

Abstract
Base excision repair (BER) is the major pathway employed to excise oxidized DNA lesions. Human Neil1, a versatile glycosylase in the BER pathway, repairs a diverse array of oxidative lesions; however, the most prevalent 8-oxo-7, 8-dihydroguanine (8-oxoG) is only weakly excised. The structural origin of hNeil1’s ability to repair a variety of lesions but not 8-oxoG is a model system for connecting enzyme structure and lesion-recognition specificity. To elucidate structural properties determining hNeil1’s substrate specificities, we have investigated it in complex with four representative well-repaired substrates: the R and S-Sp stereoisomers, non-planar further oxidation products of guanine, and the 5R,6S and 5S,6R-Tg stereoisomers, the most prevalent oxidative lesions of thymine. We also investigate the poorly-repaired 8-oxoG. We employed molecular modeling and 10 ns molecular dynamics (MD) simulations. The results of our investigations provide structural explanations for the ability of hNeil1 to excise a variety of oxidative lesions: they possess common chemical features, namely, a pyrimidine-like ring and shared hydrogen bond donor-acceptor properties, which allow the lesions to fit well in the binding pocket, which is somewhat flexible. However, the planar 8-oxoG is not as well accommodated in the shallow and comparatively cramped recognition pocket; it has fewer hydrogen bonding interactions with the enzyme and a solvent exposed six-membered ring, consistent with its poor repair susceptibility by this enzyme.
The base excision repair (BER) pathway is employed to excise certain oxidized DNA lesions, using several concerted steps to cleave and remove the damaged DNA base from the duplex, and replace it with a normal base (1–3). BER is initiated by the glycosylase scanning the DNA and recognizing the damaged site (4–7). The base lesion, together with its sugar residue, is then extruded out of the duplex into the active site of the glycosylase, and the enzyme catalyzes the cleavage of the glycosidic bond (8–11). In short patch BER, a monofunctional or bifunctional glycosylase breaks the glycosidic bond through a Schiff-base intermediate (12). In the case of bifunctional glycosylases, the enzyme lyase activity catalyzes either β-elimination of the 3′ phosphodiester bond, or β,δ-elimination of the 3′ and 5′ phosphodiester bonds, depending on the enzyme(13, 14). Subsequently, an AP (apurinic/apyrimidinic) endonuclease removes the 3′ terminal 4-hydroxypentenal phosphate (formed by β-elimination) (10), or a polynucleotide kinase removes the 3′ terminal phosphate (formed by β,δ-elimination) that blocks repair synthesis in human cells (14). The resulting single nucleotide gap with a 3′-OH terminus is then filled with a normal incoming nucleotide by DNA polymerase β (15). Finally, the nick is sealed by DNA ligase I or a ligase III/XRCC1 complex (1, 2, 10). Here, we are interested in the first BER step, namely the specificity of DNA glycosylases for recognizing different oxidative lesions.
The human glycosylase hNeil1 plays an important role in the human BER pathway (10). hNeil1, which is a bifunctional glycosylase with AP lyase activity, belongs to the endonuclease VIII/Fpg (Nei/Fpg) family. Its DNA binding motifs include a H2tH (helix-two turn-helix) motif, and a zinc-less finger motif (a zinc finger-like structure lacking the Zn2+ ion) with a conserved arginine (16–18). The N-terminal proline is employed as the nucleophile by hNeil1 (17). It primarily recognizes and repairs ring-saturated or oxidized pyrimidines including thymine glycol (Tg), dihydrothymine, dihydrouracil, 5-hydroxyuracil, and 5-hydroxycytosine. Interestingly, hNeil1 can also repair certain oxidized purines, including formamidopyrimidines (Fapy-A and Fapy-G), guanidinohydantoin (Gh), and spiroiminodihydantoin (Sp), but 8-oxoG is removed very inefficiently (10, 16, 19).
Oxidative DNA base damage, is caused by reactive oxygen species that are formed under conditions of oxidative stress, and has been recognized as a major cause of cell death and mutagenesis in aerobic organisms (20, 21). Endogenous reactive oxygen species are byproducts of natural aerobic respiration, environmental chemicals found in cigarette smoke, chemotherapeutic drugs, UV light, and ionizing radiation (9, 22, 23). Oxidized lesions are believed to play a role as promoters in cancer development and neurological disease, and have been implicated in the normal progression of aging (20, 22, 24). The most prevalent oxidized base found in cellular environments is 8-oxoG (Figure 1) (24), detected in human cells under normal conditions (25) and also in cancer cells (26–28). Further oxidation of 8-oxoG, arylamine-G adducts, or the direct oxidation of guanine, can produce the R and S stereoisomers of spiroiminodihydantoin (Sp) (29–32) (Figure 1). The Sp lesions have been detected in Nei-deficient Escherichia coli following chromate exposure (33) and cause G to C and G to T transversions in E. coli and in vitro (34, 35). They are removed by hNeil1 (36). The stereoisomeric thymine glycols (Tg) are the most common thymine lesions (Figure 1) produced by oxidation and by ionizing radiation (37). The 5R,6S and 5S,6R-Tg lesions cause mainly T to C mutations in E. coli (38), and are repaired by hNeil1 and mNeil1 (39–41). However, 8-oxoG is only weakly removed by hNeil1 (19, 36).
Figure 1.

(A) Structures of deoxyribonucleosides 8-oxoG, Sp R and S stereoisomers, and Tg 5R,6S and 5S,6R stereoisomers. (B) DNA sequences investigated and originally present in the bstFpg crystal structure. Glycosidic torsion angle χ is O4′ – C1′ – N9 – C4 for 8-oxoG and Sp, and O4′ – C1′ – N1 – C2 for Tg.
The structural origin of hNeil1’s ability to repair the diverse set of oxidative lesions is not understood, and more broadly, is a model system for connecting enzyme structure and lesion-recognition specificity (10). The goal of our work is to obtain structural insights on the ability of hNeil1 to repair a variety of oxidative lesions, but 8-oxoG only very poorly. For this purpose, we have investigated the interactions of four representative substrates, the R and S-Sp stereoisomers, and the 5R,6S and 5S,6R-Tg stereoisomers, as well as 8-oxoG, with hNeil1 using molecular modeling and 10 ns molecular dynamics (MD) simulations. In the case of the stereoisomeric Tg lesions, we selected the cis epimers in which the bulky methyl group is on the opposite face of the Tg ring from the 6-positioned hydroxyl group in aqueous solution (42). The results of our investigations provide structural explanations for the ability of hNeil1 to excise a variety of oxidative lesions: they possess common chemical features, namely, a pyrimidine-like ring and shared hydrogen bond donor-acceptor properties, which allow the lesions to fit well in the binding pocket, which is somewhat flexible. However, 8-oxoG is not as well accommodated in the shallow and comparatively cramped recognition pocket; it has fewer hydrogen bonding interactions with the enzyme and a solvent exposed six-membered ring, consistent with its poor repair susceptibility by this enzyme.
