

| entry | ketone | reagent | eq. | base | eq. | solvent | time | yield (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1c | mesyl azide | 1.5 | DBU | 1.5 | CH2Cl2 | 2 h | 79 |
| 2 | 1e | mesyl azide | 3 | DBU | 3 | CH2Cl2 | 3 h | 13 |
| 3 | 1e | PNBSA | 3 | DBU | 3 | CH3CN | 8 h | 15 |
| 4 | 1e | BSA | 1.2 | DBU | 1.5 | CH3CN | 0.5 h | 26 |
| 5 | 1e | AABSA | 2 | DBU | 1.5 | CH3CN | 1 h | 38 |
| 6 | 1e | TIBSA | 1.0 | DBU | 1.2 | CH3CN | 2 h | 53 |
| 7 | 1e | TIBSA | 1.1 | DBU | 1.4 | toluene | 3 h | 68 |
| 8 | 1e | TIBSA | 1.0 | DBU | 3 | toluene | 3 h | 80 |
Optimization of the C-H insertion
We screened conditions for the C-H insertion reaction using diazo ketones 2c and 2f. The results are summarized in Table 3. We found out that as the solvent, toluene gave better yields than dichloromethane (entries 4, 6). The addition sequence is critical to this reaction (entries 4, 5). Addition of the rhodium catalyst into the solution of diazo ketone led to more dimer and so less of the cyclized product than the reversed addition. The reaction was fast at room temperature, going to completion usually within seconds. A reaction run at −78°C gave no cyclized product (entry 7). We decided to use the Hashimoto (Rh2(pttl)4)18 catalyst in our further studies since it consistently gave the highest yields. The results of the diazo transfer reaction and the C-H insertion reaction under these optimized conditions are summarized in Table 4. Entry 2 is particularly noteworthy, as the cyclization had previously been reported not to proceed.9
TABLE 3.
Optimization of the C-H Insertion Reaction

| entry | diazo ketone |
Rh catalyst | mol % | solvent | order of addition |
temp °C |
yield % |
|---|---|---|---|---|---|---|---|
| 1 | 2f | Rh2(tbsp)4a | 1 | CH2Cl2 | Ab | 0 | 43 |
| 2 | 2f | Rh2(ptpa)4c | 1 | CH2Cl2 | A | 0 | 38 |
| 3 | 2c | Rh2(oct)4d | 2 | CH2Cl2 | A | 40 | 50 |
| 4 | 2c | Rh2(oct)4 | 2 | CH2Cl2 | A | rt | 57 |
| 5 | 2c | Rh2(oct)4 | 2 | CH2Cl2 | Be | rt | 30 |
| 6 | 2c | Rh2(oct)4 | 2 | toluene | A | rt | 69 |
| 7 | 2c | Rh2(oct)4 | 2 | CH2Cl2 | A | −78 | 0 |
| 8 | 2c | Rh2(ptpa)4 | 1 | CH2Cl2 | A | rt | 53 |
| 9 | 2c | Rh2(ptpa)4 | 1 | toluene | A | rt | 74 |
| 10 | 2c | Rh2(pttl)4f | 1 | CH2Cl2 | A | rt | 72 |
| 11 | 2c | Rh2(pttl)4 | 1 | toluene | A | 45 | 77 |
| 12 | 2c | Rh2(pttl)4 | 1 | toluene | A | rt | 79 |
| 13 | 2f | Rh2(pttl)4 | 1 | toluene | A | rt | 81 |
Tetrakis[1-[(4-tert-butylphenyl)sulfonyl]-(2S)-pyrrolidinecarboxylate]dirhodium(II).
Diazo ketone was added into rhodium catalyst.
Tetrakis[N-phthaloyl-(S)-phenylalaninato]dirhodium ethyl acetate adduct.
Rhodium(II) octanoate, dimer.
Rhodium catalyst was added into diazo ketone.
Tetrakis[N-phthaloyl-(S)-tert-leucinato]dirhodium bis(ethyl acetate) adduct.
TABLE 4.
Preparation and Cyclization of α-Aryl-α-diazo Ketones
| entry | α-diazo ketone | yield (%)a | product | yield (%) |
|---|---|---|---|---|
| 1 |

