

Abstract
Early during de novo infection of human microvascular dermal endothelial (HMVEC-d) cells, Kaposi's sarcoma-associated herpesvirus (KSHV) (human herpesvirus 8 [HHV-8]) induces the host cell's preexisting FAK, Src, phosphatidylinositol 3-kinase (PI3-K), Rho-GTPases, Diaphanous-2 (Dia-2), Ezrin, protein kinase C-ζ, extracellular signal-regulated kinase 1/2 (ERK1/2), and NF-κB signal pathways that are critical for virus entry, nuclear delivery of viral DNA, and initiation of viral gene expression. Since several of these signal molecules are known to be associated with lipid raft (LR) domains, we investigated the role of LR during KSHV infection of HMVEC-d cells. Pretreatment of cells with LR-disrupting agents methyl β-cyclo dextrin (MβCD) or nystatin significantly inhibited the expression of viral latent (ORF73) and lytic (ORF50) genes. LR disruption did not affect KSHV binding but increased viral DNA internalization. In contrast, association of internalized viral capsids with microtubules (MTs) and the quantity of infected nucleus-associated viral DNA were significantly reduced. Disorganized and disrupted MTs and thick rounded plasma membranes were observed in MβCD-treated cells. LR disruption did not affect KSHV-induced FAK and ERK1/2 phosphorylation; in contrast, it increased the phosphorylation of Src, significantly reduced the KSHV-induced PI3-K and RhoA-GTPase and NF-κB activation, and reduced the colocalizations of PI3-K and RhoA-GTPase with LRs. Biochemical characterization demonstrated the association of activated PI3-K with LR fractions which was inhibited by MβCD treatment. RhoA-GTPase activation was inhibited by PI3-K inhibitors, demonstrating that PI3-K is upstream to RhoA-GTPase. In addition, colocalization of Dia-2, a RhoA-GTPase activated molecule involved in MT activation, with LR was reduced. KSHV-RhoA-GTPase mediated acetylation and aggregation of MTs were also reduced. Taken together, these studies suggest that LRs of endothelial cells play critical roles in KSHV infection and gene expression, probably due to their roles in modulating KSHV-induced PI3-K, RhoA-GTPase, and Dia-2 molecules essential for postbinding and entry stages of infection such as modulation of microtubular dynamics, movement of virus in the cytoplasm, and nuclear delivery of viral DNA.
The gamma-2 herpesvirus Kaposi's sarcoma-associated herpesvirus (KSHV) or human herpesvirus 8 (HHV-8) is etiologically associated with Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease (25, 34, 45, 75). KSHV has a broad in vivo tropism, and KSHV DNA and RNA has been detected in human B cells, macrophages, keratinocytes, and endothelial and epithelial cells (25, 29, 83). In vitro, KSHV has been shown to infect many types of human cells, such as B cells; epithelial, endothelial, and foreskin fibroblasts; and keratinocytes (10, 54, 74, 83). In contrast to α- and β-herpesviruses, KSHV in vitro infection of target cells does not lead to the typical cascade of its immediate early, early, and late gene expression and progeny virus formation. Instead, KSHV establishes latency, and the viral genome is lost during successive passages of the infected cells (10, 32). Another novel feature of this in vitro latency in human microvascular dermal endothelial (HMVEC-d) and human foreskin fibroblast (HFF) cells is our demonstration of concurrent expression of a limited set of lytic cycle genes with antiapoptotic and immunomodulation functions, including the lytic cycle switch open reading frame 50 (ORF50) or RTA gene (39). While the expression of latent ORF72, ORF73, and K13 genes continues, nearly all lytic genes decline (10, 39). This characteristic expression of limited lytic cycle genes could be allowing KSHV to evade the immune system and to provide the necessary factors and time to establish and/or maintain latency during the initial phases of infection.
KSHV enters HFF cells, BJAB (KSHV- and Epstein-Barr virus [EBV]-negative B lymphoma) cells, and human embryonic kidney epithelial cells (293) by endocytosis (2, 5, 36), and this uptake is severely attenuated in cells pretreated with inhibitors affecting endosomal functions (2, 36). KSHV utilizes ubiquitous cell surface heparan sulfate (HS) to bind to several in vitro target cells such as BJAB, BCBL-1, 293, HFF, and HMVEC-d cells (5), and this interaction is mediated by the virion envelope glycoprotein gB and gpK8.1A (3, 78). Treatment of virus with heparin blocked the binding of radiolabeled virus to both adherent and nonadherent target cells, suggesting that HS probably serves as an initial contact receptor for the target cells. KSHV interacts with α3β1 integrin molecule of HMVEC-d and HFF cells via its envelope gB (4). Anti-α3 or -β1 antibodies or soluble α3β1 integrin blocked KSHV infection of HMVEC-d and HFF cells with ca. 50 to 70% reduction in infection as measured by green fluorescent protein (GFP) expression (4). A role of integrin in KSHV infection of B and other cells has not been demonstrated, and integrin could be one of the KSHV receptors in some adherent target cells and not in all cells.
KSHV also utilizes the transporter protein xCT for entry into adherent cells but not into the B cells (37). The xCT molecule is part of the 125-kDa disulfide-linked heterodimeric membrane glycoprotein CD98 (4F2 antigen) complex containing a common glycosylated heavy chain (80 kDa) and a group of 45-kDa light chains (19, 59). It has also been shown that CD98 constitutively associates with β1 integrin and is involved in membrane clustering and β1 integrin-mediated signal cascades (26, 27). KSHV also utilizes the dendritic cell-specific ICAM-3-grabbing nonintegrin (DC-SIGN; CD209) as a receptor for the infection of myeloid dendritic cells and macrophages (53). These studies suggest that KSHV utilizes multiple receptors for infection of target cells, and these receptors differ according to the cell type. This is not surprising since the related γ-1 EBV enters primary B cells via endocytosis and human epithelial cells via fusion at the cell membrane (12). EBV binds to the B cells via its gp350/220 interaction with the CR2 receptor and subsequently binds to HLA class II via its gp42 in the gH/gL complex (79). In contrast, infection of epithelial cells occurs via gH/gL complex interaction with an unknown receptor and/or via the interaction of the α5β1 integrin with the EBV-BMRF2 glycoprotein (72).
Integrin interactions with extracellular matrix proteins lead to the assembly of integrins, numerous signaling molecules including focal adhesion kinase (FAK), Src, and p130cas, and cytoskeletal proteins such as talin, paxillin, and vinculin into aggregates on each side of the membrane, creating the formation of focal adhesions (22, 30, 55, 81). KSHV-integrin interactions lead to the phosphorylation of FAK, which subsequently leads to the activation of Src, phosphatidylinositol 3-kinase (PI3-K), protein kinase C-ζ (PKC-ζ), Rho-GTPases, mitogen-activated protein kinase kinase (MEK), and extracellular signal regulated kinase 1/2 (ERK1/2) (46, 61, 62). Soluble gB induced extensive cytoskeletal rearrangement in the target cells via the induction of a FAK/Src/PI3-K/RhoGTPase signal pathway (62). KSHV-induced phosphorylation of FAK is essential for the entry of KSHV into adherent target cells, since entry of KSHV is greatly reduced in FAK-null fibroblasts (40). Inhibitors of cellular tyrosine kinases, Src kinases, PI3-K, and RhoA-GTPases blocked the entry of KSHV into target cells, suggesting a crucial role for Src, PI3-K, RhoA-GTPase, and other tyrosine kinases in KSHV entry (62). RhoA is also involved in the activation of microtubules (MTs) in HFF cells and is essential for the nuclear delivery of KSHV DNA (47).
In cell membranes, glycosphingolipids, cholesterol, and proteins show specific interactions to form microdomains, known as lipid rafts, which are resistant to solubilization in cold, nonionic detergents (63). Lipid rafts have been known to function as sorting centers for the delivery of lipids and proteins to the apical surface (13, 63, 65). Lipid rafts have also been attributed to a number of cellular functions such as signal transduction (64) and kinase activity modulation (24, 48, 82). Recent studies have suggested that lipid rafts play roles in a wide range of cellular events such as signal transduction, apoptosis, cell adhesion, migration, synaptic transmission, cytoskeletal organization, and protein sorting during endocytosis and exocytosis (13, 33, 64, 71). Since many of the host cell preexisting signaling cascade molecules induced by KSHV during its binding and entry into HFF and HMVEC-d cells are known to be associated with lipid rafts, we examined the role of lipid rafts during de novo KSHV infection of HMVEC-d cells.
Here, we show that KSHV viral gene expression is significantly reduced by lipid raft disruption in HMVEC-d cells. Lipid raft disruption did not affect the binding of KSHV to HMVEC-d cells. Although lipid raft disruption resulted in an increase of internalized viral DNA, nuclear delivery of viral DNA was significantly reduced in these cells. Methyl β-cyclo dextrin (MβCD) treatment resulted in the reduced association of KSHV capsids with MTs. An increase in KSHV induced Src phosphorylation and no alteration in FAK and ERK activation were observed in these cells. In contrast, significant reduction in KSHV-induced activation of PI3-K, RhoA-GTPase, and NF-κB; lipid raft association of PI3-K, RhoA-GTPase, and Diaphanous-2 (Dia-2; a RhoA-GTPase-activated adaptor molecule involved in MT activation); and reduction in acetylation of MTs were observed in lipid raft-disrupted cells. These studies suggest that lipid rafts of microvascular dermal endothelial cells are essential for KSHV infection and gene expression, and this could be due to their potential roles in the modulation of KSHV-induced PI3-K, RhoA-GTPase, and Dia-2 signal molecules playing roles in postbinding and entry stages of infection.