METHODS
Molecular modeling of initial structures
Since there is no crystal structure of a hNeil1/DNA complex available, we created a model, based on two crystal structures: an apo hNeil1 in its closed conformation (proposed to be the DNA binding state) (PDB (43) ID: 1TDH) (16) and a Bacillus stearothermophilus Fpg/DNA complex crystal structure (PDB ID: 1R2Y) (44). The two enzymes are structural homologs within the endonuclease VIII/Fpg superfamily. We superimposed the H2tH DNA binding motif of the two enzymes (Figure S1) including the 12mer DNA double helix with the 8-oxoG lesion extruded out of the duplex in Fpg. Then the Fpg coordinates were deleted, leaving the DNA docked into hNeil1 (Figure S1). At this stage, there were modest steric close-contacts between the DNA backbone and amino acid side chains in the region of the zinc-less finger which is distant from the binding pocket where the glycosidic bond cleavage occurs. The steric close-contacts were relieved through small torsional amino acid sidechain adjustments, and an ~ 5° whole-body pivot of the DNA while keeping the lesion fixed. The original DNA sequence was replaced by the ones actually used in the repair experiments of Sp and 8-oxoG (36), and Tg (39) (Figure 1). A co-crystallized molecule of tris(hydroxymethyl)aminomethane (TMN) was removed. The missing loop, about 30 Å from the binding pocket region, containing residues 203 to 207 was modeled with the program MODELLER (45–47) on the ModLoop web server (45, 48), whose rotamer library provide the side chain conformations.
Crystal water molecules were retained unless they were in collision with the DNA residues, which were modeled as described above. The N-terminal proline, which acts as the nucleophile in hNeil1, was modeled as being neutral in order to mimic the system preceding the glycosylase reaction (17, 49) and following the proton transfer step. This de-protonated Pro1 models initial proton transfer to solvent, a mechanism that has been proposed for the E. coli homolog Nei (50). Mutated residues were replaced by those that occur in the wild type. Side chains were modeled using the rotamer library in AMBER (51). The 8-oxoG lesion was retained in the syn conformation that it adopted in bstFpg (PDB ID: 1R2Y) (44) for the 10 ns 8-oxoG simulation. For the 10 ns Sp simulations, 8-oxoG was replaced by R or S-Sp in the syn conformation, using our quantum mechanically computed geometries (52). Our earlier studies showed that the syn conformation is favored in DNA duplexes (53). Preliminary model building with 3 ns molecular dynamics simulations showed that the Sp in the anti conformation cannot be well accommodated in the binding pocket causing it to be markedly enlarged and distorted. In fact, the S isomer flipped from anti to syn in the preliminary work (Figure S2A). Similar preliminary studies also showed that the bulky thymine methyl group is unfavorably placed in the binding pocket when the Tg lesions are syn, and the 5R,6S isomer flipped from syn to anti (Figure S2B). A recent crystal structure of 5R,6S-Tg in DNA polymerase RB69 shows a cis epimer with anti glycosidic torsion (54) and an NMR solution structure in a DNA duplex shows the anti conformation (55). We therefore implemented the 10 ns simulations with Tg 5R,6S and 5S,6R stereoisomers in the anti conformation in the hNeil1 enzyme. We used the crystal structures of the 5R,6S and 5S,6R-Tg (cis epimers) (56, 57) to replace the 8-oxoG lesion in the damaged DNA. The glycosidic torsions (58) of the initial models are listed in Table S1. Finally, hydrogen atoms were added with the LEaP module in AMBER 8.0 (51). The protein-DNA complexes were then energy-minimized with implicit solvent molecules (dielectric constant = 4.0) for 400 steps of steepest descent (SD) followed by 600 steps of conjugate gradient (CG) minimizations using the SANDER module of AMBER 8.0 (51).
Force field
Computations were carried out with the AMBER 8.0 suite of programs (51), the Cornell et al. force field (59), and the PARM99 parameter set (60). The force field parameters for the R and S-Sp were obtained from our previous work (53). The force field was also parametrized for the 8-oxoG lesion, the neutralized N-terminal proline, and the Tg stereoisomers as described previously (53): partial charges were computed, atom types were defined, and missing bond length and angle, and torsional parameters were obtained, based on the PARM99 and GAFF (61) parameter sets. All of the added force field parameters, atom types, and topology assignments are listed in Tables S2-S9.
Molecular dynamics protocol
The system was reoriented with SIMULAID (62) to minimize the number of water molecules needed for solvation. The LEaP module of AMBER 8.0 was then employed to add counterions for neutralization and to solvate with a rectangular box of TIP3P water molecules (63) A buffer distance of 10Å between each wall and the closest solute atom in each direction was employed. The number of counterions and water molecules added to the system and the sizes of the solvation boxes are given in Table S10.
Our protocol follows recommendations of the AMBER development team (http://amber.scripps.edu/tutorials/basic/tutorial1/section5.htm). All systems employed the same equilibration and MD treatment: (1) minimization of the counterions and solvent molecules (including crystallized waters) for 2000 steps of SD followed by 3000 steps of CG with 50 kcal/mol restraints on the solute atoms; (2) 30 ps initial MD at 10 K with 25 kcal/mol restraints on solute molecules allowing the solvent to relax; (3) 80 ps constant volume MD simulation to heat the system up from 10 K to 310 K followed by 20 ps constant volume MD at 310 K with 10.0 kcal/mol restraints on solute molecules under constant volume; (4) 30 ps, 40 ps, and 50 ps MD with decreasing restraints of 10.0, 1.0, and 0.1 kcal/mol, respectively, on solute molecules under constant pressure; (5) 10 ns MD production at 310 K under constant pressure of 1 atm. Temperature and pressure coupling constants were both 1 ps.