|
99 |

|
61b |
| 2 |

|
89 |
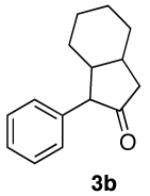
|
40d |
| 3 |

|
95 |

|
79 |
| 4 |

|
95 |

|
71b |
| 5 |

|
80 |

|
42b |
| 6 |

|
95 |

|
81b |
Yield of the diazo ketone.
Yield after equilibration of the epimeric products with DBU.
Previously reported ( Ref. 12).
The product is a mixture of ring fusion diastereomers.
Limitations
The diazo transfer reaction conditions (TIBSA/DBU/toluene) worked extremely well for each of the ketones. Our attempted preparation of a 4-methoxyphenyl diazo ketone failed, however. Diazo transfer at low temperature followed by low-temperature flash chromatography had been reported to deliver such a diazo ketone.19 We did not pursue this, as a variety of derivatives, including methoxy, can be prepared from the 4-bromo substituent.20
For C-H insertion reactions, in the cases of 3a, 3d, 3e and 3f, both cis and trans products were formed after the reaction. In order to simplify analysis, we epimerized the cis cyclopentanones to trans by the addition of a catalytic amount of DBU before work up. In the case of 3b, the product is a mixture of ring fusion diasteromers.21 The two ketones were not separable by column chromatography, so they are reported here as a mixture.
The efficiencies for Rh2(pttl)4 catalyzed intramolecular C-H insertion on α-aryl-α-diazo ketones were allylic C-H insertion ≈ tertiary aliphatic C-H insertion > secondary aliphatic C-H insertion (entries 6, 3, 1), which is consistent with previously reported4,5,21 electronic effects. Electronic effects induced by the substitution on the benzene ring also influenced the C-H insertion reaction. Substituents on the para position affected the yields more than did meta substituents. A 4-methyl substituent, moderately electron donating, reduced the yield almost one third compared to bromo (entries 1, 5). In contrast, a 3-methoxy group did not show any influence on either the diazo transfer reaction or the C-H insertion reaction (entry 4).
Since Rh2(pttl)4 is enantiomerically pure, and was developed18 to effect enantioselective C-H insertion, we assessed the enantiomeric purities of ketone 3d and of ketone 3f. To this end, we converted 3d (Scheme 3) into the diastereomeric mixture of ketals 6d/7d. These were not separated, and the relative configurations were not assigned. The ratio of the two, easily determined by integrating the methines at δ 2.92 (minor) and δ 3.04 (major) in the 1H NMR spectrum, was 2.9, indicating an enantiomeric excess of 49%. Similarly, the ratio of 6f/7f (methines at δ 3.04 (minor) and δ 3.16 (major) in the 1H NMR spectrum) was 2.6, indicating an enantiomeric excess of 44%.
SCHEME 3.

