

Abstract
Prostaglandin E2 (PGE2) and prostacyclin are lipid mediators produced by cyclooxygenase and implicated in the regulation of vascular function, wound repair, inflammatory processes, and acute lung injury. Although protective effects of these prostaglandins (PGs) are associated with stimulation of intracellular cAMP production, the crosstalk between cAMP-activated signal pathways in the regulation of endothelial cell (EC) permeability is not well understood. We studied involvement of cAMP-dependent kinase (PKA), cAMP-Epac-Rap1 pathway, and small GTPase Rac in the PGs-induced EC barrier protective effects and cytoskeletal remodeling. PGE2 and PGI2 synthetic analog beraprost increased transendothelial electrical resistance and decreased dextran permeability, enhanced peripheral F-actin rim and increased intercellular adherens junction areas reflecting EC barrier-protective response. Furthermore, beraprost dramatically attenuated thrombin-induced Rho activation, MLC phosphorylation and EC barrier dysfunction. In vivo, beraprost attenuated lung barrier dysfunction induced by high tidal volume mechanical ventilation. Both PGs caused cAMP-mediated activation of PKA-, Epac/Rap1- and Tiam1/Vav2-dependent pathways of Rac1 activation and EC barrier regulation. Knockdown of Epac, Rap1, Rac-specific exchange factors Tiam1 and Vav2 using siRNA approach, or inhibition of PKA activity decreased Rac1 activation and PG-induced EC barrier enhancement. Thus, our results show that barrier-protective effects of PGE2 and prostacyclin on pulmonary EC are mediated by PKA and Epac/Rap pathways, which converge on Rac activation and lead to enhancement of peripheral actin cytoskeleton and adherens junctions. These mechanisms may mediate protective effects of PGs against agonist-induced lung vascular barrier dysfunction in vitro and against mechanical stress-induced lung injury in vivo.
Keywords: permeability, lung endothelium, small GTPases, cytoskeleton, PKA, GEFs
INTRODUCTION
Acute respiratory distress remains a major cause of morbidity and mortality with an overall mortality rate of 30–40% [1, 2]. The acute phase of lung injury is characterized by increased permeability of the blood-gas barrier, which allows an influx of protein-rich fluid into the air spaces, causing pulmonary edema.
Prostaglandins (PG) are products of arachidonic acid metabolic pathway and synthesized by many tissues including vascular endothelial cells. The role of prostaglandins and their receptors in inflammation and regulation of vascular permeability is complicated. Although prostaglandins may be involved in the generation of acute lung inflammation in part via vasodilatory effects, PGI2 and PGF2α exhibited protective effects in the resolution phase of inflammation [3]. Stable PGI2 analogs beraprost and iloprost currently widely used as treatment of pulmonary hypertension, showed potent anti-inflammatory and endothelium-dependent anti-edemagenic effects in several models of acute lung injury [4, 5]. PGE2 has been also shown to regulate endothelial permeability in vivo and in vitro [6, 7]. However, molecular mechanisms of pulmonary endothelial barrier protection by prostaglandins remain largely unexplored.
Cytoskeletal remodeling, cell contact reorganization and actomyosin contractility are essential mechanisms of dynamic endothelial permeability regulation, which are controlled by protein kinases such as myosin light chain kinase (MLCK), Ca2+/calmodulin-dependent kinase II, protein kinase C, cAMP-dependent protein kinase A (PKA), and protein tyrosine kinases (reviewed in [8]). In addition, both barrier-protective and barrier-disruptive processes in EC are differentially regulated by small GTPases Rac and Rho, which induce distinct patterns of cytoskeletal and cell contact remodeling leading to EC barrier protection or compromise [9–13].
Prostaglandins PGE2 and PGI2 mediate their effects in target cells by binding to specific G-protein-coupled prostanoid receptors EP1-4 and IP. In addition, PGI2-mediated activation of PPAR beta/delta and gamma and PGE2-dependent PPAR delta activation has been reported [14, 15]. All type of these receptors are expressed in endothelium [14], and both EP and IP receptors are expressed in lung tissue [16]. Gq-coupled EP1 belongs to “contractile” group of prostanoid receptors and activates PLC, leading to intracellular calcium increase. Both PGE2 and PGI2 can bind EP1 receptor [17]. The “inhibitory” Gi-coupled EP3 receptor decreases the levels of intracellular cAMP [15]. Thus, organ- or tissue-specific patterns of EP/IP receptor expression may determine organ-specific responses to prostaglandins. Prostaglandin binding to Gs-coupled EP2, EP4 and IP, which represent “relaxant” type of receptors leads to Gs-dependent activation of adenylate cyclase and elevation of intracellular cAMP levels [18]. Increases in intracellular cAMP levels have been associated with increased endothelial barrier integrity and linked to activation of PKA, which reduces endothelial MLCK activity, decreases pool of phosphorylated MLC, and leads to relaxation of actomyosin complex, stabilization of F-actin filaments and strengthening of cell-matrix adhesions [19–22]. In contrast, inhibition of basal cAMP/PKA activity increases pulmonary EC leak in part via activation of MAP kinase Erk1,2 [19].
Besides effects on MLCK activity, PKA may also differentially regulate small GTPases Rac and Rho. One potential mechanism of PKA-dependent barrier protection is PKA-mediated phosphorylation of Rho-GDP dissociation inhibitor, a negative regulator of small GTPase Rho, which results in Rho inactivation and blocks Rho-dependent mechanism of EC hyper-permeability [21]. Activation of cAMP/PKA-mediated signaling also has an inhibitory effect on RhoA activity [23] by direct phosphorylation of RhoA [23, 24]. In contrast to RhoA, Rac and Cdc42 can be activated by PKA without direct phosphorylation [25, 26], but via activation of guanine nucleotide exchange factors (GEFs)Tiam1 and Trio, which have consensus PKA phosphorylation sites [27]. Another GEF Vav2 demonstrates strong GEF exchange activity toward Rac1 and Cdc42 [28]. Phosphorylation of Vav2 by Src family tyrosine kinases at Y174 induces conformational change and makes Vav2 DH domain available for interaction with Rac [29–31]. Recent studies demonstrated other possible mechanisms of Vav2 activation/phosphorylation via Rap1 and PI3 kinase [32, 33].
Recent studies described a novel mechanism of cAMP-mediated endothelial barrier regulation by small GTPases. Increased intracellular cAMP levels directly activate the nucleotide exchange proteins directly activated by cAMP (Epacs or cAMP-GEFs) [34], which helps explain PKA-independent effects of cAMP on endothelial permeability. Epac1 and Epac2 splice variants contain a cAMP-binding domain and are capable of activating small GTPase Rap1 in a cAMP-dependent, PKA-independent manner [35]. Rap1 is a member of Ras family of small GTPases. Activated Rap1 may function in diverse processes, including integrin-mediated cell adhesion [35], cadherin-mediated cell junction formation and regulation of endothelial barrier [7, 36]. Rap1 activation induces localization of Rac-specific GEFs Tiam1 and Vav2 to the membrane areas at the cell periphery [35, 37], which causes local activation of small GTPase Rac. In turn, activated Rac stimulates effector signaling kinase PAK1 as well as cytoskeletal and cell contact-associated downstream effectors and induces rearrangements of actin cytoskeleton, focal adhesions and adherens junctions, which determine increased EC monolayer barrier properties [7, 10, 13, 38, 39].
In this study we investigated PKA-dependent and PKA-independent signaling pathways induced by PGE2 and therapeutic PGI2 analog beraprost and tested their involvement in Rac activation leading to cytoskeletal remodeling, increased cell-cell interactions, and lung endothelial barrier enhancement.