We implemented all production MD simulations at 1 atm constant pressure and 310 K. Figure S5 shows pressure, temperature and volume data for the simulations, revealing stable fluctuations around target values as described by the AMBER development team (http://amber.scripps.edu/tutorials/basic/tutorial1/section5.htm). A 9 Å cutoff was applied to the non-bonded Lennard-Jones interactions. No switching function is applied to the Lennard-Jones interactions since these have values of essentially 0 at 9 Å (64). Long-range electrostatic interactions were treated with the Particle Mesh Ewald (PME) method (65, 66). The SHAKE algorithm (67) was applied to constrain all bonds involving hydrogen atoms with relative geometrical tolerance of 10−5 Å. A 2 fs time step was used, and the translational/rotational center-of-mass motion was removed every 1 ps (68).
Stability of the molecular dynamics simulation
Plots of the all-atom root-mean-square deviations (RMSD) of the current relative to the starting structure as a function of time for the binding pocket (residues within 5 Å from any lesion atom), the whole complex, the helix αF region (residues 199 to 223), and the complex without this region, are shown in Figure 2. The αF region, far from the binding pocket (Figure S1), is distinctly more flexible than the rest of the complex. The enzyme complex without this region is quite stable after about 2 ns, and the binding pocket itself is stable sooner. Our analyses were carried out for the 5.0–10.0 ns time frame to ensure that fully equilibrated structures were employed.
Figure 2.

Plots of the all-atom root-mean-square deviations (RMSD) of the current relative to the starting structure as a function of time for the binding pocket (residues within 5 Å from any lesion atom), the whole complex, the helix αF region (residues 199 to 223), and the complex without this region. The αF region is distinctly more flexible than the rest of the complex.
Structural analyses
Snapshots of the structures during the simulations and the last frame of simulations with solvent and counterions removed were obtained with the PTRAJ module of the AMBER 8.0 suite (51). PTRAJ was also employed to determine the time-dependence of the RMSD, and the glycosidic torsion angle χ. Detailed hydrogen bonding analyses were carried out with the CARNAL module of the AMBER 7.0 suite (69). Hydrogen bonding criteria are 3.4 Å between the heavy atoms and a 135° angle for the heavy-hydrogen-heavy atoms. The electrostatic potential surfaces of the enzymes were calculated and created by APBS (70) as a plug-in to PyMOL (71). PyMOL generated the surface views with APBS providing the color-coded electrostatic information. The trajectory average solvent accessible surface area (SASA) was calculated with the Connolly algorithm (72) in the INSIGHT II program. Default parameters were used: Atom Radius Scale = 1.0 Å, Probe Radius = 1.4 Å.
INSIGHT II from Accelrys, INC. and PyMOL from DeLano Scientific LLC. were employed for visualization and model building. Computations were carried out on our own cluster of Silicon Graphic Origin and Altix high performance computers.
RESULTS
The objectives of this work are to elucidate structural reasons for the diverse substrate specificities of the human BER glycosylase hNeil1. We investigated four lesions that are excised by hNeil1, namely, the 5R,6S and 5S,6R-Tg stereoisomers and the R and S-Sp stereoisomers. In addition, we investigated the poorly excised 8-oxoG (10, 16). We have carried out molecular modeling and molecular dynamics simulations of the five lesions embedded in a short DNA fragment in complexes with hNeil1 (Figure 1). We analyzed the MD trajectories to assess the accommodation and interactions of the lesions in the binding pocket of the enzyme. Our underlying hypothesis is that common structural features of the lesions together with enzyme flexibility permit hNeil1’s functional diversity.
The hNeil1-damaged-DNA binding complex
In order to investigate the accommodation and recognition of the Sp and 8-oxoG lesions in hNeil1, we first created reasonable lesion-containing models based on the bstFpg/8-oxoG-DNA complex (44) (PDB ID: 1R2Y) and the hNeil1 apo structures followed by 10 ns simulations, as detailed in Methods. Common features of the enzyme complexes are revealed from these five simulations. In all our simulated structures, the lesions (8-oxoG, R and S-Sp, 5R,6S and 5S,6R-Tg) are extruded and stabilized in the extra helical position. As presented in Figure 3A the electrostatic surface clearly shows that the negatively charged DNA backbone contacts the positively charged DNA binding groove of hNeil1. The zinc-less finger motif (Figure S1) contacts the major groove of the DNA duplex, and hydrogen bonds from the conserved Arg276 interact with the DNA bases. In addition, several arginine residues (Arg33, Arg94, Arg117, Arg118, Arg132, Arg158, Arg256, Arg273, and Arg276) interact with the DNA bases and backbone (Figure 3B). We observed that two important residues at the binding pocket, Asn175 and Tyr262, form multiple hydrogen bonds to the DNA backbone and hold the extruded nucleotide in place (Figure 3C). Met80, conserved in both hNeil1 and Fpg, fills the gap created by the lesion extrusion in the Fpg/DNA complex and also fills the gap in our hNeil1/DNA complex model. Furthermore, two additional residues fill the gap to stabilize the extruded conformation: Arg117 mimics an unextruded base at the damaged position by hydrogen bonding with the estranged cytosine at its Watson-Crick edge and stacking with the base 5′ neighbor of the damaged base, and Phe119 stacks with the 3′ base pair neighboring the damaged base (Figure 3C). This stacking interaction is considered to play an important role in the recognition of the lesion during the search process (6).
Figure 3.

Stereo views of (A) Electrostatic surface of hNeil1 with damaged DNA double strands (white and yellow) showing the positively charged DNA binding groove of hNeil1 (see text). Blue color is positive and red is negative. (B) DNA binding features of hNeil1 as described in text. The arginines are colored by atom with main chain and hydrogen atoms not shown. (C) DNA binding features of hNeil1 at the damaged site (see text). The lesion and protein residues are colored by atom. Hydrogen bonding interactions are marked with black lines. Damaged base and its base partner are in cyan. Structures from the last frame of the 10 ns simulations are presented. All stereo figures are prepared for viewing with a stereo viewer available at http://www.berezin.com/3D/viewers1.htm.
Lesion accommodation and recognition in hNeil1
The human Neil1 enzyme recognizes and repairs oxidized pyrimidines (5-hydroxyuracil and 5-hydroxycytosine) and ring-saturated pyrimidines (thymine glycol, dihydrothymine, and dihydrouracil) (10, 16). However, 8-oxoG, an oxidized purine, is only weakly repaired (16). Moreover, hNeil1 exhibits activity in removing a racemic mix of spiroiminodihydantoin lesions (36), purine oxidation products. Interestingly, the Sp A-ring resembles an oxidized and partially saturated pyrimidine when it is in the generally favored syn conformation (53). The hNeil1 apo crystal structure as well as our model indicate that the binding pocket of the enzyme is shallow and comparatively cramped, but is still suited to accommodate a one-ring lesion (Figure 4).