MATERIALS AND METHODS
Cells and viruses.
The HMVEC-d cells (CC-2543; Clonetics, Walkersville, MD) and BCBL-1 cells (KSHV carrying human B cells) used in the present study were propagated and maintained as described previously (2-5, 46). Induction of lytic cycle of KSHV from BCBL-1 cells, collection of virus from the supernatant, and viral purification were also carried out as described previously (46). Virus purity was assessed by general guidelines established in our laboratory (4, 39, 46). KSHV DNA was extracted from the virus, and copy numbers were quantitated by real-time DNA PCR using primers amplifying the KSHV ORF73 gene as described previously (39).
Antibodies and reagents.
MβCD, nystatin, heparin, LY294002 [20(4-morphodinyl)-8-phenyl-1(4H)-benzopyran-4-one], cytochalasin D, wortmannin, chlorpromazine, NH4Cl, tetradeconyl-phorbol acetate, lysophosphatidic acid, Triton X-100, and monoclonal antibodies to acetylated tubulin, total tubulin, and β-actin were obtained from Sigma, St. Louis, MO. Antibody to pERK, pNFκB was from Cell Signaling, Denver, MA. U0126 (1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenlythio butadiene) and PP2 were obtained from Calbiochem, La Jolla, CA. Mouse anti-phospho FAK (Y397), Mouse anti-caveolin and anti-FAK (total) antibodies were from BD Biosciences, San Jose, CA. Anti-phospho Src (PY418) and anti-Src (total) antibodies were obtained from Upstate Biotechnology, Lake Placid, NY. Anti-PI3-K, RhoA, mouse antihuman transferrin receptor, and Dia-2 antibodies were from Santa Cruz Biotechnology, Inc., Santa Cruz, CA. Anti-goat, anti-rabbit, and anti-mouse antibodies linked to horseradish peroxidase, alkaline phosphatase, fluorescein isothiocyanate, Alexa 488, and Alexa 594 were purchased from KPL, Inc., Gaithersburg, MD, and Molecular Probes, Eugene, OR. The Amplex Red Cholesterol assay kit and Vybrant lipid raft labeling kit were purchased from Molecular Probes. The FACE PI3-Kinase ELISA kit was purchased from Active Motif, Carlsbad, CA. The G-LISA-RhoA activation assay kit was purchased from Cytoskeleton, Denver, CO.
Cytotoxicity assays.
Target cells were incubated with Dulbecco modified Eagle medium containing different concentrations of various inhibitors for 4 h. Supernatants were collected and assessed for cellular toxicity by using an LDH cytotoxicity assay kit (Promega, Madison, WI) in accordance with the manufacturer's recommendations.
Preparation of DNA and RNA.
Isolation of total DNAs from the viral stocks and HMVEC-d cells by DNeasy tissue kit (QIAGEN, Inc., Valencia, CA) and isolation of total RNA from infected or uninfected cells using an RNeasy kit were carried out as described previously (39, 61).
Measurement of KSHV DNA internalization and nuclear delivery.
Untreated HMVEC-d cells or HMVEC-d cells incubated with nontoxic concentrations of different inhibitors for 1 h at 37°C were infected with GFP-KSHV at an MOI of 10 viral DNA copies per cell. After different time points of incubation with virus, cells were washed twice with HBSS to remove unbound virus. Cells were treated with 0.25% trypsin-EDTA for 5 min at 37°C to remove the bound, noninternalized virus. Detached cells were washed twice to remove the virus and trypsin-EDTA. Total DNA isolated from cells or from nuclear fractions was tested by real-time PCR for ORF73 as described previously (47).
Real-time RT-PCR.
The ORF50 and ORF73 transcripts were detected by real-time reverse transcription-PCR (RT-PCR) using gene-specific real-time primers and specific TaqMan probes as described previously (39).
Amplex Red Cholesterol assay.
HMVEC-d cells were washed twice with phosphate-buffered saline (PBS) and then incubated at 37°C for 1 h with different concentrations of MβCD in Dulbecco modified Eagle medium. After two washes with PBS, cells were harvested for cholesterol measurement assays as described by the manufacturer (Molecular Probes). In brief, 106 cells in 80 μl of PBS were lysed by three cycles of freeze-thawing, followed by ultrasonication (three bursts of 20 s each at room temperature). Cholesterol was extracted from the cell lysate by the addition of chloroform (200 μl) and methanol (200 μl) to the sonicated lysate (50 μl). The bottom chloroform layer was collected and evaporated under a vacuum. The residual cholesterol was dissolved in ethanol (50 μl) and assayed using the Amplex Red reagent, and the fluorescence intensity was measured in the microplate reader (Synergy HT Multi-Detection Microplate Reader) using excitation in the range of 530 to 560 nm and emission detection at ∼590 nm. For each point, the background fluorescence from the no-cholesterol control was subtracted, and the percentage of remaining cholesterol after MβCD treatment was calculated by dividing the fluorescence value of treated cells by the total fluorescence of untreated cells.
Radiolabeled KSHV binding assay.
HMVEC-d cells were preincubated with nontoxic doses of various inhibitors before the addition of [3H]thymidine-labeled, density gradient-purified KSHV (5,000 cpm) (3, 5). As a control, labeled KSHV was incubated with 100 μg of heparin/ml for 90 min at 4°C, added to HMVEC-d cells, and incubated for 90 min at 4°C. After incubation, the cells were washed five times and lysed with 1% sodium dodecyl sulfate (SDS) and 1% Triton X-100, and the radioactivity was precipitated with trichloroacetic acid and counted in a scintillation counter.
Western blot analysis.
Total cell lysates prepared from HMVEC-d cells were resolved on SDS-polyacrylamide gels, transferred onto nitrocellulose paper, and immunoblotted with primary antibodies (anti-pSrc, pFAK p-ERK, and pNF-κB). To confirm equal protein loading, blots were reacted with antibodies against human β-actin. Horseradish peroxidase- or alkaline phosphatase-conjugated secondary antibodies were used for detection. Immunoreactive bands were developed by either an enhanced chemiluminescence reaction (NEN Life Sciences Products, Boston, MA) or CDP-Star (Roche Diagnostics Corp., Indianapolis, IN) and quantified by following standard protocols (62).
Immunofluorescence assay (IFA) for ORF73 protein detection.
Confluent HMVEC-d cells grown in eight-well chamber slides were either treated with 5 mM MβCD or 50 μg of nystatin for 1 h or left untreated, washed, and infected with 10 multiplicities of infection (MOI) (DNA copies per cell) of KSHV for 2 h; complete medium was added, followed by incubation for 72 h. The cells were fixed in 3.7% paraformaldehyde, permeabilized in 0.5% Triton X-100, and incubated overnight with rabbit anti-ORF73 antibodies (1:80) in 5% bovine serum albumin (BSA). Cells were washed with PBS, incubated with anti-rabbit Alexa Fluor 488 antibodies for 1 h at room temperature, washed, mounted with antifade reagent containing DAPI (4′,6′-diamidino-2-phenylindole), and visualized under a Nikon fluorescent eclipse 80i microscope. Images were processed by using Metamorph imaging software.
IFA for acetylated tubulin detection.
HMVEC-d cells (90% confluence) in eight-well chamber slides were serum starved for 6 to 8 h. These were untreated or treated with 5 mM MβCD alone for 1 h or treated with 5 mM MβCD for 1 h and then infected with 10 MOI KSHV for various time points. Cells were washed, fixed in 3.7% paraformaldehyde, permeabilized with 0.1% Triton X-100, labeled with a 1:100 dilution of primary monoclonal antibody for acetylated tubulin for 1 h, washed, incubated with 1:500 anti-mouse secondary Alexa Fluor 488 for 1 h at room temperature, washed again, mounted with antifade reagent containing DAPI, and visualized under a Nikon fluorescent eclipse 80i microscope. Images were processed by using Metamorph imaging software.
Colocalization studies with lipid raft marker GM1.
Confluent HMVEC-d cells in eight-well chamber slides were uninfected or infected with KSHV or treated with various inhibitors at 37°C for 1 h and then infected with KSHV for the required time points. These cells were washed and labeled for lipid raft marker GM1 (lipid raft labeling kit) according to the manufacturer's instructions. Briefly, a 1:1,000 dilution of CT-B conjugate at 4°C was added for 10 min and washed thrice with PBS, and a 1:200 dilution of anti-CT-B antibody was added for 15 min at 4°C. These were washed, fixed with 3.7% paraformaldehyde, permeabilized in 0.5% Triton X-100, and stained with primary phospho-Src, anti-P-85, and anti-RhoA antibodies overnight at 4°C. The cells were washed, incubated with secondary anti-mouse Alexa Fluor 488 for 1 h at room temperature, washed, mounted with antifading agent containing DAPI, and examined under a fluorescence microscope.
Cytoplasmic trafficking of KSHV capsid.