MATERIALS AND METHODS
Reagents and cell culture
Primary antibodies to Rac, Rap1, VE-cadherin, β-catenin were purchased from BD Transduction Laboratories (San Diego, CA). Tiam1, phospho-Vav2, p115Rho-GEF, and Epac1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Cortactin antibodies were obtained from Upstate Biotechnology (Lake Placid, NY). HRP-linked anti-mouse and anti-rabbit IgG, phospho-PAK1, and PAK1 antibodies were obtained from Cell Signaling Inc. (Beverly, MA). Texas Red phalloidin and Alexa Flour 488 conjugated secondary antibodies were purchased form Molecular Probes (Eugene, OR). GGTI-298, PKA inhibitory peptide (PKI), and NSC-23766 were purchased from Calbiochem (La Jolla, CA). Rac inhibitor NSC-23766 inhibits in vitro Rac activation via reduced binding to Tiam1 and Trio, but not Vav, Lbc, or intersectin. As for today, no evidence of NSC-23776-mediated inhibition between Rac1 and Vav2 has been reported. PKA inhibitory peptide (PKI) was purchased from Promega (Madison, WI). Myristoylated cell-permeable heat-stable protein kinase inhibitor (PKI) peptide sequence (14–22) is a highly specific inhibitor of PKA (Ki for non-myristoylated form = 36nM). Unless otherwise specified, biochemical reagents were obtained from Sigma (St. Louis, MO). Human pulmonary artery endothelial cells (HPAEC) were obtained from Cambrex (Walkersville, MD), cultured according to the manufacturer’s protocol, and used at passages 5–9.
Measurements of cAMP or cGMP accumulation
Cells were seeded in 12-well culture plates. At the day of assay, the cells were stimulated with the PGE2 or beraprost for desired periods of time. Cyclic AMP or GMP accumulation was quantified by competitive immunoassay (cAMP or cGMP Biotrak Enzymeimmunoassay (EIA), Amersham Biosciences, Little Chalfont, UK) and optical density was determined using plate reader at 630 nm. Each sample/standard was analyzed in duplicate and the resulting OD values were averaged.
Non-radioactive in vitro assay for PKA activity in cell lysates
EC were grown in 12-well plates to 100% confluence. After stimulation cells were lysed in lysis buffer (0.1 ml/well) containing 25 mM Tris-HCl pH 7.5, 150 mM sodium chloride, 5 mM magnesium chloride, 1% Nonidet P-40, 1 mM DDT, 5% Glycerol, and protease and phosphatase inhibitor cocktails (Sigma, St. Louis, MO). The lysates were cleared by centrifugation at 20,000 x g, 10 min, and 5 μl of cleared lysates were subjected to a kinase reaction with the fluorescence-labeled PKA substrate, kemptide (Promega, Madison, WI) following the manufacturer’s protocol. The reaction was stopped by boiling the samples for 10 min. The phosphorylated kemptide was separated by 0.8% agarose gel electrophoresis, and the fluorescence of the phosphorylated kemptide was detected and quantified by EagleEye Image System (Stratagene, La Jolla, CA).
Depletion of endogenous Epac1, Rap1, GEF-H1, Tiam1, Vav2, and Rac
To deplete endogenous GEF-H1, Tiam1, Vav2, or Rac HPAEC were treated with gene-specific siRNA duplexes, as described elsewhere [13, 40–42]. To deplete endogenous Epac1 and Rap1, Stealth™ Select 3 RNAi (Homo sapiens) sets were used. Pre-designed Stealth siRNAs of standard purity were ordered from Invitrogen (Carlsbad, CA), and transfection of EC with siRNA was performed as previously described [13, 40]. Nonspecific, non-targeting siRNA was used as a control treatment. After 48 or 72 hrs of transfection cells were used for experiments or harvested for western blot verification of specific protein depletion.
Rac and Rap activation assays were performed using commercially available assay kits purchased from Upstate Biotechnology (Lake Placid, NY), as previously described [12, 13]. In brief, cells were treated with vehicle or stimulated with PGE2 or beraprost for various periods of time. After the incubation with agonists, cell lysates were collected, and GTP-bound Rap or Rac were captured using pull-down assays with Ral GDS-RBD agarose or PAK-1 PBD agarose, respectively, according to the manufacturers protocols. The levels of activated small GTPases as well as total Rap and Rac content were evaluated by Western blot analysis and quantified by scanning densitometry of the autoradiography films. The levels of activated proteins Rap or Rac were normalized to total Rap or Rac level for densitometry evaluations.
Measurement of transendothelial electrical resistance
The cellular barrier properties were analyzed by measurements of transendothelial electrical resistance (TER) across confluent human pulmonary artery endothelial monolayers using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY) as previously described [12, 13, 40].
Transwell permeability assays
Permeability for FITC-labeled dextran was assessed in transwell assays using in Vitro Vascular Permeability Assay Kit (Chemicon International), according to the manufacturer’s instructions.
Mechanical ventilation protocol
Adult male C57BL/6J mice, 8–10 week old, with average weight 20–25 grams (Jackson Laboratories) were anesthetized with an intraperitoneal injection of ketamine (75 mg/kg) and acepromazine (1.5 mg/kg). Tracheotomy was performed and the trachea was cannulated with a 20-gauge-one inch long catheter (Johnson and Johnson), which was tied into place to prevent air leak. The animals have been placed on mechanical ventilation (Harvard Apparatus, Boston, MA) for 4 hours with high tidal volume (30 ml/kg, HTV) ventilation. Mice were randomized to concurrently receive sterile saline solution or beraprost (2μg/kg, intraperitoneal administration) at three time points (0, 40, and 80 min) during mechanical ventilation. After the experiment, animals were sacrificed by exsanguination under anesthesia. BAL was performed using 1 ml of sterile Hanks Balanced Salt Buffer. The BAL protein concentration was determined by a modified Lowrey colorimetric assay using a Bio-Rad DC protein assay kit (Bio-Rad Laboratories, Hercules, CA).
Immunofluorescent staining
Endothelial cells plated on glass coverslips were grown to confluence, stimulated with PGs or left untreated, and immunofluorescent staining of proteins of interest using corresponding antibodies was performed as described elsewhere [12, 13, 43]. Images were processed with Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) software.
Differential protein fractionation
Confluent HPAEC were stimulated with the agonist of interest, and after rapid wash with ice cold PBS, cytosolic fraction was isolated by centrifugation using extraction buffer containing 50 mM Tris-HCl pH 7.4, 100 mM sodium chloride, 0.01% digitonin, and protease/phosphatase inhibitor cocktail. Next, pellets were resuspended in extraction buffer containing 0.05 mol/L Tris-HCl pH 7.4, 2% Triton X-100, 100 mM sodium chloride and protease/phosphatase inhibitor cocktail and incubated on ice for 30 min. The membrane fraction was isolated by centrifugation (5 min, 16,000 g). Pellets containing cytoskeletal fraction were dissolved in 1xSDS sample buffer.
Western blot analysis
Immunoblotting detection of proteins of interest was performed as described previously [12, 43]. In brief, protein extracts were subjected to 12.5% or 7.5% SDS-PAGE, transferred to nitrocellulose membrane in semidry transfer apparatus (BioRad, Hercules, CA) 10 V for 1 hour, and probed with antibody of interest. Immunoreactive proteins were visualized by SuperSignal West Dura chemiluminescence reagent according to the manufacturer’s protocol (Pierce, Rockford, IL).
Statistical analysis
Results are expressed as means ± SE of three to ten independent experiments. Stimulated samples were compared to controls by unpaired Student’s t-test. For multiple-group comparisons, a one-way variance analysis (ANOVA), followed by the post hoc Fisher’s, test was used. P<0.05 was considered statistically significant.
RESULTS
Effects of prostaglandins on EC permeability and cytoskeletal remodeling
Effects of PGE2 and PGI2 analog beraprost on human pulmonary EC barrier properties were examined by the measurements of the changes in transendothelial electrical resistance (TER) of cell monolayers grown on gold electrodes and stimulated with various concentrations of PGs. Both, PGE2 and beraprost caused dose-dependent TER increases with maximal response to 200 ng/ml PGE2 and 500 ng/ml beraprost (Figure 1A). Maximal barrier protective response was observed by 15 min of stimulation, declined in the next 1–2 hrs, but remained elevated above baseline. Analysis of EC cytoskeletal remodeling in response to PG stimulation revealed increased accumulation of actin-binding protein cortactin and adherens junction protein β-catenin in the membrane fraction (Figure 1B). Consistent with barrier-protective effects on human pulmonary EC monolayers, treatment with PGE2 or beraprost dramatically increased areas covered by adherens junctions, as detected by VE-cadherin-positive immunofluorescent staining (Figure 1C, upper panels). Analysis of actin remodeling revealed that both PGs induced significant reduction in central stress fibers and rapid accumulation of peripheral F-actin at the areas covered by adherens junctions visible after 15 min of treatment (Figure 1C, middle and lower panels) and still preserved after 30 min of stimulation (data not shown).