Figure 4.

Stereo views of electrostatic surface (color code same as in Figure 3A) of the binding pocket of hNeil1 with Sp(syn) and Tg(anti) stereoisomers, and 8-oxoG(syn). Lesions are colored by atom. The sugar connected to the lesions is shown in yellow. Note the variable sizes and shapes of the binding pockets, as well as electrostatic interactions mainly indicating hydrogen bonds. Structures from the last frame of the 10 ns simulations are presented.
R and S-Sp and 8-oxoG in hNeil1
The pyrimidine-like Sp(syn) lesions can be accommodated in the binding pocket of this enzyme. The Sp A-ring interacts with the binding pocket of hNeil1 through hydrogen bonds (Table S11) as does the perpendicular B-ring. For S-Sp, the mainchain oxygen of Met80 forms a strong bifurcated hydrogen bond to the amino and imino groups of Sp. In the case of R-Sp, the amino group only weakly hydrogen bonds to the sidechain oxygen atom of Tyr262 (Figure 5 and Table S11). The S-Sp is better housed in the binding pocket with more favorable electrostatic interactions due to the strong hydrogen bond to the Met80; however, O6 of R-Sp interacts repulsively with this residue, being anchored by the hydrogen bond with Tyr262 (Figure 5). As a result, the binding pocket is enlarged. These hydrogen bonding interactions are unique to Sp because of its propeller-like structural features. However, the 8-oxoG is not well accommodated in the same binding pocket. Although there are hydrogen bonds between the 8-oxoG five-membered ring and the enzyme (Table S11), there is no interaction between the enzyme and the six-membered ring (corresponding to the Sp B-ring), which is exposed to the solvent (Figure 4). Trajectory average solvent accessible surface areas for the lesions are given in Table 1. These show distinctly greater solvent exposure for the 8-oxoG than for the other lesions, as well as a significantly greater SD, indicating more flexibility.
Figure 5.

Stereo views of the binding pocket of hNeil1 with Tg and Sp stereoisomers, and 8-oxoG, highlighting hydrogen bonding interactions. See text for details. The enzyme is rendered in cartoon. Lesions and key residues are colored by atom. The sugar connected to the lesions is shown in yellow. Hydrogen bonding interactions are marked with black lines. Structures from the last frame of the 10 ns simulations are presented.
Table 1.
Solvent accessible surface area (SASA) of base lesionsa
| 8-oxoG | R-Sp | S-Sp | 5R,6S-Tg | 5S,6R-Tg | |
|---|---|---|---|---|---|
| SASA(Å2) | 93.8 | 85.1 | 83.8 | 78.6 | 87.8 |
| SD(Å2) | 13.9 | 8.4 | 7.8 | 9.9 | 12.6 |
Trajectory average values and standard deviation (SD) are given for the 5–10 ns time frame of the 10 ns simulation.
5R,6S and 5S,6R-Tg in hNeil1
In order to further evaluate our hNeil1 model, we also investigated a pair of stereoisomeric thymine glycol lesions which are excised by this enzyme; for this study the cis epimers of 5R,6S and 5S,6R – Tg (Figure 1), predominant in solution (42) and observed in a recent crystal structure in a DNA polymerase (54), were employed. The two Tg stereoisomers can be accommodated in the binding pocket of hNeil1. The O2 and N3H3 in the Tg ring interact with the binding pocket of hNeil1 by hydrogen bonding, in a manner analogous to the interactions of the O8 and N7H7 groups in the Sp A-ring. Table S11 gives hydrogen bonds between the damaged bases and hNeil1 and their occupancies (the percent of structures with hydrogen bond intact over the 5 – 10 ns time frame analyzed). In addition, due to the non-planarity of the Tg stereoisomers, other favorable interactions occur with hNeil1 which could contribute to the recognition of the Tg lesions by this enzyme. In the case of 5R,6S-Tg, the O2 atom forms a bifurcated hydrogen bond to the imino group of the nucleophile Pro1 and its neighboring amino acid Glu2 (total occupancy 83%). Met80, which forms a hydrogen bond to S-Sp, forms a weak hydrogen bond with the hydroxyl group at the 6-position (occupancy 20%) (Figure 5 and Table S11). In the case of 5S,6R-Tg, the same hydroxyl group forms a weak hydrogen bond with the sidechain oxygen atom of Tyr176 (occupancy 32%). The O2 atom also forms a weak hydrogen bond to the imino group of Glu2 (Figure 5 and Table S11). Furthermore, the bulky methyl group of 5R,6S-Tg is in hydrophobic contact with the phenyl group of Tyr176; however, the analogous group in 5S,6R-Tg does not form any favorable interactions with hNeil1.
hNeil1 Binding Pocket Features
From our simulations of R and S-Sp and 5R,6S and 5S,6R-Tg, we can infer binding pocket features of hNeil1 that accommodate and recognize these pyrimidinic lesions. Figure 4 shows that the binding pocket adapts both sterically and electrostatically to the specific lesions. In addition, the results suggest that the binding pocket contains residues Pro1 (the nucleophile), Glu2 and Glu5 (these hydrogen bond with the planar part of the Tg ring and the planar Sp A-ring), Tyr262/Tyr176 (these interact with the exocyclic groups of Tg and the Sp B-ring), and Met80 (this inserts into the gap created by the extrusion of the lesion and hydrogen bonds with the exocyclic groups of Tg and the S-Sp B-ring). Figure 5 shows hydrogen bonding interactions between the lesions and the binding pocket.
DISCUSSION
Lesion accommodation and recognition by hNeil1
Our hNeil1 structures, based on modeling employing the hNeil1 apo enzyme structure (PDB ID: 1TDH) (16) and a structurally relevant bstFpg/DNA complex crystal structure containing 8-oxoG (PDB ID: 1R2Y) (44), with 10 ns MD simulations share certain overall structural features with the hNeil1/DNA model constructed by Doublie et al. based on an EcoNei/DNA complex without lesion (16): DNA in proximity of the catalytic N-terminal proline, the Met that occupies the position of the averted damaged base, and the H2TH and zinc less finger motifs.