Serum-starved HMVEC-d cells in chamber slides were preincubated with 10 μg of nocodazole or 5 mM MβCD or LY294002 (PI3-K inhibitor) for 1 h and infected with a high MOI of KSHV (100 viral DNA copies/cell) to facilitate visualization of viral capsids. These cells were washed, fixed for 15 min with 3.7% formaldehyde in PBS at room temperature, permeabilized for 3 min with 0.5% Triton X-100, and blocked for 30 min with 10% normal goat serum (Sigma) in PBS. The cells were incubated for 1 h at room temperature with a 1:100 dilution of rabbit polyclonal immunoglobulin G (IgG) against KSHV capsid ORF65 protein (42) and 1:500 dilution of a mouse anti-tubulin monoclonal antibody. After being washed with PBS, the cells were incubated with goat anti-mouse IgG-Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 594 (10 μg/ml) for tubulin and ORF65, respectively, for 1 h at room temperature, washed, and mounted with antifade reagent containing DAPI before examination.
Detergent-free method of raft microdomain isolation.
Raft microdomains were purified from HMVEC-d cells by a previously described method (67) with modifications. Briefly, HMVEC-d cells grown to 80 to 90% confluence in a 150-cm2 flask were washed twice with Hanks balanced salt solution. HMVEC-d cells were left uninfected, infected with 10 MOI KSHV for 30 min, or treated with 5 mM MβCD for 1 h; washed thoroughly; and then infected with 10 MOI of KSHV for 30 min. After infection, cells were washed with ice-cold PBS. Cells were then scraped into 2 to 4 ml of PBS and collected into cold 5-ml tubes. The cells were spun at 250 × g for 5 min. The supernatant was removed, and the cell pellet was resuspended with 1 ml of 500 mM Na2CO3 per tube. The content of tube was transferred to the Dounce homogenizer (kept on ice), and cells were lysed with 20 strokes using the tight-fitting pestle. The cell lysate was collected into a 5-ml tube, and further disruption of the membranes was achieved by passing the lysate 10 times through a 23-gauge needle. These cell lysates were sonicated three times for 15 s each time, and 1 ml of cell lysate was mixed with 1 ml of cold 90% sucrose-MBS {25 mM MES [2-(N-morpholinoethanesulfonic acid) plus 0.15 M NaCl [pH 6.5]}. This was overlaid onto 2 ml of cold 35% sucrose-MBS-Na2CO3 and 1.5 to 2 ml of cold 35% sucrose-MBS-Na2CO3 and centrifuged for 16 to 18 h at 45,000 rpm in SW-55 rotor at 4°C. Then, 500-μl fractions from the top to the bottom of the gradient were collected and stored at −80°C until processed. The pH of the fractions was adjusted with 1 N HCl, and protein levels in the isolated fractions were estimated by the microBCA method. A total of 20 μg of the lysate from the fractions was immunoblotted for caveolin as a raft marker and for transferrin as a nonraft marker.
Immunoprecipitation of raft fractions for PI3-K phosphorylation.
Portions (150 μg) of raft fraction lysates were immunoprecipitated by incubation with PI3-K p85α (Z-8) antibody and probed with anti-phosphotyrosine monoclonal antibody PY20. The total p85 (PI3-K) level was measured by testing the whole-cell lysate in Western blot assays with PI3-K p85α (Z-8) antibody. Immunoreactive bands were visualized scanned and quantitated by using Alphaease FC (Fluor Chem) software from Alpha Innotech, San Leandro, CA.
Measurement of PI3-K induction by FACE PI3-K ELISA.
The FACE (for fast activated cell-based) ELISA kit is designed to detect activated proteins in mammalian cells. The phospho-PI3-K p85 antibody raised against a synthetic phospho-(Tyr) p85 motif binding peptide (preferentially recognizes peptides and proteins containing phosphotyrosine at the consensus YXXM motif). The total PI3-K p85 antibody recognizes the total level of PI3-K p85 proteins regardless of the phosphorylation state. The FACE PI3-kinase kits can be used to study phosphorylated PI3-K relative to the cell number and also to determine PI3-K phosphorylation relative to the total PI3-K protein found in the cells. After the phospho-PI3-K and total PI3-K signals have been normalized for cell number, a comparison of the ratio of phosphorylated PI3-K to total PI3-K for each of the cell growth conditions is made. Briefly, HMVEC-d cells (90% confluence) serum starved for 6 to 8 h were left untreated or treated with 5 mM MβCD for 1 h, infected with 10 MOI KSHV for various time points, fixed in 4% formaldehyde, and assayed for PI3-K according to the manufacturer's instructions.
Determination of RhoA activity by G-LISA RhoA activation assay.
This assay is based on the principle that a Rho-GTP-binding protein is linked to the 96-well plates. The active GTP-bound Rho in the cell lysates binds to the wells, while the inactive GDP-bound Rho is removed during the washing steps. The bound active RhoA is detected with a RhoA specific antibody and quantitated by absorbance. The degree of RhoA activation is determined by comparing readings from the activated cell lysates versus the nonactivated cell lysates. The inactivation of RhoA is achieved by serum starvation of cells prior to the experiment for 6 to 8 h. HMVEC-d cells (90% confluence) serum starved for 6 to 8 h were left untreated or were treated with 5 mM MβCD for 1 h and infected with KSHV (10 DNA copies/cell) for various time points, and lysates were collected and tested for RhoA activation according to the manufacturer's instructions. To determine RhoA activity after PI3-K inhibition, HMVEC-d cells were untreated or pretreated with PI3-K inhibitors LY294002 (100 μM) and wortmannin (500 nM) and then infected with 10 MOI of KSHV for various time points. The RhoA-GTPase inhibitor, Clostridium difficile toxin B, was used as a control.
RESULTS
Lipid raft disruption inhibits KSHV gene expression in HMVEC-d cells.
As a first step toward understanding the role of lipid rafts in KSHV infection, HMVEC-d cells were treated with nontoxic doses of MβCD and nystatin, which sequester and chelate cholesterol from lipid rafts, respectively. After treatment, cells were washed and infected with KSHV (10 DNA copies/cell) for 2 h, and the ORF73 and ORF50 gene expression levels at different time points were measured by real-time RT-PCR. We have previously shown that in vitro infection of HMVEC-d cells by KSHV leads to concurrent persistent expression of latency associated ORF73, ORF72, and K13 genes and transient expression of limited lytic cycle genes with antiapoptotic and immunomodulation functions, including the lytic cycle switch ORF50 or RTA gene (39). Similar to our earlier finding, an increase in ORF50 gene expression was observed as early as 2 h postinfection (p.i.), which decreased by 24 h p.i. (Fig. 1A). In contrast, ORF73 latent gene expression slowly increased from 2 to 24 h p.i. (Fig. 1A). Although we observed differences in the copy numbers of the transcripts with different batches of KSHV, the patterns of expression were similar and were highly reproducible (Fig. 1A). When cells were pretreated with 10 μM MEK-ERK1/2 inhibitor U0126 for 1 h, similar to our earlier studies (61), we observed a significant reduction in both ORF73 and ORF50 gene expression (Fig. 1B).
FIG. 1.

Effect of lipid raft-disrupting agents on KSHV gene expression. HMVEC-d cells were infected with KSHV alone (A) or pretreated with 10 μM U0126 for 1 h prior to infection (B) or pretreated with 5 mM MβCD for 1 h and then infected (C) or cells treated with 50 μg of nystatin for 1 h and infected (D). After infection with KSHV (10 DNA copies/cell), total RNA was isolated at 2, 8, and 24 h p.i. and 50 ng of DNase-treated RNA/μl was subjected to real-time RT-PCR with ORF73 and ORF50 gene specific primers and TaqMan probes. Known concentrations of DNase-treated in vitro-transcribed ORF50 and ORF73 transcripts were used in a real-time RT-PCR to construct a standard graph from which the relative copy numbers of viral transcripts were calculated and normalized with GAPDH. Panels B, C, and D show histograms depicting the percent inhibition in RNA copy numbers for KSHV ORF73 and ORF50 genes in the presence of 10 μM U0126, 5 mM MβCD, and 50 μg of nystatin, respectively. Each reaction was done in duplicate, and each point represents the average standard deviation (SD) of three independent experiments. (E) Immunofluorescence observation of KSHV-ORF73 expression in lipid raft-disrupted cells. HMVEC-d cells (90% confluent) grown in eight-well chamber slides were infected with 10 DNA copies of KSHV (subpanels a and b)/cell or pretreated with 5 mM MβCD for 1 h and infected (subpanels c and d) or pretreated with 50 μg of nystatin for 1 h and infected (subpanels e and f). After infection for 2 h, complete medium was added, followed by incubation for 72 h. The cells were fixed in 3.7% paraformaldehyde for 15 min at room temperature, permeabilized, and incubated overnight with rabbit anti-ORF73 antibodies (1:80) in 5% BSA. Cells were washed with PBS, incubated with anti-rabbit Alexa Fluor 488 antibodies for 1 h at room temperature, washed, mounted with antifade agent containing DAPI, and visualized under a fluorescence microscope. Arrows show nuclear staining of ORF73 protein (LANA-1) (subpanels b, d, and f). Scale bars, 10 μm.