Figure 1. Effect of PGE2 and beraprost on EC permeability, intracellular redistribution of cortactin and β-catenin and EC cytoskeletal remodeling.
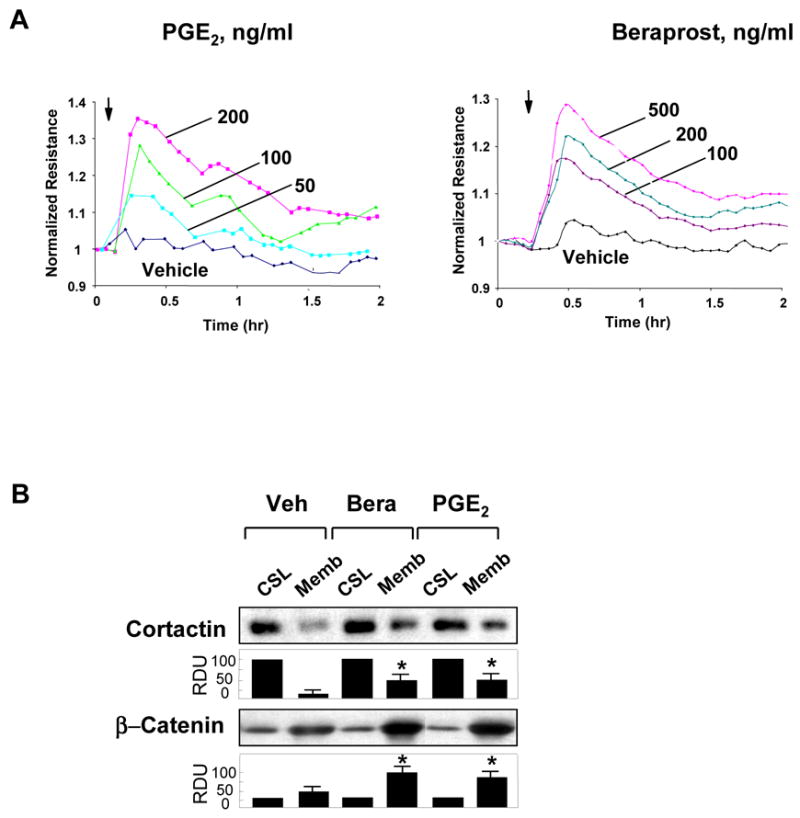
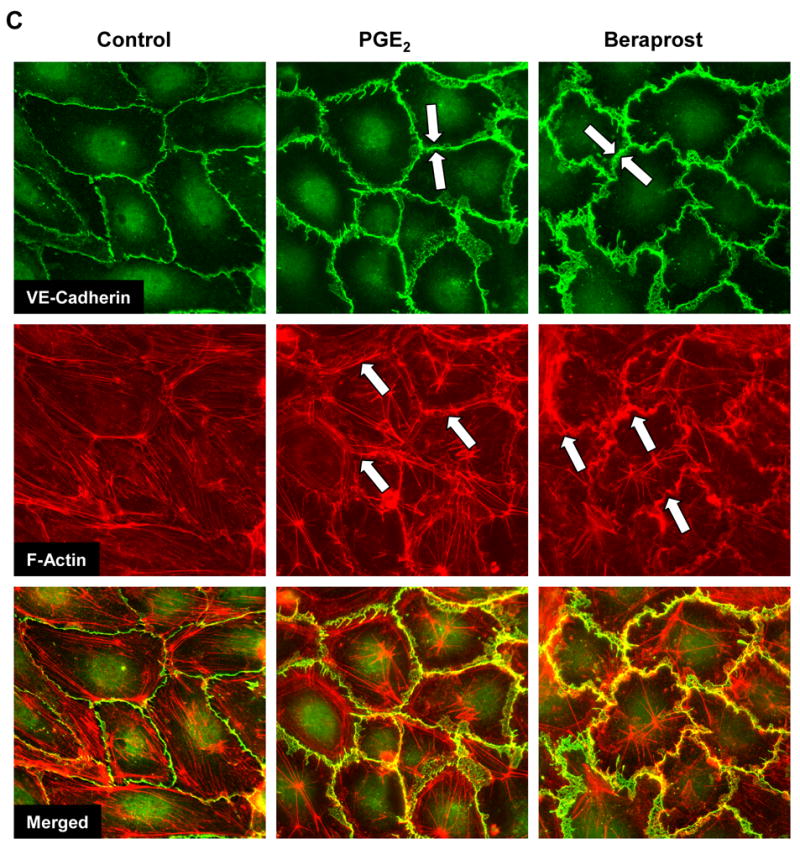
A: Human pulmonary EC monolayers were grown on gold microelectrodes. At the time point indicated by arrow, cells were treated with 50 ng/ml, 100 ng/ml, or 200 ng/ml PGE2 (left panel) or 100 ng/ml, 200 ng/ml, or 500 ng/ml beraprost (right panel) followed by measurements of TER reflecting EC monolayer barrier properties. B: EC were stimulated with PGE2 (200 ng/ml) or beraprost (500 ng/ml) for 15 min, and translocation of cortactin and β-catenin to the membrane fraction was detected with specific antibodies. Results are representative of three independent experiments. C: EC monolayers grown on glass coverslips were treated with PGE2 (200 ng/ml, 15 min) or beraprost (500 ng/ml, 15 min) followed by double immunofluorescent staining for VE-cadherin (upper panels) and F-actin (middle panels). Lower panel shows merged images of F-actin and VE-cadherin staining.
Activation of small GTPases and guanine nucleotide exchange factors by PGs
Direct measurements of small GTPase activation showed that TER increases and unique cytoskeletal remodeling induced by PGE2 and beraprost depicted in Figure 1 were associated with rapid activation of Rac GTPase (Figure 2A, upper panels) and activation of Cdc42 (Figure 2A, lower left panel). In contrast, Rho activity was not affected by beraprost (Figure 2A, lower right panel) or PGE2 stimulation (data not shown). Facilitation of GDP-GTP exchange in the nucleotide binding site of the small GTPases by guanine nucleotide exchange factors is a key mechanism of the regulation of small GTPase activity. In the next experiments we examined whether Tiam1 and Vav2 are involved in the PG-mediated barrier protection in human lung endothelium. Stimulation of EC with PGE2 or beraprost induced site-specific tyrosine phosphorylation of Vav2 in a time-dependent manner (Figure 2B, left panels), which along with Vav2 membrane translocation (Figure 2B, right panels) is essential for a full activation of Vav2 nucleotide exchange activity [33, 37]. PGs stimulation also induced membrane translocation of Tiam1 (Figure 2C), which reflects stimulation of its GEF activity towards Rac [37, 44]. Control experiments showed that PGs did not change subcellular distribution of Rho-specific p115Rho-GEF (Figure 2C, lower panel), which is consistent with the notion that Rho is not stimulated by PGs and p115Rho-GEF is not involved in PG-mediated Rac signaling.
Figure 2. Effect of PGE2 or beraprost on activation of Rac-dependent signaling.

EC were stimulated with PGE2 (200 ng/ml) or beraprost (500 ng/ml) for indicated periods of time. A: GTPase activation assays. Effects of PGE2 and beraprost on activation of Rac (upper panels), Cdc42 and Rho (lower panels) were evaluated using GTPase pulldown assays and normalized to the total GTPase content in cell lysates. B: Phosphorylation of Vav2 in control and PG-stimulated EC was determined in the total lysates using phospho-Vav2 specific antibody (left panels), or by immunofluorescent staining of beraprost-stimulated EC with phospho-Vav2 antibody (right panels). C: Time-dependent translocation of Tiam1 and p115-RhoGEF to the membrane fraction was detected by western blot with corresponding antibodies. D: Phosphorylation of PAK1 was determined in the total lysates using phospho-PAK1 specific antibody. Results are representative of three independent experiments.