Our results with lesions reveal common structural features of the variety of damages that are recognized by hNeil1. These structural features together with enzyme active site flexibility help explain the enzyme’s versatility. hNeil1 is capable of repairing oxidative damages which include the thymine glycol stereoisomers, dihydrothymine, dihydrouracil, 5-hydroxyuracil, the spiroiminodihydantoin stereoisomers, guanidinohydantoin, and formamidopyrimidines (16, 36, 39, 73). Specifically, these lesions share the endocyclic amide group (O=C-N-H) (Figure 1) and contain or mimic features of single, pyrimidine–like rings. Some also have exocyclic groups containing hydrogen bond donors and acceptors. We have simulated four such lesions, two pairs of stereoisomers (R and S-Sp and 5R,6S and 5S,6R-Tg). From these simulations, we suggest features of the binding pocket that accommodate and recognize these lesions: these include the presence of Pro1, Glu2, Glu5, Tyr176/Tyr262, and Met80. These features appear well suited for the recognition of other substrates of hNeil1: Glu2 and Glu5 interact with the endocyclic amide group (Figure 6), Tyr262/Tyr176 and/or Met80 interact with exocyclic groups, and Met80 inserts into the gap created by the extrusion of the lesions (Figure 5). Our simulations of the Tg and Sp damages suggest that the hNeil1 binding pocket may be somewhat flexible, which is reasonable in light of the broad substrate specificity of this enzyme. The binding pockets adapt both sterically and electrostatically to the specific lesions (Figure 4).
Figure 6.
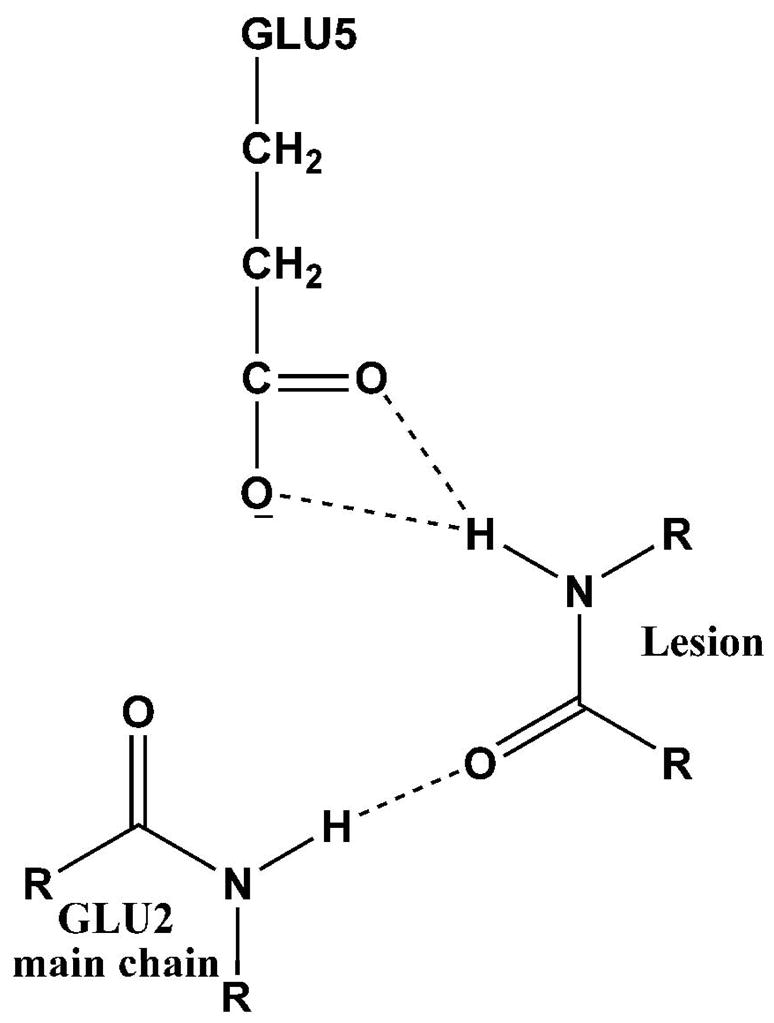
Hydrogen bonding interactions of hNeil1 residues Glu2 and Glu5 proposed for lesion recognition.
The residues Glu2 and Glu5 in the binding pocket of hNeil1 are conserved in two other enzymes, Fpg and Nei, which also belong to the Endonuclease VIII/Fpg (Nei/Fpg) family (73, 74). These residues play key roles in lesion recognition and catalysis. The crystal structures of Fpg and Nei in complex with damaged DNA reveal that the two residues interact with lesions via hydrogen bonding in order to recognize and accommodate them in the active site (50, 75, 76). Mutation data show that in hNeil1, as well as Nei and Fpg, mutation of Glu2 significantly reduces enzyme activity (50, 73, 77). Mutation studies suggest that Glu2 may be a putative base which accepts a proton during the cleavage process in Nei and Fpg (50, 76), but solvent water may also be the base (50) as implicit in the present simulations investigating a de-protonated proline. Mutation data for Glu5 does not appear to be available, and our simulations suggest that mutating this residue would reduce the enzyme activity.
Our results show that the Sp can be well accommodated in the hNeil1 binding pocket and that Sp is recognized via favorable hydrogen bond interactions (Table S11) between its A-ring and residues in the binding pocket. This A-ring mimics oxidized and partially ring-saturated pyrimidines (Figure 1). In addition, the uniquely oriented amino and imino groups on the B-ring also have hydrogen bond interactions with the binding pocket. This is possible due to the perpendicular orientation of the Sp A and B-ring, stemming from the sp3 hybridization of C4. However, in the case of the planar 8-oxoG, although the five-membered ring is in the binding pocket, the six-membered ring is exposed to solvent without interactions with the enzyme (Table 1 and Figure 4). This may be one reason why the R and S Sp stereoisomers are excised by hNeil1 while 8-oxoG is only very weakly excised (36).