In MβCD-treated cells, ORF73 expression was inhibited by ca. 70 and 95% at 8 and 24 h p.i., respectively, and ORF50 expression was inhibited by ca. 80 and 85% by 8 and 24 h p.i., respectively (Fig. 1C). Similarly, in cells pretreated with 50 μg of nystatin, ORF73 levels were inhibited by ca. 80, 95, and 90%, and ORF50 levels were inhibited by ca. 90, 87, and 85% at 2, 8, and 24 h p.i., respectively (Fig. 1D). In MβCD-treated cells, we did not observe any alteration in host GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene expression (data not shown), demonstrating the specificity of reduction in viral gene expression and suggesting that lipid rafts play a significant role in KSHV viral gene expression.
To confirm these observations, we next examined the infected cells by immunofluorescence techniques. In HMVEC-d cells infected with 10 MOI of KSHV/cell, ca. 50% of cells exhibited the characteristic nuclear staining of ORF73 protein at 72 h p.i. (Fig. 1Ea and b). In contrast, in cells pretreated with lipid raft inhibitors, a dramatic reduction in the number of ORF73-expressing cells were observed (Fig. 1Ec to f), with ca. 86% reduction in 5 mM MβCD-treated cells (Fig. 1Ec and d) and ca. 80% reduction in 50 μg of nystatin-treated cells (Fig. 1Ee and f). The expression of ORF50 protein was also inhibited by ca. 80 to 90% with 5 mM MβCD or 50 μg of nystatin treatments (data not shown). These results further validated our observation that lipid rafts play an important role in KSHV infection.
Lipid raft disruption does not affect KSHV binding to HMVEC-d cells but enhances KSHV entry into HMVEC-d cells.
To determine whether treatment of HMVEC-d cells with various concentrations of MβCD and nystatin efficiently extracted cholesterol and to check whether the cholesterol levels remain depleted even after 24 h, we next assayed the cellular cholesterol levels. As shown in Fig. 2A and B, the cellular cholesterol levels decreased in a dose-dependent manner at both 1 h and 24 h after treatment with nontoxic doses of drugs. With 25-, 50-, and 100-μg concentrations of nystatin, we observed a reduction of ca. 40, 50, and 70%, respectively (Fig. 2A and B). Similarly, ca. 50, 60, and 70% reduction in cholesterol levels was observed with 2.5, 5, and 10 mM concentrations of MβCD, respectively (Fig. 2A and B). The depleted cholesterol levels remained low even after 24 h (Fig. 2B). In contrast, no significant change in cellular cholesterol levels was observed when cells were treated with 10 μg of nocodazole (MT-disrupting agent) or 1 mM NH4Cl (endocytic vesicle acidification inhibitor) for 1 h at 37°C (Fig. 2A). These results demonstrated that under our testing conditions both MβCD and nystatin treatment were efficient in sequestering or chelating cholesterol from lipid rafts of HMVEC-d cells and that depleted cellular cholesterol levels remained low even after 24 h of drug treatment.
FIG. 2.

Effect of MβCD and nystatin on KSHV infection. (A and B) Cholesterol depletion/chelation by MβCD and nystatin. HMVEC-d cells were treated with various concentrations of MβCD and nystatin for 1 h, and cholesterol levels were measured at 1 h (A) and 24 h (B) after treatment. The percentage reduction in the cholesterol upon drug treatment was calculated with respect to the total cholesterol in the untreated cells taken as 100%. Each reaction was done in duplicate, and each point represents the mean ± the SD of three independent experiments. (C) Disruption of lipid raft does not affect KSHV binding. HMVEC-d cells were either untreated or pretreated with various nontoxic concentrations of LY294002, NH4Cl, chlorpromazine, U0126, cytochalasin D, MβCD, or nystatin for 1 h. Cells were kept at 4°C for 1 h and incubated with a fixed concentration of [3H]thymidine-labeled virus in RPMI 1640 for 1 h with gentle rotation. As a control, [3H]thymidine-labeled KSHV was preincubated with 100 μg of heparin/ml for 1 h at 37°C before being added to the cells. After incubation, cells were washed, lysed, and precipitated with trichloroacetic acid, and the cell-associated virus radioactivity (in cpm) was counted. The cell-associated virus cpm in the presence of each different treatment was calculated as the percentage inhibition of virus binding. Each reaction was done in triplicate, and each point represents the average ± the SD of three independent experiments. (D) Chlorpromazine and microfilament depolymerizing agents does not affect KSHV DNA internalization in HMVEC-d cells. HMVEC-d cells grown in six-well plates were either untreated or preincubated with various nontoxic concentrations of nocodazole, chlorpromazine, U0126, LY294002, or NH4Cl. Cells were incubated with KSHV for 2 h, washed twice with PBS to remove unbound virus, treated with trypsin-EDTA for 5 min at 37°C to remove the bound noninternalized virus, and washed, and the total DNA was isolated. As a control, KSHV was preincubated with 100 μg of heparin/ml for 1 h at 37°C before being added to the cells. KSHV ORF73 DNA copies were estimated by real-time DNA PCR. The data are represented as the percentage of the inhibition of KSHV DNA internalization obtained when the cells were incubated with virus alone. Each reaction was done in duplicate, and each bar represents the average ± the SD of three experiments. (E and F) Lipid raft-disrupting agents increase the internalization of KSHV DNA in HMVEC-d cells. HMVEC-d cells after treatment with 5 mM MβCD (E) or 50 μg of nystatin (F) for 1 h were washed and infected with 10 MOI of KSHV. At different time points, internalized viral DNA was quantitated as described for Fig. 3B. The total viral DNA detected at 120 min in virus alone is considered as 100%. *, P < 0.01; **, P < 0.001.
Since the reduction in KSHV ORF73 and ORF50 gene expression in lipid raft-disrupted cells as shown in Fig. 1 could be due to interference in virus binding or viral DNA internalization or at postentry steps such as transport of the virus capsid to the cell nucleus and delivery of viral DNA to the nucleus, we next investigated the stage at which lipid rafts play a role in KSHV infection. KSHV has been shown to interact with adherent and nonadherent cell surface HS during the initial attachment stage of infection (5). To determine whether the inhibition in KSHV gene expression was due to the inability of KSHV to bind to endothelial cells, radiolabeled KSHV binding assay was carried out. Similar to our previous observation (2), heparin at a concentration of 100 μg/ml inhibited >80% of [3H]thymidine-labeled KSHV binding to HMVEC-d cells (Fig. 2C). In contrast, no significant effect on KSHV binding was seen with 5 mM MβCD and 50 μg of nystatin (Fig. 2C). Similarly, as shown before, no inhibition of binding was observed with PI3-K inhibitor (LY294002), MEK/ERK inhibitor (U0126), clathrin endocytosis inhibitor (chlorpromazine), endocytic vesicle acidification inhibitor (NH4Cl), or actin polymerization inhibitor (cytochalasin D) (Fig. 2C). These results demonstrated that the effect of lipid raft inhibition on KSHV gene expression was probably at a postattachment step of infection.
To determine whether reduction in KSHV gene expression in lipid raft-disrupted cells was due to interference in viral entry, total viral DNA internalization (cytoplasmic and nuclear) was measured by real-time DNA PCR. Similar to our earlier studies (62), we observed ca. 70 to 80% reduction in viral DNA internalization by preincubating virus with heparin or by pretreating cells with the PI3-K inhibitor LY294002 (Fig. 2D). No significant inhibition in KSHV-DNA internalization was seen when cells were treated with chlorpromazine or NH4Cl or cytochalasin D or U0126 (Fig. 2D). Surprisingly, we observed an increase in KSHV internalization in cells pretreated with 5 mM MβCD and 50 μg of nystatin (Fig. 2E and F). We compared the entry kinetics of KSHV internalization in untreated or MβCD- or nystatin-treated cells at 15, 30, 60, and 120 min p.i. When KSHV entry in untreated cells at each time point was taken as 100%, we observed a significant increase in viral DNA internalization at all time points in lipid raft-disrupted cells (Fig. 2E and F). KSHV-DNA internalization was maximal at the 2-h time point, with ca. 140 and 250% in MβCD- and nystatin-treated cells, respectively (Fig. 2E and F). These results suggested that intact lipid rafts probably play a critical checkpoint role in the orderly entry of KSHV, and their disruption probably affected the equilibrium of KSHV entry into HMVEC-d cells.
Lipid raft disruption decreases the nuclear delivery of KSHV-DNA in HMVEC-d cells.
To determine whether the observed reduction in KSHV gene expression despite the increased KSHV entry into the lipid raft disrupted cells was due to interference at the postentry steps such as delivery of KSHV-DNA to the nuclei, we next examined the kinetics of KSHV- DNA delivery into the infected cell nuclei. For this, the nuclear fractions of KSHV-infected HMVEC-d cells at various time points were isolated by cell lysis and gradient purification (47). Nuclear fractions were positive for the nuclear marker, lamin B (Fig. 3A, upper panel, lane 2), while the cytoskeletal marker tubulin was absent in these nuclear fractions (Fig. 3A, lower panel, lane 2). These results demonstrated that our nuclear fractions were mostly free of cytoskeletal contamination.
FIG. 3.