A serine/threonine p21-activated kinase (PAK) is an important mediator of Rac and Cdc42 GTPase activity. Active Rac and Cdc42 induce catalytic activity of PAK via direct binding to its PBD domain. That rearranges kinase active site of PAK into catalytically competent state and leads to phosphorylation at Thr423 within the catalytic domain. This phosphorylation contributes to full catalytic activity of PAK and also prevents against PAK1 auto inhibition [45]. In accordance with PG-induced Rac activation, PGE2 and beraprost caused time-dependent phosphorylation of Rac effector PAK1 at Thr423 detected by western blot (Figure 2D). PAK autophosphosphorylation at this site results from PAK activation induced by Rac and Cdc42 GTPases [45].
Involvement of cAMP and PKA in PG-induced Rac activation and EC barrier regulation
Previous studies indicate that PG effects on endothelial permeability and vascular tone may be mediated in part by cAMP-dependent protein kinase [4, 46]. However, PKA-independent signaling pathways activated by PGI2 are much less investigated. Treatment of EC with PGE2 and beraprost markedly increased intracellular cAMP levels (Figure 3A, upper panels) without significant changes in cGMP levels (Figure 3A, lower panels). Elevation of cAMP was associated with increased enzymatic activity of PKA (Figure 3B) in a time scale corresponding to dynamics of cAMP increase. Importantly, experiments with pharmacological inhibitor of Rap1 processing GGT1 (30 μM, 1 hr) have shown that PGs-induced PKA activation was independent on Epac/Rap1 signaling (Figure 3C). As expected, PG-induced PKA activation was abolished by cell permeable PKA inhibitory peptide (PKI, 20 μM, 1 hr) (Figure 3C). Interestingly, inhibition of PKA activity by PKI also attenuated PGE2- or beraprost-induced Rac activation (Figure 4A). Control pretreatment with pharmacological Rac inhibitor NSC-23766 (200 μM, 1 hr) prior to PGE2 or beraprost stimulation completely abolished Rac activation. In addition, inhibition of PKA and Rac-dependent signaling significantly decreased PGI2-induced PAK1 phosphorylation and activation (Figure 4B). These data clearly demonstrate the involvement of PKA-mediated signaling in PG-induced Rac activation. To delineate a role of PKA-dependent mechanism in PG-induced EC barrier protective response, we measured PGE2- and beraprost-induced changes in transendothelial resistance (TER) in cells preincubated with PKA inhibitory peptide (PKI, 20 μM, 1 hr). PGE2- and beraprost-induced TER increases were attenuated by PKI (Figure 4C).
Figure 3. Effect of PGE2 and beraprost on cAMP, cGMP, and PKA activation.

A: EC were stimulated with PGE2 (200 ng/ml) or beraprost (500 ng/ml) for indicated periods of time, and intracellular cAMP and cGMP levels were determined using a non-radioactive immunoassay, as described in Materials and Methods. Results are mean ± SD of three independent experiments. *P<0.001. B and C: Cell lysates were analyzed for PKA activity by non-radioactive in vitro PKA assay. EC were stimulated with PGE2 (200 ng/ml) or beraprost (500 ng/ml) for indicated periods of time followed by determination of PKA activity (B). HPAEC were pretreated with GGT1 (30 μM, 1 hr) or PKI (20 μM, 1hr) prior to PGE2 or beraprost challenge for 5 min (C). The insets represent fluorescent phosphorylated form of PKA substrate kemptide separated from non-phosphorylated form by 0.8% agarose gel electrophoresis. The fluorescence intensity was detected and quantified by EagleEye Image System. Results are mean ± SD of three independent experiments. *P<0.001.
Figure 4. Effect of PKA inhibition on PGE2- and beraprost-induced Rac activation and EC barrier enhancement.
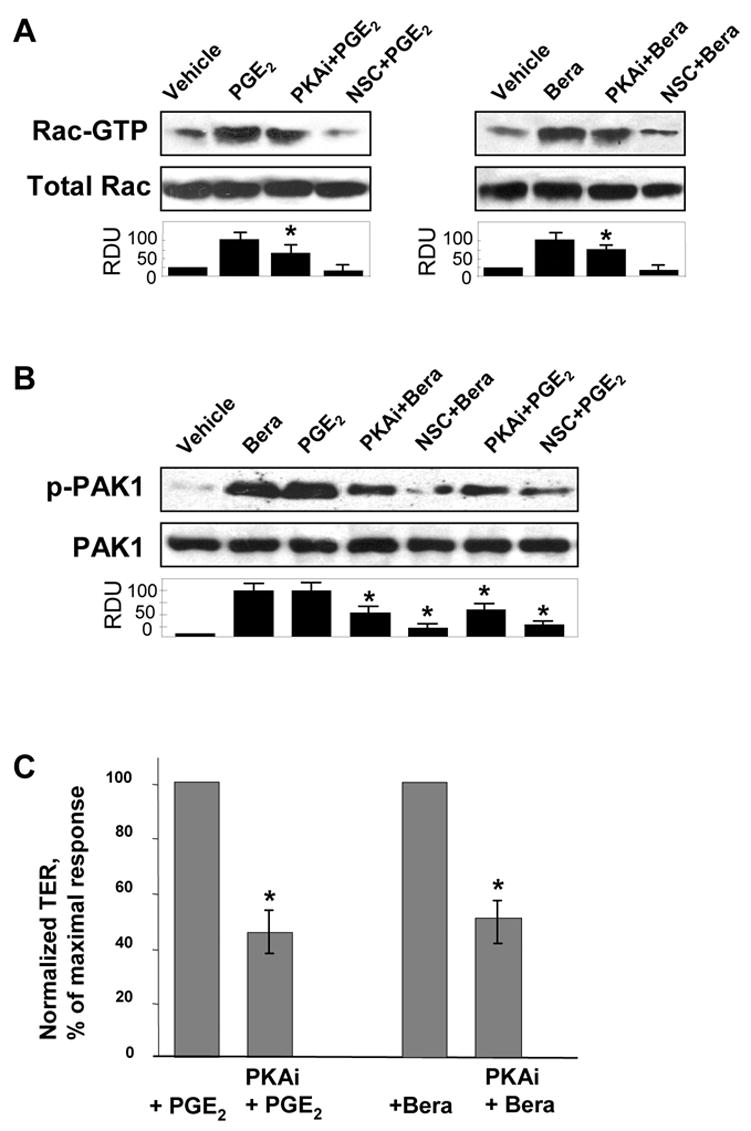
A: Pulmonary EC were pretreated with PKA inhibitory peptide PKI (20 μM, 1hr) or Rac inhibitor NSC-23766 (200 μM, 1hr) prior to PGE2 (200 ng/ml) or beraprost (500 ng/ml) stimulation. Measurements of Rac activity were performed using pull-down assays. B: PAK1 phosphorylation was detected by immunoblotting with phospho-specific antibodies. C: Pulmonary EC were pretreated with PKI (20 μM, 1hr) prior to PGE2 (200 ng/ml) or beraprost (500 ng/ml) stimulation, and TER changes after 15 min corresponding to maximal EC response were measured. Results are representative of three to five independent experiments.
Involvement of Epac and Rap1 in PG-induced Rac activation and pulmonary EC barrier protective responses
Recent reports described a novel mechanism of cAMP-dependent regulation of Rac pathway via cAMP-dependent nucleotide exchange factor Epac, which activates small GTPase Rap1 [7, 37]. We examined a role of Epac/Rap1 mechanism in the PG-induced activation of Rac pathway. Treatment of pulmonary EC with PGE2 or beraprost induced rapid activation of Rap1 (Figure 5A). Moreover, a pool of activated Rap (Rap-GTP) as a result of EC stimulation with PGE2 was associated of Rap-specific GEF Epac1, which was detected after re-probing the membranes containing GTP-bound Rap with anti-Epac1 antibody (Figure 5A, upper panel).
Figure 5. Effect of PGE2 and beraprost on Epac-Rap1 and Tiam1/Vav2-dependent Rac activation.