Possible stereoisomeric effects of the Sp and Tg lesions
We hypothesize that sterically better accommodation of the lesion and stronger interactions between the lesion and enzyme residues in the binding pocket are the conditions that facilitate the formation of excision-ready lesion–enzyme complexes. Our results indicate the possibility of some differential activity of hNeil1 toward members of stereoisomer pairs. The structural differences of the R and S-Sp lesions, reflected in their oppositely oriented B-ring (Figure 1), suggest that the R and S forms might be excised at somewhat different rates. Differences in hydrogen bonding interactions within the binding pocket are observed in our MD simulations. Specifically, the amino and imino groups on the B-ring of the S-Sp form a strong bifurcated hydrogen bond to the main-chain oxygen atom of Met80. However, only the amino group of the R-Sp stereoisomer weakly hydrogen bonds to the sidechain oxygen atom of Tyr262 (Table S11), placing O6 in a repulsive position vis a vis Met80 which enlarges the binding pocket (Figure 5). Thus, the R-Sp stereoisomer appears less favorably placed for hydrolysis of the glycosidic bond. Experimental studies with separated stereoisomers may be useful in this connection.
Our simulations also indicate that the 5R,6S-Tg has stronger interactions with the binding pocket than the 5S,6R-Tg, due to the oppositely oriented methyl and hydroxyl groups. The methyl group of 5R,6S-Tg is in favorable hydrophobic contact with the phenyl ring of Tyr176 only in this stereoisomer, and this stereoisomer has a stronger bifurcated hydrogen bond between the lesion O2 and Pro1/Glu2 of the enzyme (Table S11); together, these produce a better accommodated 5R,6S-Tg stereoisomer (Figure 4). Neil1 repair experiments have shown the possibility of a modestly greater relative excision activity toward the 5R,6S-Tg than toward the 5S,6R stereoisomer in the human (39). and the mouse homolog (40). Recently, another study has shown that stereoselectivity of Tg excision by hNeil1 is not manifest when the enzyme concentration is much greater than the substrate; however, modest selectivity for the 5R,6S stereoisomer was mentioned when the concentration of the substrate approached that of the enzyme (41).
In these studies, we investigated the cis epimers, which predominate in solution (42) and have been observed crystallographically (54, 56, 57). The cis epimers are likely preferred because there is less crowding between the 6-hydroxyl and the methyl groups, which are on opposite faces of the thymine ring in the cis epimers but on the same face in the trans case. It is therefore plausible that the cis epimer preference remains in the hNeil1 enzyme as well; it has been observed in a DNA polymerase recently (54). It has been inferred that only one epimer is excised by hNeil1, whose identity is not determined (41).
We note that the MD simulations are, of course, limited by the problem of sampling sufficiency, together with issues relating to force field quality, which are on-going frontier problems. Nonetheless, the ensembles of structures derived from these MD trajectories can be acquired only by simulation and simulations can provide structural insights where crystal structures are not yet available. Thus, this computational approach is proving valuable in elucidating structure-function relationships and providing suggestions for new experimental directions (78–80).
CONCLUSION
We have modeled and carried out 10 ns MD simulations for hNeil1 in complex with four oxidative lesions that are excised by this BER glycosylase, namely, R and S-Sp and 5R,6S and 5S,6R-Tg. In addition, we similarly investigated the 8-oxoG damage which hNeil1 excise only very weakly (19, 36). Our results provide structural insights into the recognition mechanisms for diverse oxidative base damages by this human enzyme. Specifically, hNeil1’s ability to recognize a variety of pyrimidinic lesions is connected to the flexible binding pocket of this enzyme and the common chemical features of lesions that contain a pyrimidine-like ring, including the purine-derived R and S Sp stereoisomers in their syn conformations. The binding pocket is however shallow and comparatively cramped and hence the planar purine 8-oxoG lesion fits poorly, with fewer hydrogen bonding interactions and greater solvent exposure.
Supplementary Material
Figure S1 shows superposition of hNeil1 and Fpg. Figure S2 shows results of 3 ns preliminary simulations for R and S-Sp(anti), and 5R,6S and 5S,6R-Tg(syn). Figure S3 shows RMSD vs time plot for each preliminary 3ns simulation. Figures S4 shows glycosidic torsion χ vs time plots for each 10 ns simulation. Figure S5 shows pressure, volume and temperature vs time plots for the 10 ns simulations. Table S1 shows initial glycosidic torsion χ values. Tables S2-S10 show added force field parameters, torsion angles, simulation box sizes, and numbers of added counterions and waters of initial models. Tables S11 shows hydrogen bonds and occupancies between damaged base and hNeil1. This material is available free of charge via the Internet at http://pubs.acs.org.
ABBREVIATIONS
- BER
base excision repair
- AP
apurinic/apyrimidinic
- Neil
endonuclease VIII-like
- Nth
endonuclease III
- Fpg
formamidopyrimidine glycosylase
- Nei
endonuclease VIII
- H2tH
helix-two turn-helix
- 8-oxoG
8-oxo-7, 8-dihydroguanine
- Fapy
formamidopyrimidine
- Gh
guanidinohydantoin
- Sp
spiroiminodihydantoin
- Tg
thymine glycols
- TMN
tris(hydroxymethyl)aminomethane
- MD
molecular dynamics
- SD
steepest descent
- CG
conjugate gradient
- RESP
restrained electrostatic potential fitting algorithm
- PME
particle mesh Ewald
- RMSD
root-mean-square deviations
- SASA
solvent accessible surface area
Footnotes
This work is supported by NIH Grant 2R01 CA75449 to SB, 1R01 ES11589 to VS, and 1R01 CA99194 to NEG.