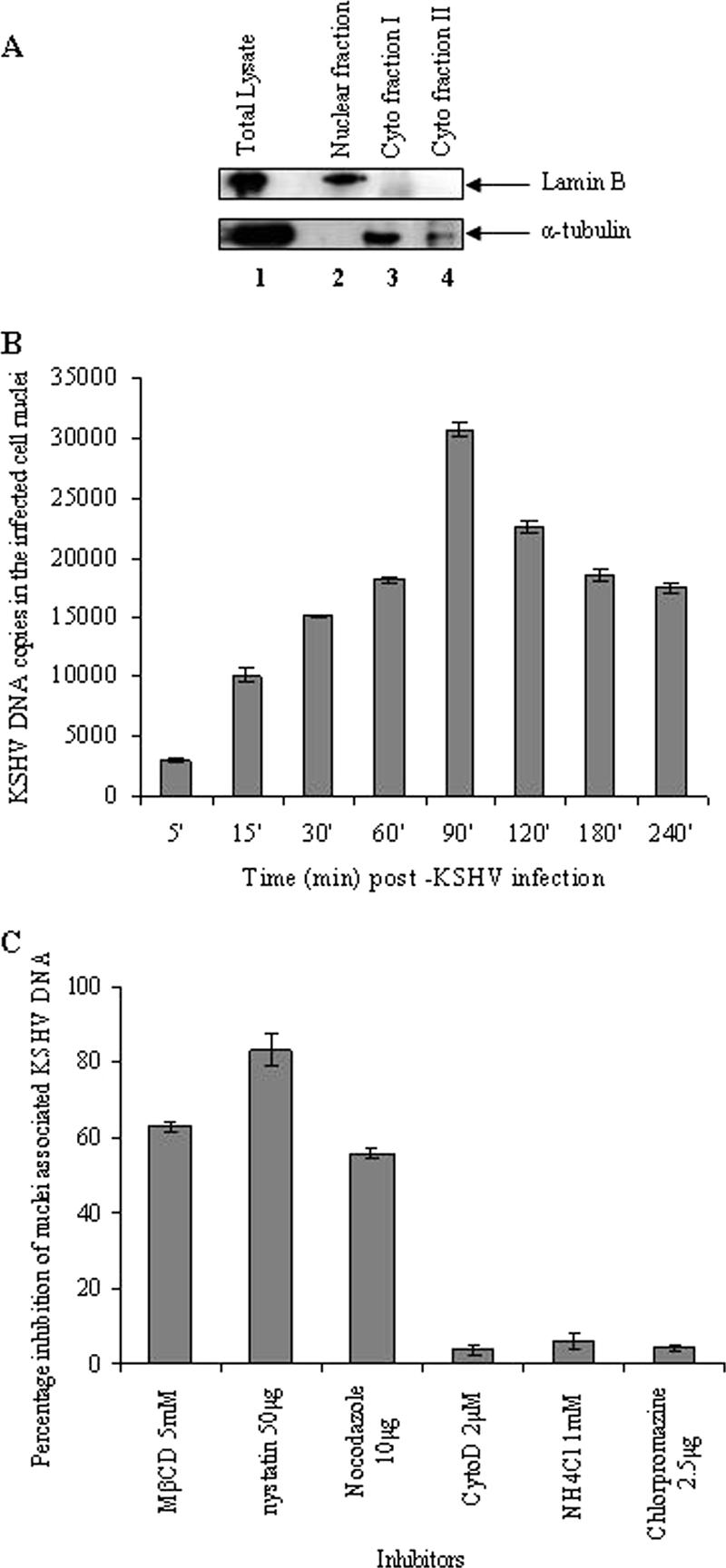
The lipid raft is crucial for the nuclear delivery of KSHV DNA. (A) Kinetics of KSHV DNA delivery into the infected cell nuclei. Uninfected and KSHV-infected HMVEC-d cells were washed, suspended in PBS, treated with 0.25% trypsin-EDTA to remove noninternalized virus, washed, suspended in lysis buffer, and allowed to swell on ice for 5 min. Nuclei and cytoplasmic fractions were prepared (44). (A) Total cell lysate (lane 1), purified nuclear fractions (lane 2), and cytoplasmic fractions I and II (lanes 3 and 4, respectively) were tested with monoclonal antibodies against lamin B and α-tubulin. (B) Kinetics of KSHV DNA delivery into the infected cell nuclei. Nuclear fractions from HMVEC-d cells infected with KSHV at 10 MOI for the indicated time points were isolated, and the total DNA was isolated, normalized to 100 ng/5 μl, and analyzed by real-time PCR with KSHV ORF73 primers. Each reaction was done in duplicate, and each bar represents the mean ± the SD for three experiments. (C) Lipid raft disruption reduces the nuclear delivery of KSHV DNA. HMVEC-d cells preincubated with nontoxic doses of MβCD, nystatin, nocodazole, cytochalasin D, NH4Cl, and chlorpromazine for 1 h were infected with KSHV (10 DNA copies/cell) in the presence of inhibitors. The purification of the nuclear fractions and real-time DNA PCR estimating the numbers of KSHV ORF73 copies was done as described in Materials and Methods. The data are presented as the percentage of inhibition of infected cell nucleus-associated KSHV DNA relative to that of cells infected with virus alone. Each reaction was done in duplicate, and each bar represents the mean ± the SD for three experiments.
When DNA extracted from untreated infected cell nuclei was quantified by real-time DNA PCR, a rapid significant nuclear delivery of KSHV-DNA as early as 5 to 15 min p.i. was observed, which peaked at 90 min p.i., gradually reduced thereafter between 120 to 240 min p.i. (Fig. 3B). Even though infected cell nucleus-associated KSHV DNA increased steadily over time, after 120 min p.i., we also observed a reduction in nucleus-associated viral DNA (Fig. 3B). The reason for this is not clear and could be due either to degradation of viral DNA by host intrinsic defenses in the nucleus and/or to host cell gene products modulated early during infection by KSHV (39).
When cells were pretreated with 5 mM MβCD and 50 μg of nystatin, we observed ca. 60 and 80% inhibition of nucleus-associated KSHV-DNA, respectively (Fig. 3C). Similar to our earlier observation in HFF cells (47), incubation of HMVEC-d cells with 10 μg of nocodazole blocked ca. 50% of nuclear viral DNA delivery (Fig. 3C). In contrast, pretreatment with cytochalasin D, NH4Cl, or chlorpromazine did not inhibit the nuclear delivery of KSHV-DNA (Fig. 3C). These studies demonstrated that despite increased KSHV entry in lipid raft-disrupted endothelial cells, there were defects in the delivery of KSHV-DNA to the infected cell nuclei, suggesting that these defects were probably responsible for the observed inhibition of viral gene expression.
Lipid raft disruption reduced the association of KSHV capsids with MTs and cytoplasmic trafficking in HMVEC-d cells.
To further verify these observations, we monitored KSHV capsid movement in the cytoplasm using our well-characterized antibodies against KSHV capsid protein ORF65 (42) and anti-α-tubulin antibodies to visualize MTs. At 1 h p.i. in the absence of drug treatments, abundant accumulation of KSHV capsids was detected as fine red dots that colocalized with MTs and near the infected cell nuclei (Fig. 4Aa to c). In contrast, in endothelial cells pretreated with 5 mM MβCD for 1 h at 37°C, although KSHV capsids were distributed throughout the cytoplasm, the majority of these did not colocalize with the MTs (Fig. 4Ad to f). Interestingly, KSHV-induced MT aggregation was comparatively less prominent in MβCD-treated cells and, instead, less-organized disrupted MTs and thickened rounded plasma membranes were prominently visible (Fig. 4Ad). Cells treated with 5 mM MβCD alone for 1 h at 37°C similarly exhibited less-organized disrupted MTs and thickened rounded plasma membranes (Fig. 4Bb and see Fig. 9Be). We have shown previously that treatment with the MT-disrupting agent nocodazole inhibited the MT-dependent transport of KSHV via dynein motor toward the nuclei of infected HFF cells (47). Similarly, in endothelial cells treated with nocodazole as a control, we observed the absence of MT aggregation, localization of KSHV capsid near the periphery of infected cell plasma membranes, and significant reduction in perinuclear accumulation of viral capsids (Fig. 4Ag to i). The specificity of these observations was shown by the absence of staining with preimmune IgG antibodies (data not shown).
FIG. 4.

Disruption of the lipid raft affects the trafficking of KSHV capsid. (A) Untreated HMVEC-d cells (Aa to c), cells preincubated with 5 mM MβCD for 1 h (Ad to f), and cells preincubated with 10 μg of nocodazole for 1 h (Ag to i) were infected with 100 MOI of KSHV for 1 h at 37°C, washed, fixed, permeabilized, reacted with rabbit anti-KSHV ORF65 antibodies and mouse anti-tubulin monoclonal antibodies, and detected by goat anti-rabbit IgG-Alexa Fluor 594 and goat anti-mouse Alexa Fluor 488 antibodies, respectively. The nuclei were stained with DAPI (blue). Arrows indicate ORF65 capsid staining, and arrowheads indicate MTs. Magnification, ×80. Scale bar, 20 μm. (B) Untreated HMVEC-d cells (Ba) and cells preincubated with 5 mM MβCD for 1 h (Bb) were stained with mouse anti-tubulin monoclonal antibodies and goat anti-mouse Alexa Fluor 594 antibodies. Scale bar, 10 μm.
FIG. 9.