A: Rap1 activation pull-down assay. EC were stimulated with PGE2 (200 ng/ml) or beraprost (500 ng/ml) for indicated periods of time. Upper panels depict Epac1 bound to activated Rap1 (Rap-GTP). Lower panel shows total Rap1 content in EC lysates. B: EC were preincubated with inhibitor of Rap1 processing GGT1 (30 μM, 1 hr) or vehicle followed by the measurements of PGE2-induced Rac activation. C: Pulmonary EC were transfected with siRNA specific to Epac1, Rap1, Tiam1, or Vav2. Depletion of target proteins induced by specific siRNA duplexes was confirmed by immunoblotting with appropriate antibody, as compared to treatment with non-specific RNA. Immunoblot with β-actin antibody was used as normalization control. Results are representative of three to five independent experiments. D: Pulmonary EC were transfected with specific siRNAs followed by agonist stimulation and measurements of Rac activity. Control cells were treated with non-specific RNA. Results are representative of three to six independent experiments.
Because activated Rap1 may stimulate Rac-specific GEFs Tiam1 and Vav2 [37], in the next experiments we evaluated the involvement of Rap pathway in PG-induced activation of Rac. First, EC monolayers were preincubated with inhibitor of Rap1 processing GGT1 (30 μM, 1 hr) or vehicle followed by the measurements of PGE2-induced Rac activation. GGT1 dramatically reduced Rac activation in response to PGE2 (Figure 5B). In the next experiments, we sequentially depleted the signaling proteins mediating signal transduction from Epac to Rac. Using EC transfection with appropriate specific siRNA, we depleted endogenous Epac1, Rap1, Tiam1, or Vav2 in pulmonary EC. Specific protein depletion upon treatment with corresponding siRNA was confirmed by western blot (Figure 5C). Control EC was transfected with nonspecific RNA duplexes. After 72 hrs of transfection EC were stimulated with PGE2 or beraprost, and Rac activity was determined using in vitro pull-down assays. Knockdown of Epac1, Rap1, Tiam1, or Vav2 significantly reduced PGE2-induced Rac activation in comparison to PGE2-stimulated cells transfected with nonspecific RNA duplexes (Figure 5D, upper panels). Similar results were obtained in beraprost-stimulated EC (Figure 5D, lower panels). These results strongly suggest involvement of Epac1-Rap1-Tiam1/Vav2 signaling cascade in PG-induced activation of Rac. To further delineate the role of Epac/Rap1-dependent mechanisms in PG-induced EC barrier protective responses, we measured PG-induced TER changes in cells with depleted Rap1, Tiam1, Vav2 or Rac. Control cells were transfected with non-specific RNA duplexes. Knockdown of Rap1, Tiam1, Vav2, or Rac significantly attenuated PGE2-induced TER increases, whereas depletion of Rho-specific GEF-H1 was without effect (Figure 6A). Consistent with the data indicating the involvement of Rap1 signaling in the PG-mediated increase in TER, siRNA-based knockdown of Epac1 also significantly reduced protective effects of PGs on EC barrier (Figure 6B). Importantly, combined down-regulation of PKA (PKI, 20 μM, 1 hr) and Epac1 (siRNA-based depletion of Epac1) abolished EC barrier enhancement in response to prostaglandins (Figure 6B). Thus, involvement of PKA (Figure 4) and Epac (Figure 5) pathways in PG-induced Rac activation strongly suggests that Rac may serve as convergence point between PKA- and Epac1-Rap1 signaling mediating PG-induced endothelial cell barrier-protective responses.
Figure 6. Involvement of Rac, Epac1, Rap1, Tiam1, Vav2, and PKA in prostaglandin-mediated EC barrier enhancement.

A: EC monolayers were transfected with siRNA specific to Rap1, Tiam1, Vav2, Rac, or GEF-H1. Control cells were transfected with non-specific RNA. After 72 hrs of transfection, cells were stimulated with PGE2 (200 ng/ml), and TER changes after 15 min corresponding to maximal EC response were measured. B: Cells were transfected with Epac1-specific or non-specific siRNA. Measurements of EC permeability were performed in EC stimulated with PGE2 (200 ng/ml) or beraprost (500 ng/ml), or pre-treated with PKI (20 μM, 1hr) prior to PGs stimulation. Bar graphs depict changes in TER determined after 15 min of PG stimulation. Results are representative of three to seven independent experiments.
Role of Rap1 and Rac in PG-induced cytoskeletal and adherens junction remodeling
EC barrier enhancement induced by oxidized phospholipids, sphingosine 1-phosphate, hepatocyte growth factor [10, 13, 47] or prostaglandins (this study) is tightly associated with actomyosin remodeling characterized by Rac-dependent formation of sub-cortical F-actin rim and disappearance of central stress fibers. In the next experiments we explored a role of small GTPases Rap1 and Rac in PGE2-induced actin cytoskeletal remodeling using transfection of pulmonary EC with specific siRNAs. Control cells were transfected with non-specific RNA duplexes. Similarly to non-transfected cells (Figure 1C), EC transfected with non-specific RNA responded to PGE2 (200 ng/ml, 15 min) treatment by increased spreading, pronounced peripheral accumulation of F-actin (red) and enlarged adherens junction areas (green) monitored by VE-cadherin staining (Figure 7A). Overlapping areas of adherens junctions and actin filaments appeared in yellow. EC transfection with Rap1- or Rac-specific siRNA abolished PGE2-induced remodeling of peripheral actin cytoskeleton and adherens junctions (Figure 7B,C). Taken together, these results strongly suggest a critical role of Rap1/Rac-dependent pathway in the pulmonary EC cytoskeletal remodeling and barrier enhancement by PGs.
Figure 7. Effect of Rap1 and Rac knockdown on PGE2-induced cytoskeletal and adherens junction remodeling.

EC grown on glass coverslips and transfected with non-specific RNA (A), with Rap1-specific siRNA (B) or with Rac-specific siRNA (C) were stimulated with PGE2 (200 ng/ml, 15 min). Analysis of actin cytoskeletal remodeling was performed by double immunofluorescent staining with VE-cadherin and Texas Red phalloidin, as described in Methods section. Lower panel shows merged images of F-actin and VE-cadherin staining.
Protective effects of PG against lung vascular barrier dysfunction
To investigate the role of PGs in the regulation of lung barrier function we utilized in vitro and in vivo models of acute lung injury. First, we evaluated protective effects of PGs on endothelial barrier function using cell culture model of thrombin-induced EC hyper-permeability. Pretreatment of pulmonary EC monolayers with PGE2 or beraprost significantly attenuated permeability increase in response to thrombin (Figure 8A). In addition to TER measurements, PGs protective effects against thrombin-induced EC hyper-permeability we examined using the solute flux assay. EC monolayers grown on semi-permeable membranes were pretreated with PGE2 or beraprost alone or followed by thrombin challenge, and permeability for FITC-labeled dextran was assessed in transwell assays. Similar to TER results, PGE2 or beraprost markedly reduced basal EC monolayer permeability and significantly attenuated thrombin-induced permeability for FITC-labeled dextran (Figure 8B). Immunofluorescence analysis of cytoskeletal remodeling revealed that barrier-protective effects of PGs were accompanied by dramatic reduction of thrombin-induced stress fiber and paracellular gap formation, and preservation of adherens junctions reflecting protective effects of PGs against thrombin-induced disruption of pulmonary EC monolayer integrity (Figure 8C). Because thrombin-induced EC barrier dysfunction is associated with activation of Rho pathway [12], we next examined effects of PGs on the thrombin-induced Rho signaling. Pretreatment of EC with beraprost dramatically attenuated thrombin-induced Rho activation (Figure 8D) and MLC phosphorylation (Figure 8E).
Figure 8. Protective effects of prostaglandins in pulmonary cell culture model of thrombin-induced hyper-permeability and murine model of VILI.