References
- 1.Fromme JC, Verdine GL. Base excision repair. Adv Protein Chem. 2004;69:1–41. doi: 10.1016/S0065-3233(04)69001-2. [DOI] [PubMed] [Google Scholar]
- 2.Scharer OD. Chemistry and biology of DNA repair. Angew Chem, Int Ed. 2003;42:2946–74. doi: 10.1002/anie.200200523. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee A, Yang W, Karplus M, Verdine GL. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature. 2005;434:612–8. doi: 10.1038/nature03458. [DOI] [PubMed] [Google Scholar]
- 5.David SS. Structural biology: DNA search and rescue. Nature. 2005;434:569–70. doi: 10.1038/434569a. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee A, Santos WL, Verdine GL. Structure of a DNA glycosylase searching for lesions. Science. 2006;311:1153–7. doi: 10.1126/science.1120288. [DOI] [PubMed] [Google Scholar]
- 7.Yang W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair (Amst) 2006 doi: 10.1016/j.dnarep.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 8.David SS, Williams SD. Chemistry of Glycosylases and Endonucleases Involved in Base-Excision Repair. Chem Rev. 1998;98:1221–1262. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 9.Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: new twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Ide H, Kotera M. Human DNA glycosylases involved in the repair of oxidatively damaged DNA. Biol Pharm Bull. 2004;27:480–5. doi: 10.1248/bpb.27.480. [DOI] [PubMed] [Google Scholar]
- 11.McCullough AK, Dodson ML, Lloyd RS. Initiation of base excision repair: glycosylase mechanisms and structures. Annu Rev Biochem. 1999;68:255–85. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- 12.Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–66. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 13.Izumi T, Wiederhold LR, Roy G, Roy R, Jaiswal A, Bhakat KK, Mitra S, Hazra TK. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology. 2003;193:43–65. doi: 10.1016/s0300-483x(03)00289-0. [DOI] [PubMed] [Google Scholar]
- 14.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–20. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Fan J, Wilson DM., 3rd Protein-protein interactions and posttranslational modifications in mammalian base excision repair. Free Radical Biol Med. 2005;38:1121–38. doi: 10.1016/j.freeradbiomed.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 16.Doublie S, Bandaru V, Bond JP, Wallace SS. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc Natl Acad Sci U S A. 2004;101:10284–9. doi: 10.1073/pnas.0402051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A. 2002;99:3523–8. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takao M, Kanno S, Kobayashi K, Zhang QM, Yonei S, van der Horst GT, Yasui A. A back-up glycosylase in Nth1 knock-out mice is a functional Nei (endonuclease VIII) homologue. J Biol Chem. 2002;277:42205–13. doi: 10.1074/jbc.M206884200. [DOI] [PubMed] [Google Scholar]
- 19.Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–84. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 20.Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res. 2003;531:37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 22.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 23.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Dizdaroglu M. Chemistry of Free Radical Damage to DNA and Nucleoproteins. Ellis Horwood; London: 1993. [Google Scholar]
- 26.Malins DC, Haimanot R. Major alterations in the nucleotide structure of DNA in cancer of the female breast. Cancer Res. 1991;51:5430–2. [PubMed] [Google Scholar]
- 27.Olinski R, Zastawny T, Budzbon J, Skokowski J, Zegarski W, Dizdaroglu M. DNA base modifications in chromatin of human cancerous tissues. FEBS Lett. 1992;309:193–8. doi: 10.1016/0014-5793(92)81093-2. [DOI] [PubMed] [Google Scholar]
- 28.Wiseman H, Kaur H, Halliwell B. DNA damage and cancer: measurement and mechanism. Cancer Lett. 1995;93:113–20. doi: 10.1016/0304-3835(95)03792-U. [DOI] [PubMed] [Google Scholar]
- 29.Joffe A, Geacintov NE, Shafirovich V. DNA lesions derived from the site selective oxidation of Guanine by carbonate radical anions. Chem Res Toxicol. 2003;16:1528–38. doi: 10.1021/tx034142t. [DOI] [PubMed] [Google Scholar]
- 30.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-Oxo-7,8-dihydroguanosine. Org Lett. 2000;2:613–6. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 31.Sugden KD, Campo CK, Martin BD. Direct oxidation of guanine and 7,8-dihydro-8-oxoguanine in DNA by a high-valent chromium complex: a possible mechanism for chromate genotoxicity. Chem Res Toxicol. 2001;14:1315–22. doi: 10.1021/tx010088+. [DOI] [PubMed] [Google Scholar]
- 32.Stover JS, Ciobanu M, Cliffel DE, Rizzo CJ. Chemical and Electrochemical Oxidation of C8-Arylamine Adducts of 2′-Deoxyguanosine. J Am Chem Soc. 2007 doi: 10.1021/ja066404u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hailer MK, Slade PG, Martin BD, Sugden KD. Nei deficient Escherichia coli are sensitive to chromate and accumulate the oxidized guanine lesion spiroiminodihydantoin. Chem Res Toxicol. 2005;18:1378–83. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–62. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 35.Kornyushyna O, Berges AM, Muller JG, Burrows CJ. In vitro nucleotide misinsertion opposite the oxidized guanosine lesions spiroiminodihydantoin and guanidinohydantoin and DNA synthesis past the lesions using Escherichia coli DNA polymerase I (Klenow fragment) Biochemistry. 2002;41:15304–14. doi: 10.1021/bi0264925. [DOI] [PubMed] [Google Scholar]
- 36.Hailer MK, Slade PG, Martin BD, Rosenquist TA, Sugden KD. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair (Amst) 2005;4:41–50. doi: 10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Wallace SS. Biological consequences of free radical-damaged DNA bases. Free Radic Biol Med. 2002;33:1–14. doi: 10.1016/s0891-5849(02)00827-4. [DOI] [PubMed] [Google Scholar]
- 38.Basu AK, Loechler EL, Leadon SA, Essigmann JM. Genetic effects of thymine glycol: site-specific mutagenesis and molecular modeling studies. Proc Natl Acad Sci U S A. 1989;86:7677–81. doi: 10.1073/pnas.86.20.7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katafuchi A, Nakano T, Masaoka A, Terato H, Iwai S, Hanaoka F, Ide H. Differential specificity of human and Escherichia coli endonuclease III and VIII homologues for oxidative base lesions. J Biol Chem. 2004;279:14464–71. doi: 10.1074/jbc.M400393200. [DOI] [PubMed] [Google Scholar]