Effect of lipid raft disruption on KSHV-induced MT acetylation. (A) Lipid raft disruption decreases hyperacetylation of tubulin in endothelial cells. Serum-starved HMVEC-d cells were either mock infected or infected with 10 MOI of KSHV for the indicated times and then lysed with RIPA lysis buffer. A total of 10 μg of lysate was resolved by SDS-10% polyacrylamide gel electrophoresis and immunoblotted with monoclonal antibody to acetylated tubulin (upper panel) or total tubulin (lower panel). Immunoreactive bands were developed by standard enhanced chemiluminescence reactions. The middle panel shows the fold phosphorylation of acetylated tubulin calculated with mock-infected HMVEC-d cells taken as 1. (B) Lipid raft disruption reduces the hyperacetylation of tubulin in endothelial cells. Serum-starved HMVEC-d cells were either mock treated (a) or infected with 10 MOI of KSHV (b to d). At different time points, the cells were washed, fixed in 4% formaldehyde, washed, and permeabilized in 0.5% Triton X-100 at room temperature. The cells were incubated for 30 min with 1% BSA and then with mouse anti-acetylated tubulin monoclonal antibody (diluted 1:500 in 1% BSA) at room temperature for 1 h. Cells were washed, incubated with anti-mouse Alexa Fluor 488 for 1 h, washed, mounted with slow fade agent containing DAPI, and visualized under a fluorescence microscope. Narrow arrowheads indicate bundles of acetylated tubulin upon infection. Scale bar, 20 μm. The insets at the bottom represents enlarged views of panels c, e, and g, respectively.
These results suggested that decreased association of KSHV capsid in lipid raft-disrupted cells may partially be responsible for the (i) decreased trafficking of internalized virus toward the nucleus, (ii) decreased KSHV-DNA nuclear delivery, and (iii) subsequent reduction in viral gene expression. The data also suggested that intact lipid rafts are critical for MT dynamics and that disruption affected the KSHV-induced MT modulation and association of viral capsid with MT.
Lipid raft disruption does not influence KSHV-induced FAK activation in HMVEC-d cells.
Early during the infection of adherent HMVEC-d and HFF cells, KSHV induced the integrin-dependent host cell preexisting FAK, Src, PI3-K, and Rho-GTPase signal pathways, as well as microtubular acetylation (46, 47). Inactivation of Rho-GTPases by Clostridium difficile toxin B significantly reduced microtubular acetylation and the delivery of viral DNA to the nucleus (47). Nuclear delivery of viral DNA increased in cells expressing a constitutively active RhoA mutant and decreased in cells expressing a dominant-negative mutant of RhoA (47, 73). KSHV capsid colocalization with MT was also abolished by PI3-K inhibitor affecting the Rho-GTPases (47). These results suggested that KSHV induced the Rho-GTPases and, in doing so, modulates the MTs and promotes the trafficking of viral capsid and the establishment of infection. These observations, coupled with the results presented above demonstrating the effect of lipid raft disruption on MT dynamics and trafficking of KSHV, suggested that KSHV-induced signaling pathways may depend upon intact lipid rafts. Hence, we next examined the activation of signaling molecules by KSHV in lipid raft-disrupted cells.
KSHV-induced FAK has been shown to be essential for virus entry into the adherent target cells (40). Similar to our earlier observations, KSHV induced robust FAK phosphorylation by ∼5-fold at 5 min p.i. of HMVEC-d cells, which then stabilized at ∼3-fold at 30, 60, and 120 min p.i (see supplemental Fig. 1A at http://66.99.255.20/cms/micro/Chandran.cfm). MβCD alone did not alter FAK phosphorylation in the uninfected cells. In cells pretreated with MβCD for 1 h at 37°C and then infected with KSHV, no significant change in FAK phosphorylation levels was observed (see supplemental Fig. 1A at http://66.99.255.20/cms/micro/Chandran.cfm). Equal loading of total lysates was confirmed with β-actin antibodies. These results suggested that the lipid raft does not play an active role in KSHV-induced FAK phosphorylation and that increased virus entry seen in lipid raft-disrupted cells (Fig. 4d-h) was not due to enhanced FAK activation.
Lipid raft disruption does not influence KSHV-induced ERK1/2 activation.
KSHV infection induces a rapid transient MEK1/2 and ERK1/2 phosphorylation in HMVEC-d and HFF cells, and ERK1/2 plays a critical role in the initiation of ORF50 and ORF73 gene expression, as well as host gene expression (61). Similar to our earlier observations, KSHV induced a robust ERK1/2 phosphorylation by about 2-, 4-, and 3-fold at 15, 30, and 60 min p.i., respectively, which returned to the basal level at 120 min p.i. (see supplemental Fig. 1 at http://66.99.255.20/cms/micro/Chandran.cfm). Interestingly, when uninfected cells were treated with MβCD alone for 1 h at 37°C, we observed a twofold induction of ERK1/2 phosphorylation. This is similar to other studies (17), demonstrating that the activation of ERK1/2 by MβCD alone in COS-1 and NIH 3T3 cells is due to cross-linking and ligand-independent activation of epidermal growth factor receptor. When cells were pretreated with MβCD for 1 h at 37°C and then infected with KSHV, we observed a moderate augmentation in ERK1/2 activation with about 3-, 4-, 4-, and 3-fold inductions at 15, 30, 60, and 120 min p.i., respectively (see supplemental Fig. 1 at http://66.99.255.20/cms/micro/Chandran.cfm). These results suggested that the observed reduced viral gene expression in lipid raft-disrupted cells (Fig. 1) was probably not due to the absence and/or reduction in KSHV-induced ERK1/2 necessary for viral gene expression.
Lipid raft disruption decreased the KSHV-induced NF-κB.
We have recently shown that NF-κB also plays a role in influencing viral gene expression (56). When HMVEC-d cells were infected with KSHV (10 MOI/cell) there was a rapid NF-κB activation as early as 15 min p.i. (see supplemental Fig. 1 at http://66.99.255.20/cms/micro/Chandran.cfm). As previously reported (56), there was a sustained activation of NF-κB until 120 min of our observation (see supplemental Fig. 1 at http://66.99.255.20/cms/micro/Chandran.cfm). When the cells were pretreated with 5 mM MβCD, there was a 50% reduction in NF-κB phosphorylation at 15 min p.i. and nearly 25 to 35% reduction at 30, 60, and 120 min p.i., respectively. Equal loading of total lysates was confirmed with β-actin antibodies (see supplemental Fig. 1 at http://66.99.255.20/cms/micro/Chandran.cfm). These results suggest that the observed reduced viral gene expression in lipid raft disrupted cells could be due to the reduction in the KSHV-induced NF-κB which is necessary for viral gene expression.
Lipid raft disruption increased KSHV-induced Src phosphorylation in HMVEC-d cells.
To check whether lipid raft disruption affected Src phosphorylation, lysates prepared from HMVEC-d cells before and after treatment with 5 mM MβCD were probed with antibodies against phospho-Src PY418. Src phosphorylation in uninfected cells was taken as onefold (Fig. 5B, upper panel, lane 1). Similar to our earlier observations, KSHV induced a robust Src phosphorylation by about 2-, 3-, and 4-fold at 15, 30, and 60 min p.i., respectively (Fig. 5B, upper panel, lanes 2 to 4). Cholesterol depletion by MβCD has been shown to constitutively phosphorylate Src in NIH 3T3 cells (49). Similarly, as seen for ERK1/2 activation (unpublished data), we also observed a twofold Src phosphorylation in uninfected cells treated with 5 mM MβCD alone (Fig. 5B, upper panel, lane 8). When MβCD-pretreated cells were infected with KSHV, we observed about 5-, 4-, and 4-fold Src activation at 15, 30, and 60 min p.i., respectively (Fig. 5B, upper panel, lanes 5 to 7). These results demonstrated the augmentation of KSHV-induced Src by lipid raft disruptions and suggested that, since Src is critical for KSHV entry (73), this increased Src activation in the presence of MβCD may potentially be responsible for the observed increased KSHV entry in lipid raft-disrupted cells (Fig. 2D).
FIG. 5.

(A) Src colocalizes with lipid rafts in KSHV-infected endothelial cells. HMVEC-d cells grown in eight-well chamber slides were serum starved for 6 to 8 h and infected for 10 min with or without 5 mM MβCD treatments. Cells were washed, labeled for lipid raft marker GM1, fixed, permeabilized, and blocked with 5% BSA for 1 h. These cells were washed, labeled with anti-p-Src antibodies for 1 h at room temperature, washed, and stained for 1 h with Alexa Fluor 488-conjugated secondary antibodies (p-Src) and Alexa Fluor 594 (GM1) at room temperature. Subpanels: a to c, uninfected cells; d to f, cells infected for 10 min; g to i, cells pretreated with PP2 (Src inhibitor) for 1 h and infected with KSHV for 10 min; j to l, cells pretreated with 5 mM MβCD alone for 60 min; m to o, cells pretreated with 5 mM MβCD for 60 min and infected with KSHV for 10 min. Stained cells were viewed with appropriate filters under a fluorescence microscope. The insets in panels c, f, i, l, and o show an enlarged view of colocalization. Arrows indicate colocalization of p-Src with GM1. Arrowheads indicate colocalization of p-Src with GM1 after MβCD treatment. Scale bars, 10 μm. (B) Effect of lipid raft disruption on KSHV-induced Src phosphorylation. Serum-starved HMVEC-d cells were pretreated with 5 mM MβCD for 1 h and infected with KSHV (10 MOI) for the indicated time points. Cells were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer containing protease inhibitors and cellular debris removed by centrifugation at 15,000 × g for 20 min at 4°C. Equal amounts of protein samples were resolved by SDS-7.5% polyacrylamide gel electrophoresis, transferred onto nitrocellulose membrane, and probed with anti-phospho Src (PY418) antibodies and total β-actin antibodies. Immunoreactive band intensities were assessed and are expressed as increased fold phosphorylation of Src over uninfected cells. Each blot is representative of a minimum of three separate experiments.