A: Effect of prostaglandins on thrombin-induced barrier dysfunction. Pulmonary EC were pre-incubated with PGE2 (200 ng/ml) or beraprost (500 ng/ml) for 15 min followed by thrombin (0.5 U/ml) challenge, and TER changes were monitored over the time. B: Effect of prostaglandins on thrombin-induced EC permeability. Pulmonary EC were pre-incubated with PGE2 (200 ng/ml) or beraprost (500 ng/ml) followed by thrombin (0.5 U/ml) challenge, and measurements of permeability for FITC-labeled dextran. C: Effect of PG (beraprost, 500 ng/ml, 15 min) on thrombin (0.5 U/ml, 15 min)-induced cytoskeletal remodeling and adherens junction integrity. Double immunofluorescence staining was performed using VE-cadherin antibodies and Texas-Red phalloidin. Paracellular gaps are marked by arrows. D and E: Effect of PG on thrombin-induced activation of Rho-dependent pathway. HPAEC were pre-incubated with beraprost followed by treatment with thrombin for 5 min or 30 min and determination of Rho activity using pull-down assay (D). Phosphorylation of MLC in EC pretreated with beraprost followed by thrombin challenge was detected by western blot with specific antibodies (E). F: − 57BL/6J mice were subjected to mechanical ventilation at high tidal volume (HTV, 30 ml/kg, 4 hrs) or left spontaneously ventilated. Intravenous administration of beraprost (2 μg/kg) was performed at three time points (0, 40, and 80 min) during mechanical ventilation. Cell count (upper panel) and measurement of protein concentration (lower panel) were performed in bronchoalveolar lavage fluid taken from control and experimental animals. Results are represented as mean + SE; *p < 0.01; **p < 0.05; n=6 per group.
Protective effects of beraprost were further tested in the murine model of ventilator induced lung injury (VILI). Experimental animals were treated with beraprost (2 μg/kg, intraperitoneal administration, 3 times) or sterile PBS at the onset of high tidal volume mechanical ventilation (30 ml/kg, HTV). After 4 hours, bronchoalveolar lavage was performed, and protein concentration and cell counts were analyzed as described in the Methods. Mechanical ventilation at high tidal volume significantly increased BAL cell counts and BAL protein (Figure 8F). These effects were attenuated by injections of beraprost. There was no significant difference in BAL cell counts and BAL protein between PG-treated animals and controls in the absence of HTV.
Taken together, our results strongly suggest the protective effects of prostaglandins against agonist-induced lung vascular barrier dysfunction in vitro and against mechanical stress-induced lung injury in vivo.
DISCUSSION
Acute lung injury is one of the major causes of morbidity and mortality, and the search for lung barrier-protective agonists and investigations into molecular mechanisms of their action continue. We and others have previously described mechanisms of Rac activation by various groups of barrier-protective agonists such as sphingosine 1-phosphate, HGF and oxidized phosphocholine and characterized a role of Rac in EC cytoskeletal remodeling and pulmonary EC barrier protective responses [10, 11, 13, 47, 48].
Prostaglandins represent another important group of lipid mediators with barrier-protective potential towards vascular endothelium [6, 7]. However, the role of PGs and their receptors in inflammation is not well understood. Diverse effects of COX products on progression and resolution of acute lung injury remain to be investigated and represent an open field for the further studies. Recent reports demonstrate species-specific splice variants of prostanoid receptors and differential activation of prostanoid receptors in various cell types and organs. For example, at least eight EP3 receptor splice variants have been identified in humans [15, 16]. Thus, a diversity of prostanoid receptor expression and differential activation upon various stimuli may explain complexity of responses to prostaglandin stimulation. For instance, studies using rabbit isolated lungs showed that platelet-activating factor-induced edema formation was partially mediated by EP3 activation and PGE2 release. On the other hand, expression of PGH synthase gene leading to increased levels of prostacyclin and PGE2 markedly attenuated endotoxin-induced pulmonary edema and release of thromboxane B2 [49]. Protective effects of PGE2 on the endothelial function have been reported in the ischemia-reperfusion model using isolated rat hearts [50]. Our studies demonstrate barrier-protective effects of PGE2 and prostacyclin in human pulmonary EC and show significant protective effect of prostacyclin in the murine model of ventilator-induced lung injury. Despite the extensive investigation of receptor splice variants, tissue specificity and differential activation, physiological significance of these differences remains to be explored
Our results clearly demonstrate barrier-enhancing effects of PG in the pulmonary EC. Although involvement of PKA-dependent mechanisms in the PG protective effects on vascular endothelium has been recognized [19, 51, 52], the role of recently discovered Epac-Rap pathway in PG-mediated barrier regulation is not well understood.
Our novel data show that both, Epac/Rap1 and PKA pathways mediate PG-induced Rac activation and EC barrier protective response. Although some cellular responses to elevation of intracellular cAMP are regulated exclusively by PKA [53] or Epac [54] signaling pathways, or PKA and Epac may even play opposing roles [55], in many processes Epac and PKA pathways are frequently interconnected [56]. Recent report indicates that Rap1 activation induced by elevation of cAMP may be regulated by both Epac- and PKA-mediated signaling pathways [57]. In turn, Rap1-induced activation of Rac by Tiam1 may be additionally modulated by PKA-dependent phosphorylation of Tiam1 at PKA consensus site [58]. Thus, precise mechanisms of crosstalk between Epac/Rap1 and PKA signaling observed in this study and involved in Rac activation and EC barrier protective responses to PG remain to be elucidated.
Our results also indicate that PG-induced activation of Rac pathway was mediated by Rac-specific GEFs Tiam1 and Vav2. Activated Tiam1 translocated to the membrane [44] may become recruited to caveolae, be associated with trans-membrane receptors or co-localized with its activator Rap1 [37]. Similar to Tiam1, Vav2 also may be activated upon membrane translocation or by Src-mediated tyrosine phosphorylation [32, 33]. Whether Tiam1 or Vav2 is involved in PG-mediated barrier protection in human lung endothelium was unknown. Our results show that PGE2 and beraprost induced activation of Tiam1 and Vav2 judged by their membrane translocation and Vav2 tyrosine phosphorylation (Figure 2). In addition to activation by Rap1, Tiam1 may be regulated by PKA phosphorylation [58, 59]. These findings suggest complementary mechanisms of PG-induced Vav2 and Tiam1 activation that may be important for fine tuning of Rac regulation by PGs. Importantly, knockdown of Tiam1 or Vav2 expression significantly reduced Rac activation in response to prostaglandins (Figure 5D) and attenuated prostaglandin-induced barrier enhancement in pulmonary EC (Figure 5E). Therefore, we conclude that both GEFs are important for the mediation of barrier protective effects by prostaglandins.
Using siRNA approach we found that PG-induced Rac activation was critically dependent on Tiam1 and Vav2, as well as on Epac and Rap1 (Figure 2). These results are consistent with previous reports, which show that Tiam1 and Vav2 activation induced by elevation of intracellular cAMP may be regulated by Epac-Rap1-dependent mechanism [35, 37]. Thus, Epac-Rap1-Tiam1/Vav2 signaling complex, which may be associated with adherens junction structures [7, 36] may locally regulate Rac activity essential for cortical actin polymerization, peripheral cytoskeletal enhancement and PG-induced endothelial barrier protection observed in this study.
This study (Figure 7) shows essential role of Rap1-Rac-dependent mechanisms in PG-mediated remodeling of peripheral actin cytoskeleton and intercellular junctions critical for EC barrier-protective responses to PG. Combined activation of Rac and Cdc42 leads to peripheral translocation of Rac/Cdc42 cytoskeletal effectors cortactin, Arp2/3, WAVE, Arc21, and N-WASP leading to enhancement of cortical actin cytoskeleton and increased width of intercellular adherens junctions essential for EC barrier maintenance [10, 60–63]. Activation of Rac/Cdc42 effector kinase PAK1 also contributes to activation of WAVE-Arp2/3 complex and initiates peripheral actin polymerization [62]. Moreover, recent findings demonstrate ability of cortactin to regulate intracellular adhesion and cell spreading via direct interactions with cadherin and β-catenin within adherens junctions [64]. Thus, cortactin-β-catenin interactions may be involved in PGs-induced EC barrier enhancement and further studies are required to explore this potential mechanism.