- 40.Miller H, Fernandes AS, Zaika E, McTigue MM, Torres MC, Wente M, Iden CR, Grollman AP. Stereoselective excision of thymine glycol from oxidatively damaged DNA. Nucleic Acids Res. 2004;32:338–45. doi: 10.1093/nar/gkh190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ocampo-Hafalla MT, Altamirano A, Basu AK, Chan MK, Ocampo JE, Cummings A, Jr, Boorstein RJ, Cunningham RP, Teebor GW. Repair of thymine glycol by hNth1 and hNeil1 is modulated by base pairing and cis-trans epimerization. DNA Repair (Amst) 2006;5:444–54. doi: 10.1016/j.dnarep.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 42.Lustig MJ, Cadet J, Boorstein RJ, Teebor GW. Synthesis of the diastereomers of thymidine glycol, determination of concentrations and rates of interconversion of their cis-trans epimers at equilibrium and demonstration of differential alkali lability within DNA. Nucleic Acids Res. 1992;20:4839–45. doi: 10.1093/nar/20.18.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fromme JC, Verdine GL. DNA lesion recognition by the bacterial repair enzyme MutM. J Biol Chem. 2003;278:51543–8. doi: 10.1074/jbc.M307768200. [DOI] [PubMed] [Google Scholar]
- 45.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–73. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 47.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 48.Fiser A, Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500–1. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- 49.Zaika EI, Perlow RA, Matz E, Broyde S, Gilboa R, Grollman AP, Zharkov DO. Substrate discrimination by formamidopyrimidine-DNA glycosylase: a mutational analysis. J Biol Chem. 2004;279:4849–61. doi: 10.1074/jbc.M310262200. [DOI] [PubMed] [Google Scholar]
- 50.Zharkov DO, Golan G, Gilboa R, Fernandes AS, Gerchman SE, Kycia JH, Rieger RA, Grollman AP, Shoham G. Structural analysis of an Escherichia coli endonuclease VIII covalent reaction intermediate. Embo J. 2002;21:789–800. doi: 10.1093/emboj/21.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER. Vol. 8. University of California; San Francisco: 2004. [Google Scholar]
- 52.Jia L, Shafirovich V, Shapiro R, Geacintov NE, Broyde S. Spiroiminodihydantoin lesions derived from guanine oxidation: structures, energetics, and functional implications. Biochemistry. 2005;44:6043–51. doi: 10.1021/bi0473657. [DOI] [PubMed] [Google Scholar]
- 53.Jia L, Shafirovich V, Shapiro R, Geacintov NE, Broyde S. Structural and thermodynamic features of spiroiminodihydantoin damaged DNA duplexes. Biochemistry. 2005;44:13342–53. doi: 10.1021/bi050790v. [DOI] [PubMed] [Google Scholar]
- 54.Aller P, Rould MA, Hogg M, Wallace SS, Doublie S. A structural rationale for stalling of a replicative DNA polymerase at the most common oxidative thymine lesion, thymine glycol. Proc Natl Acad Sci U S A. 2007;104:814–8. doi: 10.1073/pnas.0606648104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kung HC, Bolton PH. Structure of a duplex DNA containing a thymine glycol residue in solution. J Biol Chem. 1997;272:9227–36. doi: 10.1074/jbc.272.14.9227. [DOI] [PubMed] [Google Scholar]
- 56.Flippen JL. Crystal and Molecular-Structures of Reaction-Products from Gamma-Irradiation of Thymine and Cytosine - Cis-Thymine Glycol, C5h8n2o4, and Trans-1-Carbamoyl-Imidazolidone-4,5-Diol, C4h7n3o4. Acta Crystallographica Section B-Structural Science B. 1973;29:1756–1762. [Google Scholar]
- 57.Jolibois F, Voituriez L, Grand A, Cadet J. Conformational and electronic properties of the two cis (5S,6R) and (5R,6S) diastereoisomers of 5,6-dihydroxy-5,6-dihydrothymidine: X-ray and theoretical studies. Chem Res Toxicol. 1996;9:298–305. doi: 10.1021/tx9501344. [DOI] [PubMed] [Google Scholar]
- 58.Saenger W. Principles of nucleic acid structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 59.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 60.Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? . J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 61.Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 62.Mezei M. Mount Sinai School of Medicine. [Google Scholar]
- 63.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 64.Levine IN. Physical Chemistry. 5. McGraw-Hill College; New York: 2001. [Google Scholar]
- 65.Darden T, York D, Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 66.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 67.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 68.Harvey SC, Tan RKZ, Cheatham TE. The flying ice cube: Velocity rescaling in molecular dynamics leads to violation of energy equipartition. J Comput Chem. 1998;19:726–740. [Google Scholar]
- 69.Case DA, Pearlman DA, Caldwell JW, Cheatham TE, III, Wang J, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer RJ, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER. Vol. 7. University of California; San Francisco: 2002. [Google Scholar]
- 70.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeLano W. 2006 [Google Scholar]
- 72.Connolly ML. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983;221:709–13. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
- 73.Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 2002;1:517–29. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 74.Zharkov DO, Shoham G, Grollman AP. Structural characterization of the Fpg family of DNA glycosylases. DNA Repair (Amst) 2003;2:839–62. doi: 10.1016/s1568-7864(03)00084-3. [DOI] [PubMed] [Google Scholar]
- 75.Coste F, Ober M, Carell T, Boiteux S, Zelwer C, Castaing B. Structural basis for the recognition of the FapydG lesion (2,6-diamino-4-hydroxy-5-formamidopyrimidine) by formamidopyrimidine-DNA glycosylase. J Biol Chem. 2004;279:44074–83. doi: 10.1074/jbc.M405928200. [DOI] [PubMed] [Google Scholar]
- 76.Pereira de Jesus K, Serre L, Zelwer C, Castaing B. Structural insights into abasic site for Fpg specific binding and catalysis: comparative high-resolution crystallographic studies of Fpg bound to various models of abasic site analogues-containing DNA. Nucleic Acids Res. 2005;33:5936–44. doi: 10.1093/nar/gki879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lavrukhin OV, Lloyd RS. Involvement of phylogenetically conserved acidic amino acid residues in catalysis by an oxidative DNA damage enzyme formamidopyrimidine glycosylase. Biochemistry. 2000;39:15266–71. doi: 10.1021/bi001587x. [DOI] [PubMed] [Google Scholar]
- 78.Hansson T, Oostenbrink C, van Gunsteren W. Molecular dynamics simulations. Curr Opin Struct Biol. 2002;12:190–6. doi: 10.1016/s0959-440x(02)00308-1. [DOI] [PubMed] [Google Scholar]
- 79.Karplus M, Kuriyan J. Molecular dynamics and protein function. Proc Natl Acad Sci U S A. 2005;102:6679–85. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;9:646–52. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 shows superposition of hNeil1 and Fpg. Figure S2 shows results of 3 ns preliminary simulations for R and S-Sp(anti), and 5R,6S and 5S,6R-Tg(syn). Figure S3 shows RMSD vs time plot for each preliminary 3ns simulation. Figures S4 shows glycosidic torsion χ vs time plots for each 10 ns simulation. Figure S5 shows pressure, volume and temperature vs time plots for the 10 ns simulations. Table S1 shows initial glycosidic torsion χ values. Tables S2-S10 show added force field parameters, torsion angles, simulation box sizes, and numbers of added counterions and waters of initial models. Tables S11 shows hydrogen bonds and occupancies between damaged base and hNeil1. This material is available free of charge via the Internet at http://pubs.acs.org.