After activation, phosphorylated Src is known to colocalize with the plasma membranes. To confirm the increased Src activation morphologically, serum-starved HMVEC-d cells treated with or without MβCD or 4 μM PP2 for 1 h were infected with KSHV for 10 min and examined for phospho-Src colocalization with the lipid raft marker GM1. In uninfected cells, several discrete lipid raft spots were observed throughout the cells (Fig. 5Ab), and a dispersed low level of p-Src colocalization with lipid rafts was observed near the plasma membranes (Fig. 5Aa to c). In contrast, numerous very prominent spots representing both p-Src and lipid rafts were very readily observed throughout the KSHV-infected cells, and most of the p-Src spots colocalized with lipid rafts (Fig. 5Ad to f). The specificity of this reaction was shown by the near-complete abolition of p-Src/lipid raft colocalization in cells pretreated with Src inhibitor PP2 (Fig. 5Ag to i). In cells treated with MβCD alone, we observed very discrete p-Src spots (Fig. 5Aj). Although MβCD treatment reduced the number of lipid raft spots in uninfected cells (compare Fig. 5Ab with k), we observed prominent lipid raft spots colocalizing with p-Src (Fig. 5Aj to l). Since 5 mM MβCD treatment reduced the cholesterol levels only by ca. 70% (Fig. 2A), the observed lipid raft spots must be representing the residual and/or modified lipid raft region. When lipid raft-disrupted cells were infected with KSHV, although the lipid raft spots were reduced, aggregated p-Src colocalization with raft spots were readily seen (Fig. 5Am to o). Together with the biochemical studies shown in Fig. 5B, these studies suggested that MβCD treatment alone augmented the low level of Src phosphorylation that colocalized with residual lipid rafts and that KSHV-induced Src activation was increased by lipid raft disruption.
Lipid raft disruption downregulates KSHV-induced PI3-K activation in HMVEC-d cells.
PI3-K is an important regulator of cell proliferation, survival, and growth, and the phosphorylation of PI3-K leads to the downstream activation of a variety of cell signaling pathways (76). Early during the infection of target cells, PI3-K was induced by KSHV, which is critical for virus entry and induction of Rho-GTPases (61). Since PI3-K has been shown to be associated with lipid raft microdomains (24), we examined here the association of KSHV-induced PI3-K with lipid rafts and measured the phospho PI3-K and total PI3-K levels.
A rapid induction of PI3-K activity with about 2.5- and 3-fold activations at 5 min and 15 min p.i., respectively, was observed in KSHV-infected HMVEC-d cells, which peaked to 6-fold at 30 min and remained at ∼3-fold at 60 min p.i. (Fig. 6A). MβCD alone did not alter PI3-K phosphorylation significantly in the uninfected cells (data not shown). In contrast, infection of cells pretreated with MβCD resulted in the drastic reduction of PI3-K activity, with only about 1- to 1.5-fold induction throughout the 60-min period of observation (Fig. 6A). When serum-starved untreated HMVEC-d cells or cells preincubated with MβCD for 1 h were infected with KSHV and examined at 30 min (a time point of maximum PI3-K activation) (Fig. 6B), only very minimal colocalization of PI3-K with lipid rafts was observed in the uninfected cells (Fig. 6Ba to c). In contrast, very prominent PI3-K (P85α) colocalization with lipid rafts was observed throughout the untreated infected cells (Fig. 6Bd to f). As observed in Fig. 5A, lipid raft spots were vastly reduced by MβCD treatment in the uninfected cells (Fig. 6Bg to i), with negligible PI3-K and lipid raft colocalization (Fig. 6Bg-i). In lipid raft-disrupted KSHV-infected cells, we observed a substantial reduction in PI3-K and lipid raft colocalizations (Fig. 6Bj to l), which were dispersed and strikingly different from the high concentrations observed in virus-infected untreated cells (compare Fig. 6Bd to f to Fig. 6Bj to l).
FIG. 6.

Lipid raft disruption reduces KSHV-induced PI3-K activation and colocalization with lipid rafts. (A) HMVEC-d cells cultured in 96-well plates were serum starved for 6 to 8 h, treated with or without 5 mM MβCD for 1 h, and infected with 10 MOI of KSHV for the indicated time points. Total and phospho PI3-K levels were measured in triplicate by using the phospho and total PI3-K p85 antibodies and the FACE PI3-kinase kit. The data are represented as a bar graph showing fold activation of phospho PI3-K in HMVEC-d cells before and after 5 mM MβCD treatment. Each bar represents the mean ± the SD for three experiments. *, P < 0.01; **, P < 0.001. (B) HMVEC-d cells grown in eight-well chamber slides were serum starved for 6 to 8 h, treated with MβCD for 1 h or untreated, and infected for 30 min. Cells were washed, labeled for lipid raft marker GM1, fixed, permeabilized, and blocked with 5% BSA for 1 h. Cells were washed, labeled with anti-p85 antibodies overnight, washed, and stained with Alexa Fluor 488-conjugated secondary antibodies. These cells were washed, stained with DAPI, mounted, and analyzed. Subpanels: a to c, uninfected cells; d to f, cells infected for 30 min; g to i, cells pretreated with 5 mM MβCD alone for 60 min; j to l, cells pretreated with 5 mM MβCD for 60 min and infected for 30 min. Stained cells were viewed with appropriate filters under a fluorescence microscope. Arrowheads indicate colocalization of PI3-K with lipid rafts. Scale bars, 20 μm. (C) Characterization of lipid raft fractions. HMVEC-d cells were treated with 5 mM MβCD for 1 h, washed or left untreated, and then infected with 10 MOI KSHV for 30 min and washed. Cells were lysed, and raft and nonraft fractions were collected as described in Materials and Methods. Fractions were characterized as raft fractions or nonraft fractions by Western blotting with caveolin as a raft marker and transferrin as a nonraft marker. Fractions 3 to 5 were pooled as raft fractions, and fractions 6 to 8 were taken as nonraft fractions. (D) Association of PI3-K with lipid rafts upon KSHV infection. Portions (150 μg) of protein from the pooled raft and nonraft fractions were immunoprecipitated with PI3-K p85α (Z-8) antibody for 2 h at 4°C. The immune complexes were washed four times with ice-cold RIPA buffer containing protease inhibitors, and bound proteins were eluted by boiling in 50 μl of 2× Laemmli buffer for 3 min and subjected to Western blot analysis with anti-phosphotyrosine PY20 antibody (upper panel [p-P85α]) and for total PI3-K (lower panel). The percent inhibition of PI3-K activity in the raft fraction after MβCD treatment was calculated by comparison with PI3-K activity in the untreated infected lysate.
To determine whether activated PI3-K associates with lipid raft domains in endothelial cells upon KSHV infection, biochemical isolation of lipid rafts by the nondetergent method of Song et al. (67) was carried out. Fractions 3, 4, and 5, which were positive for lipid raft marker caveolin (Fig. 6C, left upper panel) and negative for transferrin (Fig. 6C, left bottom panel), were labeled as lipid rafts, and fractions 6, 7, and 8, which were positive for transferrin, a nonraft marker (Fig. 6C, right bottom panel) and negative for caveolin (Fig. 6C, right upper panel), were labeled as nonraft fractions. The PI3-K activities in the pooled raft fractions (3 to 5) and nonraft fractions (6 to 8) were examined by immunoprecipitation assays. A higher level of activated PI3-K was observed in the raft fractions than in the nonraft fractions in KSHV-infected cells (Fig. 6D, left and right upper panels). MβCD treatment decreased the association of activated PI3-K in the raft fractions by ca. 55% (Fig. 6D, right upper panel, middle lane), and less PI3-K activity was seen in the non-lipid raft fractions (Fig. 6D, right upper panel, last lane). Equal loading was confirmed by blotting with total PI3-K levels. These results suggested that activated PI3-K associates with lipid rafts in endothelial cells upon KSHV infection.
Taken together, these results suggested that lipid rafts play an active role in the modulation of KSHV-induced PI3-K activation and that the observed decreased PI3-K levels could play a significant role(s) in the postentry stages of KSHV infection in endothelial cells by modulating the downstream host cell signaling effector molecules and the associated cascades.
Lipid raft disruption downregulates KSHV-induced RhoA-GTPase activation in HMVEC-d cells.
RhoA, Rac1, and Cdc42, the best-studied members of the Rho-GTPase family, are the major downstream effector molecules of PI3-K and function as molecular switches by cycling between an inactive GDP-bound state and an active GTP-bound state. Besides their role in cytoskeleton reorganization, Rho-GTPases participate in a wide variety of cellular processes, including proliferation, differentiation, MT stabilization, endocytosis, vesicle trafficking, cytoplasmic transport, and gene expression (47). In KSHV-infected HFF cells, RhoA promoted the actin stress fibers, whereas Rac1 and Cdc42 mediated the lamellipodia and filopodial extensions, respectively (47). We have demonstrated that KSHV-induced RhoA-GTPase is critical for virus entry, microtubular acetylation, and the delivery of viral DNA to the nucleus (47, 73). These observations, together with the present study demonstrating the defective transport of virus to the nucleus by lipid raft disruption, prompted us to examine interlinks between KSHV-induced RhoA-GTPase activation and lipid rafts.