Conclusion
α-Aryl-α-diazo ketones are easily assembled. Rh-catalyzed cyclization works well, even with a substrate previously reported to be unsuccessful (2b). This approach allows readily access to α-aryl-β-alkyl cyclopentanones.
Experimental Section
General procedure for the preparation of the ketones:
1-(4-Methylphenyl)-2-octanone 1e: In a round bottom flask, 4-bromotoluene (5.00 g, 29.3 mmol), Mg (0.71 g, 29.3 mmol), iodine (trace) and 50 mL of THF were combined. The reaction was exothermic, reaching reflux after 10 min. Then the reaction mixture was kept at reflux by heating until the Mg disappeared (about 30 min). After cooling to −30°C, copper (I) bromide-dimethyl sulfide complex (0.62 g, 3.0 mmol) was added. After 5 min, 1,2-epoxyoctane (3.46 g, 27.0 mmol) in THF (10 mL) was added dropwise in one min. The cooling bath was removed, and the mixture was stirred for an additional 0.5 h. Then the reaction mixture was diluted with 500 mL of MTBE and passed through a pad of silica gel. The collected liquid was concentrated, and the residue was chromatographed (TLC Rf = 0.38, 20% MTBE/pet ether) to afford the alcohol (4.12 g) as a colorless oil.
To 100 mL of acetonitrile was added 4.48 g (197 mmol) of H5IO6, and the mixture was stirred vigorously at rt for 15 min. After cooling to 0°C, the alcohol (4.12 g, 18.7 mmol) was added followed by the addition of 81 mg (2 mol %) of PCC in 2 mL of acetonitrile. The reaction mixture was stirred for 2 h at 0 °C. Then the reaction mixture was diluted with 500 mL of MTBE and passed through a pad of silica gel. The collected liquid was concentrated, and the residue was chromatographed to afford the ketone 1e as a colorless oil (3.63 g, 16.7 mmol, 62% yield from the epoxide). TLC Rf (PE/MTBE = 8/2) = 0.64; 1H NMR δ 0.85 (3H, t, J = 7.0 Hz), 1.18-1.28 (6H, m), 1.49-1.57 (2H, m), 2.32 (3H, s), 2.41 (2H, t, J = 7.0 Hz), 3.62 (2H, s), 7.06-7.13 (4H, m); 13C NMR22 δ u 22.7, 23.9, 29.0, 31.8, 42.1, 50.0, 131.6, 136.7, 209.0; d 14.2, 21.2, 129.4, 129.6; IR (film, cm−1) 2928, 2858, 1714, 1514, 805; HRMS calcd for C15H23O (M+H) 219.1749, obsd 219.1749.
Optimized procedure for the diazo transfer reaction:
1-Diazo-1-(4-methylphenyl)-2-octanone 2e: To 3.5 mL of toluene were added ketone 3e (76 mg, 0.35 mmol), DBU (222 mg, 1.46 mmol) and 2,4,6-triisopropylbenzenesulfonylazide (108 mg, 0.35 mmol) sequentially at rt. The reaction mixture was maintained in darkness and stirred at rt for 3 h. Then the reaction mixture was directly chromatographed to afford 2e as a yellow oil (68 mg, 0.28 mmol, 80% yield). TLC Rf (PE/MTBE = 8/2) = 0.64; 1H NMR δ 0.88 (3H, t, J = 7.0 Hz), 1.26-1.36 (6H, m), 1.64-1.71 (2H, m), 2.35 (3H, s), 2.56 (2H, t, J = 7.6 Hz), 7.20-7.23 (2H, m), 7.37 (2H, d, J = 8.0 Hz); 13C NMR δ u 22.6, 24.9, 29.0, 31.7, 39.2, 122.5, 137.1, 151.0; d 14.1, 21.2, 124.2, 129.8; IR (film, cm−1) 2927, 2066, 1652, 1513, 811; HRMS calcd for C15H20O (M-N2) 216.1514, obsd 216.1517.
Optimized procedure for the C-H insertion reaction:
2-(4-Bromophenyl)-3-pentylcyclopentanone 3a: Rh2(pttl)4 (4.2 mg, 0.003 mmol) was dissolved in 2.0 mL of toluene at rt. A solution of 2a (100 mg, 0.30 mmol) in 0.8 mL of toluene was added dropwise over 2 min. The reaction was continued for an additional 10 min at rt. Then DBU (1 drop) was added before the reaction mixture was chromatographed to afford 3a as a colorless oil (56 mg, 0.18 mmol, 61% yield). TLC Rf (PE/MTBE = 8/2) = 0.21; 1H NMR δ 0.85 (3H, t, J = 7.2Hz), 1.15-1.45 (7H, m), 1.50-1.60 (2H, m), 2.15-2.35 (3H, m), 2.49-2.56 (1H, m), 2.86 (1H, d, J = 12.0Hz), 6.96 (2H, d, J = 8.4Hz), 7.45 (2H, d, J = 8.4 Hz); 13C NMR δ u 22.7, 26.8, 27.3, 32.0, 34.4, 38.5, 121.1, 137.1, 217.5; d 14.1, 45.2, 62.7, 130.6, 131.9; IR (film, cm−1) 2926, 1744, 1488, 1011, 808; HRMS calcd for C16H21 79BrO (M+) 308.0776, obsd 308.0774.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM 60287).
Footnotes
SUPPORTING INFORMATION. Experimental details and spectra for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- (1) (a).For leading references to α-aryl cyclopentanones in target-directed synthesis, see Takano S, Inomata K, Sato T, Takahashi M, Ogasawara K. J. Chem. Soc., Chem. Commun. 1990:290. Srikrishna A, Reddy T. Tetrahedron. 1998;54:8133. Kita Y, Furukawa A, Futamura J, Higuchi K, Ueda K, Fujioka H. Tetrahedron Lett. 2000;41:2133. Kita Y, Futamura J, Ohba Y, Sawama Y, Ganesh JK, Fujioka H. J. Org. Chem. 2003;68:5917. doi: 10.1021/jo034573g. Thede K, Diedrichs N, Ragot JP. Org. Lett. 2004;6:4595. doi: 10.1021/ol0479904. Kuethe JT, Marcoux J-F, Wong A, Wu J, Hillier MC, Dormer PG, Davies IW, Hughes DL. J. Org. Chem. 2006;71:7378. doi: 10.1021/jo061268x. Finke PE, Meurer LC, Levorse DA, Mills SG, MacCoss M, Sadowski S, Cascieri MA, Tsao K-L, Chicchi GG, Metzger JM, MacIntyre DE. Biorg. Med. Chem. Lett. 2006;16:4497. doi: 10.1016/j.bmcl.2006.06.035.
- (2) (a).Hamada T, Chieffi A, Ahman J, Buchwald SL. J. Am. Chem. Soc. 2002;124:1261. doi: 10.1021/ja011122+. [DOI] [PubMed] [Google Scholar]; (b) Nilsson P, Larhed M, Hallberg A. J. Am. Chem. Soc. 2003;125:3430. doi: 10.1021/ja029646c. [DOI] [PubMed] [Google Scholar]; (c) Shen Y, Wang B, Shi Y. Angew. Chem. Int. Ed. 2006;45:1429. doi: 10.1002/anie.200501520. [DOI] [PubMed] [Google Scholar]; (d) Gora M, Luczynski MK, Sepiol JJ. Synthesis. 2005;10:1625. [Google Scholar]
- (3) (a).Adams J, Spero DM. Tetrahedron. 1991;47:1765. [Google Scholar]; (b) Miller DJ, Moody CJ. Tetrahedron. 1995;51:10881. [Google Scholar]
- (4) (a).Doyle MP. Chem. Rev. 1986;86:919. [Google Scholar]; (b) Doyle MP, Forbes DC. Chem. Rev. 1998;98:911. doi: 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]; (c) Boche G, Lohrenz JCW. Chem. Rev. 2001;101:697. doi: 10.1021/cr940260x. [DOI] [PubMed] [Google Scholar]
- (5) (a).Davies HML. Comprehensive Organic Synthesis: Selectivity Strategy and Efficiency in Modern Organic Chemistry. Vol. 4. Permagon; Oxford: 1991. p. 1031. [Google Scholar]; (b) Taber DF. Comprehensive Organic Synthesis: Selectivity Strategy and Efficiency in Modern Organic Chemistry. Vol. 3. Permagon; Oxford: 1991. p. 1045. [Google Scholar]; (c) Padwa A, Krumpe KE. Tetrahedron. 1992;48:5385. [Google Scholar]; (d) Ye T, McKervey MA. Chem. Rev. 1994;94:1091. [Google Scholar]; (e) Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: from Cyclopropanes to Ylides. Wiley; New York: 1998. [Google Scholar]