Thus, our results summarized in Figure 9 suggest that both, PKA and Epac/Rap1 signaling cascades mediate PG-induced Rac activation and EC cytoskeletal remodeling leading to enhancement of EC monolayer barrier properties. In addition PG-induced activation of PKA may cause actin cytoskeletal remodeling by Rac-independent mechanisms, for example, via phosphorylation of myosin light chain kinase and reduction of its enzymatic activity [38, 65], or via phosphorylation of Rho regulatory protein RhoGDI leading to Rho inhibition [21, 38], or via PKA-mediated phosphorylation of vasodilator-stimulated phosphoprotein (VASP) leading to the structural relaxation of the actin cytoskeleton linked to tight junctions via VASP - ZO-1 complex, which result in enhanced endothelial barrier function [52]. Importantly, our studies revealed strong protective effects of PG in the pulmonary EC model of thrombin-induced barrier hyperpermeability and in the murine model of ventilator-induced lung injury (Figure 8). Our data suggest that protective effects by PG can involve Rac-mediated inactivation of Rho. Besides PKA-mediated RhoGDI phosphorylatioln, Rac may regulate RhoA activity via direct interaction with RhoGDI [66]. Alternatively, activated Tiam1/Rac may downregulate Rho activity via stimulation of Rho-specific GTPase activating protein p190-RhoGAP [67]. In addition, activation of Rac downstream target PAK1 may lead to further increase in Rac activity, therefore enhancing its inhibitory effects on Rho pathway [67]. However, precise mechanisms of crosstalk between Rac and Rho in PG-stimulated endothelial cells remain to be determined.
Figure 9. Upstream mechanisms of PG-induced Rac activation and cytoskeletal remodeling leading to endothelial barrier enhancement.

Stimulation of EC with PGE2 or PGI2 elevates intracellular cAMP levels and stimulates cAMP-dependent protein kinase (PKA), cAMP-activated guanine nucleotide exchange factor Epac1, which activates its effector small GTPase Rap1. Activated PKA and Rap1 promote Rac activation via stimulation of Rac specific GEFs Tiam1 and Vav2. Activated Rac interacts with downstream cytoskeletal and cell adhesion effectors and promotes cytoskeletal remodeling and EC barrier enhancement. In addition, PKA may directly affect EC cytoskeletal organization and monolayer barrier properties via modulation of myosin light chain kinase activity or VASP-dependent relaxation of actin cytoskeleton.
In conclusion, these studies provide comprehensive analysis of PKA- and Epac/Rap1-mediated mechanisms of Rac regulation and EC barrier protection in human pulmonary EC stimulated with PGE2 or prostacyclin analog beraprost. As molecular basis of acute lung injury is poorly understood, and no specific pharmacologic therapies are currently available, these studies advance our understanding of the molecular mechanisms underlying lung endothelial barrier regulation, and support potential therapeutic significance of PGs in the management of acute lung injury.
Acknowledgments
This work was supported by grants from National Heart, Lung, and Bl ood Institutes (HL076259 and HL075349) for KGB and the AHA Scientist Development Grant for AAB. The authors wish to thank Nurgul Moldobaeva for superb laboratory assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ricard JD, Dreyfuss D, Saumon G. Ventilator-induced lung injury. Eur Respir J Suppl. 2003;42:2s–9s. doi: 10.1183/09031936.03.00420103. [DOI] [PubMed] [Google Scholar]
- 2.Matthay MA, Zimmerman GA, Esmon C, et al. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003;167:1027–35. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- 3.Park GY, Christman JW. Involvement of cyclooxygenase-2 and prostaglandins in the molecular pathogenesis of inflammatory lung diseases. Am J Physiol Lung Cell Mol Physiol. 2006;290:L797–805. doi: 10.1152/ajplung.00513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howard LS, Morrell NW. New therapeutic agents for pulmonary vascular disease. Paediatr Respir Rev. 2005;6:285–91. doi: 10.1016/j.prrv.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Ueno Y, Koike H, Annoh S, et al. Anti-inflammatory effects of beraprost sodium, a stable analogue of PGI2, and its mechanisms. Prostaglandins. 1997;53:279–89. doi: 10.1016/s0090-6980(97)89601-3. [DOI] [PubMed] [Google Scholar]
- 6.Farmer PJ, Bernier SG, Lepage A, et al. Permeability of endothelial monolayers to albumin is increased by bradykinin and inhibited by prostaglandins. Am J Physiol Lung Cell Mol Physiol. 2001;280:L732–8. doi: 10.1152/ajplung.2001.280.4.L732. [DOI] [PubMed] [Google Scholar]
- 7.Fukuhara S, Sakurai A, Sano H, et al. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol. 2005;25:136–46. doi: 10.1128/MCB.25.1.136-146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 9.van Nieuw Amerongen GP, Draijer R, Vermeer MA, et al. Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ Res. 1998;83:1115–23. doi: 10.1161/01.res.83.11.1115. [DOI] [PubMed] [Google Scholar]
- 10.Garcia JG, Liu F, Verin AD, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wojciak-Stothard B, Potempa S, Eichholtz T, et al. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci. 2001;114:1343–55. doi: 10.1242/jcs.114.7.1343. [DOI] [PubMed] [Google Scholar]
- 12.Birukova AA, Smurova K, Birukov KG, et al. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004;67:64–77. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 13.Birukov KG, Bochkov VN, Birukova AA, et al. Epoxycyclopentenone-containing oxidized phospholipids restore endothelial barrier function via Cdc42 and Rac. Circ Res. 2004;95:892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- 14.Alfranca A, Iniguez MA, Fresno M, et al. Prostanoid signal transduction and gene expression in the endothelium: role in cardiovascular diseases. Cardiovasc Res. 2006;70:446–56. doi: 10.1016/j.cardiores.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 15.Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–66. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Breyer RM, Bagdassarian CK, Myers SA, et al. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–90. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 17.Ritchie RH, Rosenkranz AC, Huynh LP, et al. Activation of IP prostanoid receptors prevents cardiomyocyte hypertrophy via cAMP-dependent signaling. Am J Physiol Heart Circ Physiol. 2004;287:H1179–85. doi: 10.1152/ajpheart.00725.2003. [DOI] [PubMed] [Google Scholar]
- 18.Bos CL, Richel DJ, Ritsema T, et al. Prostanoids and prostanoid receptors in signal transduction. Int J Biochem Cell Biol. 2004;36:1187–205. doi: 10.1016/j.biocel.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Liu F, Verin AD, Borbiev T, et al. Role of cAMP-dependent protein kinase A activity in endothelial cell cytoskeleton rearrangement. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1309–17. doi: 10.1152/ajplung.2001.280.6.L1309. [DOI] [PubMed] [Google Scholar]
- 20.Birukova AA, Liu F, Garcia JG, et al. Protein kinase A attenuates endothelial cell barrier dysfunction induced by microtubule disassembly. Am J Physiol Lung Cell Mol Physiol. 2004;287:L86–93. doi: 10.1152/ajplung.00441.2003. [DOI] [PubMed] [Google Scholar]
- 21.Qiao J, Huang F, Lum H. PKA inhibits RhoA activation: a protection mechanism against endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2003;284:L972–80. doi: 10.1152/ajplung.00429.2002. [DOI] [PubMed] [Google Scholar]
- 22.Lampugnani MG, Giorgi M, Gaboli M, et al. Endothelial cell motility, integrin receptor clustering, and microfilament organization are inhibited by agents that increase intracellular cAMP. Lab Invest. 1990;63:521–31. [PubMed] [Google Scholar]
- 23.Dong JM, Leung T, Manser E, et al. cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J Biol Chem. 1998;273:22554–62. doi: 10.1074/jbc.273.35.22554. [DOI] [PubMed] [Google Scholar]
- 24.Lang P, Gesbert F, Delespine-Carmagnat M, et al. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. Embo J. 1996;15:510–9. [PMC free article] [PubMed] [Google Scholar]