- (6).Merlic CA, Zechman AL. Synthesis. 2003;8:1137. [Google Scholar]
- (7).Davies HML, Hansen T. J. Am. Chem. Soc. 1997;119:9075. [Google Scholar]
- (8) (a).For recent reviews: Davies HML, Beckwith REJ. Chem. Rev. 2003;103:2861. doi: 10.1021/cr0200217. Davies HML, Nikolai J. Org. Biomol. Chem. 2005;3:4176. doi: 10.1039/b509425a.
- (9) (a).For the cyclization of α-diazo-α-aryl ketones with an alkene tether between the ketone and the target C-H, see Fernández-Mateos TA, Coca G. Pascual, Alonso J. J. Pérez, González R. Rubio, Simmonds MSJ, Blaney WM. Tetrahedron. 1988;54:14989. Fernández-Mateos A, Coca G. Pascual, Alonso J. J. Pérez, González R. Rubio, Hernández CT. Synlett. 1996:1134.
- (10) (a).Several general methods have been developed for the preparation of α-aryl ketones. For example: Abramovitch RA, Barton DHR, Finet JP. Tetrahedron. 1988;44:3039. Palucki M, Buchwald SL. J. Am. Chem. Soc. 1997;119:11108. Angle SR, Neitzel ML. J. Org. Chem. 2000;65:6458. doi: 10.1021/jo000446y. Justik MW, Koser GF. Tetrahedron Lett. 2004;45:6159. Whitesell JK, Lawrence RM, Chen HH. J. Org. Chem. 1986;51:4779.
- (11).Hunsen M. Tetrahedron Lett. 2005;46:1651. [Google Scholar]
- (12) (a).Several of the substances reported here were previously described. 1a: Taber DF, Tian W. J. Am. Chem. Soc. 2006;128:1058. doi: 10.1021/ja058026j. 1b: Utimoto K, Takai K. NATO ASI Series, Series C: Mathematical and Physical Sciences. 1989;269:379. (c) 2b: ref. 9b. 4c: Quelet R. Bull. Soc. Chim. Fr. 1929:75.
- (13) (a).Hodgson DM, Glena R, Redgrave AJ. Tetrahedron Lett. 2002;43:3927. [Google Scholar]; (b) Taber DF, Ruckle RE, Hennessy MJ. J. Org. Chem. 1986;51:4077. [Google Scholar]; (c) Lombardo L, Mander LN. Synthesis. 1980:368. [Google Scholar]
- (14).Taber DF, You K, Song Y. J. Org. Chem. 1995;60:1093. [Google Scholar]
- (15).Davies HML, Hedley SJ, Bohall BR. J. Org. Chem. 2005;70:10737. doi: 10.1021/jo051747g. [DOI] [PubMed] [Google Scholar]
- (16).Yamazaki S, Kohgami K, Okazaki M, Yamabe S, Arai T. J. Org. Chem. 1989;54:240. [Google Scholar]
- (17).Baum JS, Shook DA, Davies HML, Smith HD. Synth. Commun. 1987;17:1709. [Google Scholar]
- (18).Anada M, Hashimoto S. Tetrahedron Lett. 1998;39:79. [Google Scholar]
- (19).Zeller KP. Chem. Ber. 1979;112:678. [Google Scholar]
- (20).Testaferri L, Tiecco M, Tingoli M, Chianelli D, Montanucci M. Tetrahedron. 1983;39:193. [Google Scholar]
- (21).Taber DF, Ruckle RE., Jr. J. Am. Chem. Soc. 1986;108:7686. doi: 10.1021/ja00284a037. [DOI] [PubMed] [Google Scholar]
- (22).13C multiplicities were determined with the aid of a JVERT pulse sequence, differentiating the signals for methyl and methine carbons as “d” and for methylene and quaternary carbons as “u”.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.