- 25.Fleming IN, Elliott CM, Buchanan FG, et al. Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J Biol Chem. 1999;274:12753–8. doi: 10.1074/jbc.274.18.12753. [DOI] [PubMed] [Google Scholar]
- 26.Feoktistov I, Goldstein AE, Biaggioni I. Cyclic AMP and protein kinase A stimulate Cdc42: role of A(2) adenosine receptors in human mast cells. Mol Pharmacol. 2000;58:903–10. doi: 10.1124/mol.58.5.903. [DOI] [PubMed] [Google Scholar]
- 27.O’Connor KL, Mercurio AM. Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J Biol Chem. 2001;276:47895–900. doi: 10.1074/jbc.M107235200. [DOI] [PubMed] [Google Scholar]
- 28.Abe K, Rossman KL, Liu B, et al. Vav2 is an activator of Cdc42, Rac1, and RhoA. J Biol Chem. 2000;275:10141–9. doi: 10.1074/jbc.275.14.10141. [DOI] [PubMed] [Google Scholar]
- 29.Aghazadeh B, Lowry WE, Huang XY, et al. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell. 2000;102:625–33. doi: 10.1016/s0092-8674(00)00085-4. [DOI] [PubMed] [Google Scholar]
- 30.Bustelo XR. Regulatory and signaling properties of the Vav family. Mol Cell Biol. 2000;20:1461–77. doi: 10.1128/mcb.20.5.1461-1477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Servitja JM, Marinissen MJ, Sodhi A, et al. Rac1 function is required for Src-induced transformation. Evidence of a role for Tiam1 and Vav2 in Rac activation by Src. J Biol Chem. 2003;278:34339–46. doi: 10.1074/jbc.M302960200. [DOI] [PubMed] [Google Scholar]
- 32.Fukuyama T, Ogita H, Kawakatsu T, et al. Activation of Rac by cadherin through the c-Src-Rap1-phosphatidylinositol 3-kinase-Vav2 pathway. Oncogene. 2006;25:8–19. doi: 10.1038/sj.onc.1209010. [DOI] [PubMed] [Google Scholar]
- 33.Kawakatsu T, Ogita H, Fukuhara T, et al. Vav2 as a Rac-GDP/GTP exchange factor responsible for the nectin-induced, c-Src- and Cdc42-mediated activation of Rac. J Biol Chem. 2005;280:4940–7. doi: 10.1074/jbc.M408710200. [DOI] [PubMed] [Google Scholar]
- 34.de Rooij J, Zwartkruis FJ, Verheijen MH, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–7. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 35.Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol. 2003;4:733–8. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- 36.Kooistra MR, Corada M, Dejana E, et al. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579:4966–72. doi: 10.1016/j.febslet.2005.07.080. [DOI] [PubMed] [Google Scholar]
- 37.Arthur WT, Quilliam LA, Cooper JA. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J Cell Biol. 2004;167:111–22. doi: 10.1083/jcb.200404068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 39.Dudek SM, Jacobson JR, Chiang ET, et al. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. 2004;279:24692–700. doi: 10.1074/jbc.M313969200. [DOI] [PubMed] [Google Scholar]
- 40.Birukova AA, Adyshev D, Gorshkov B, et al. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2006;290:L540–8. doi: 10.1152/ajplung.00259.2005. [DOI] [PubMed] [Google Scholar]
- 41.Malliri A, van Es S, Huveneers S, et al. The Rac exchange factor Tiam1 is required for the establishment and maintenance of cadherin-based adhesions. J Biol Chem. 2004;279:30092–8. doi: 10.1074/jbc.M401192200. [DOI] [PubMed] [Google Scholar]
- 42.Gampel A, Mellor H. Small interfering RNAs as a tool to assign Rho GTPase exchange-factor function in vivo. Biochem J. 2002;366:393–8. doi: 10.1042/BJ20020844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Birukova AA, Chatchavalvanich S, Rios A, et al. Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am J Pathol. 2006;168:1749–61. doi: 10.2353/ajpath.2006.050431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singleton PA, Dudek SM, Chiang ET, et al. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. Faseb J. 2005;19:1646–56. doi: 10.1096/fj.05-3928com. [DOI] [PubMed] [Google Scholar]
- 45.Bokoch GM. Biology of the p21-Activated Kinases. Annu Rev Biochem. 2003;72:743–81. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- 46.Moncada S. Review. Adventures in vascular biology: a tale of two mediators. Philos Trans R Soc Lond B Biol Sci. 2006;361:735–59. doi: 10.1098/rstb.2005.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu F, Schaphorst KL, Verin AD, et al. Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3beta. FASEB J. 2002;16:950–62. doi: 10.1096/fj.01-0870com. [DOI] [PubMed] [Google Scholar]
- 48.Mehta D, Konstantoulaki M, Ahmmed GU, et al. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem. 2005;280:17320–8. doi: 10.1074/jbc.M411674200. [DOI] [PubMed] [Google Scholar]
- 49.Conary JT, Parker RE, Christman BW, et al. Protection of rabbit lungs from endotoxin injury by in vivo hyperexpression of the prostaglandin G/H synthase gene. J Clin Invest. 1994;93:1834–40. doi: 10.1172/JCI117169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouchard JF, Chouinard J, Lamontagne D. Participation of prostaglandin E2 in the endothelial protective effect of ischaemic preconditioning in isolated rat heart. Cardiovasc Res. 2000;45:418–27. doi: 10.1016/s0008-6363(99)00343-0. [DOI] [PubMed] [Google Scholar]
- 51.Yuan Y, Huang Q, Wu HM. Myosin light chain phosphorylation: modulation of basal and agonist-stimulated venular permeability. Am J Physiol. 1997;272:H1437–43. doi: 10.1152/ajpheart.1997.272.3.H1437. [DOI] [PubMed] [Google Scholar]
- 52.Comerford KM, Lawrence DW, Synnestvedt K, et al. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. Faseb J. 2002;16:583–5. doi: 10.1096/fj.01-0739fje. [DOI] [PubMed] [Google Scholar]
- 53.Huang SK, Wettlaufer SH, Hogaboam CM, et al. Prostaglandin E2 inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am J Physiol Lung Cell Mol Physiol. 2006 doi: 10.1152/ajplung.00232.2006. [DOI] [PubMed] [Google Scholar]
- 54.Lorenowicz MJ, van Gils J, de Boer M, et al. Epac1-Rap1 signaling regulates monocyte adhesion and chemotaxis. J Leukoc Biol. 2006 doi: 10.1189/jlb.0506357. [DOI] [PubMed] [Google Scholar]
- 55.Mei FC, Qiao J, Tsygankova OM, et al. Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem. 2002;277:11497–504. doi: 10.1074/jbc.M110856200. [DOI] [PubMed] [Google Scholar]
- 56.Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006 doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 57.Li J, O’Connor KL, Cheng X, et al. Cyclic Amp-Stimulated Neurotensin Secretion Is Mediated through Rap1 Downstream of Both Epac and Pka Signaling Pathways. Mol Endocrinol. 2006 doi: 10.1210/me.2006-0340. [DOI] [PubMed] [Google Scholar]
- 58.Zheng Y. Dbl family guanine nucleotide exchange factors. Trends Biochem Sci. 2001;26:724–32. doi: 10.1016/s0968-0004(01)01973-9. [DOI] [PubMed] [Google Scholar]
- 59.Minard ME, Kim LS, Price JE, et al. The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast Cancer Res Treat. 2004;84:21–32. doi: 10.1023/B:BREA.0000018421.31632.e6. [DOI] [PubMed] [Google Scholar]
- 60.McVerry BJ, Garcia JG. In vitro and in vivo modulation of vascular barrier integrity by sphingosine 1-phosphate: mechanistic insights. Cell Signal. 2005;17:131–9. doi: 10.1016/j.cellsig.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 61.Weaver AM, Karginov AV, Kinley AW, et al. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol. 2001;11:370–4. doi: 10.1016/s0960-9822(01)00098-7. [DOI] [PubMed] [Google Scholar]
- 62.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J 348 Pt. 2000;2:241–55. [PMC free article] [PubMed] [Google Scholar]
- 63.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–70. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 64.van Rossum AG, Moolenaar WH, Schuuring E. Cortactin affects cell migration by regulating intercellular adhesion and cell spreading. Exp Cell Res. 2006;312:1658–70. doi: 10.1016/j.yexcr.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 65.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2002;39:213–23. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 66.Wong KW, Mohammadi S, Isberg RR. Disruption of RhoGDI and RhoA regulation by a Rac1 specificity switch mutant. J Biol Chem. 2006;281:40379–88. doi: 10.1074/jbc.M605387200. [DOI] [PubMed] [Google Scholar]
- 67.Herbrand U, Ahmadian MR. p190-RhoGAP as an integral component of the Tiam1/Rac1-induced downregulation of Rho. Biol Chem. 2006;387:311–7. doi: 10.1515/BC.2006.041. [DOI] [PubMed] [Google Scholar]