When RhoA-GTPase activation was examined by the G-LISA method, we observed a rapid 3.5-fold RhoA-GTPase activation by KSHV as early as 1 min p.i., peaking at 4-fold at 5 min p.i. and persisting at ∼2-fold during the 60 min of observation (Fig. 7A). Treatment of cells with MβCD alone did not significantly alter RhoA-GTPase phosphorylation (data not shown). When serum-starved HMVEC-d cells were pretreated with 5 mM MβCD for 1 h and then infected with KSHV for various time points, RhoA-GTPase activity was drastically inhibited between 1 and 30 min p.i. (Fig. 7A). This inhibition appears to be reversible slowly since ∼1.5-fold activation was seen at 60 min p.i. When uninfected cells were examined by IFA, only very minimal colocalizations of RhoA with lipid rafts were observed (Fig. 7Ba to c). At 5 min p.i., a time point of maximum RhoA-GTPase activation (Fig. 7A), we observed very prominent RhoA colocalization with lipid rafts throughout the untreated infected cells (Fig. 7Bd to f). Only negligible RhoA and lipid raft colocalization was seen in MβCD treated cells (Fig. 7Bg to i). In lipid raft-disrupted KSHV-infected cells, a substantial reduction in RhoA colocalization compared to untreated infected cells was seen (Fig. 7Bj to l).
FIG. 7.

Lipid raft disruption reduces KSHV-induced RhoA-GTPase activation and colocalization with lipid rafts. (A) HMVEC-d cells were serum starved for 6 to 8 h, treated with 5 mM MβCD for 1 h, and infected with 10 MOI of KSHV for the indicated time points. RhoA-GTPase levels were measured by G-LISA. A histogram depicts the fold change of RhoA activation. Each reaction was done in duplicate, and each bar represents the mean ± the SD for three experiments. **, P < 0.001. (B) HMVEC-d cells grown in eight-well chamber slides were serum starved for 6 to 8 h, treated with MβCD for 1 h or untreated, and infected for 5 min. Cells were washed, labeled for lipid raft marker GM1, fixed, permeabilized, and blocked with 5% BSA for 1 h. Cells were washed, labeled with anti-RhoA antibodies, washed, stained with Alexa Fluor 488-conjugated secondary antibodies, washed, stained with DAPI, mounted, and analyzed. Subpanels: a to c, uninfected cells; d to f, cells infected for 5 min; g to i, cells pretreated with 5 mM MβCD alone for 60 min; j to l, cells pretreated with 5 mM MβCD and infected for 5 min. Stained cells were viewed with appropriate filters under a fluorescence microscope. Arrowheads indicate colocalization of RhoA with GM1. (C) RhoA activation by KSHV in cells pretreated with PI3-K inhibitors. HMVEC-d cells were serum starved for 6 to 8 h, left untreated or pretreated with 100 μM LY294002 or with 500 nM wortmannin, washed, and infected with 10 MOI of KSHV for the indicated time points. RhoA-GTPase levels were measured by G-LISA. A histogram depicts the percent inhibition of RhoA activation compared to untreated infected cells at the respective time points. Each reaction was done in duplicate, and each bar represents the mean ± the SD for three experiments. (D) Cytoplasmic trafficking of KSHV capsid in cells pretreated with PI3-K inhibitor. HMVEC-d cells grown in eight-chamber slides were left untreated or pretreated with 100 μM LY294002 for 1 h and then infected with 100 MOI of KSHV for 1 h at 37°C washed, fixed, permeabilized, reacted with rabbit anti-KSHV ORF65 antibodies and mouse anti-tubulin monoclonal antibodies for 1 h at 4°C, and detected by goat anti-mouse IgG-Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 594 antibodies. The nuclei were stained with DAPI (blue). Arrows indicate ORF65 capsid staining, and arrowheads indicate MTs. The images were obtained on a Nikon 80i epifluorescence microscope, Magnification, ×40. Scale bar, 10 μm.
To further confirm that RhoA-GTPase is downstream to PI3-K activation, we examined RhoA-GTPase activation by KSHV in HMVEC-d cells pretreated with the PI3-K inhibitors LY294002 (100 μM) and wortmannin (500 nM). In PI-3K inhibitor-treated cells, we observed ca. 20 to 30% inhibition of RhoA-GTPase activity at 1 and 5 min p.i., 65 to 70% inhibition at 10 min p.i., 40% inhibition at 30 min p.i., and 30% inhibition at 60 min p.i (Fig. 7C). In cells pretreated for 90 min with Clostridium difficile toxin B (200 ng), a known RhoA-GTPase inhibitor, as a control, ca. 60% inhibition of RhoA-GTPase activation was observed (Fig. 7C). These results demonstrated that PI3-K is upstream of RhoA-GTPase activation.
To determine whether inhibition of PI3-K affected trafficking of KSHV capsid, HMVEC-d cells pretreated with 100 μM LY294002 were infected with 100 MOI of KSHV for 60 min. As shown in Fig. 4Aa and b, we observed the microtubular thickening and association of KSHV capsid with the MTs in untreated infected cells (Fig. 7Da to c), In contrast, the treatment with LY294002 resulted in the disruption of the MT network and, consequently, the absence of perinuclear accumulation of the KSHV capsids and lack of their MT association were observed (Fig. 7Dd to f).
These results demonstrated that lipid raft disruption significantly affected KSHV-induced activation of PI3-K and RhoA-GTPase. Since these molecules are critical for KSHV movement toward the infected cell nuclei (47), the data suggested that the observed reduced viral gene expression in lipid raft-disrupted cells (Fig. 1) could be attributed to reduced activation of PI3-K and RhoA-GTPase by KSHV in lipid raft-disrupted cells.
Lipid raft disruption downregulates KSHV-induced RhoA activation of Dia-2 molecule.
Dia-2, a diaphanous-related formin family of molecules activated by RhoA, is involved in mediating the interaction between RhoA and Src and forms part of the signal-transduction cascade leading to rearrangement of the cytoskeleton. Activation of Dia-2 by RhoA has been shown to stimulate the formation of stable MTs that are capped, and Dia-2 promotes this capping by directly binding to MTs (48). More importantly, we have demonstrated that KSHV-induced RhoA-GTPase is essential for the nuclear trafficking of viral DNA and in the regulation of MT dynamics (47, 73). KSHV infection of HFF and 293 cells increased the RhoA-dependent activation of Dia-2 early during infection (47, 73). Our studies suggested that KSHV-induced RhoA-GTPases probably control the MT dynamics via the activation of Dia-2 and thus facilitate the efficient trafficking of viral DNA to the infected cell nuclei.
To determine whether the reduced PI3-K and RhoA activation by KSHV in lipid raft-disrupted cells also results in the modulation of Dia-2 activity, we examined the association of Dia-2 with lipid rafts. When serum-starved untreated HMVEC-d cells or cells preincubated with MβCD for 1 h were infected with KSHV and examined at 10 min, a time point of maximum Dia-2 activation (47), only very minimal colocalization of Dia-2 with lipid rafts was observed in the uninfected cells (Fig. 8a to c). In contrast, very prominent Dia-2 colocalization with lipid rafts was seen in the infected cells (Fig. 8d to f). The specificity of this reaction was shown by the complete absence of Dia-2/lipid raft colocalization in cells pretreated with PP2 (Fig. 8g to i). In cells treated with MβCD alone, we observed a minimal amount of Dia-2 and lipid raft colocalization (Fig. 8j to l). In lipid raft-disrupted KSHV-infected cells, we observed a significant reduction in Dia-2 and lipid raft colocalizations (Fig. 8m to o) compared to untreated virus-infected cells (Fig. 8d to f).
FIG. 8.

Lipid raft disruption reduces KSHV-induced RhoA-GTPase-dependent Dia-2 colocalization with lipid rafts. Serum-starved HMVEC-d cells grown in eight-well chamber slides were infected for 10 min with or without 5 mM MβCD treatments. Cells were washed, labeled for lipid raft marker GM1, fixed, permeabilized, and blocked with 5% BSA for 1 h. Cells were washed, labeled with anti-Dia-2 with Alexa Fluor 488-conjugated secondary antibody for Dia-2 and Alexa Fluor 594 for GM1 for 1 h at room temperature. Subpanels: a to c, uninfected; d to f, cells infected for 10 min; g to i, cells pretreated with PP2 (Src inhibitor) for 1 h and infected; j to l, cells pretreated with 5 mM MβCD alone for 1 h; m to o, cells pretreated with 5 mM MβCD for 1 h and infected for 10 min. Arrows indicate the localization of Dia-2 with GM1. Arrowheads indicate the disruption of Dia-2 and GM1 after treatment with 5 mM MβCD. Scale bars, 10 μm.
These results further ascertain that lipid raft disruption significantly affect KSHV-induced activation of PI3-K, RhoA-GTPase, and Dia-2 molecules that are essential for KSHV movement toward the infected cell nuclei (47).
Lipid raft disruption downregulates KSHV-induced acetylation of MTs in endothelial cells.
Acetylation and deactylation are powerful and dynamic methods of controlling MT dynamics (51), and KSHV induces the acetylation of MTs in the infected HFF cells via the induction of PI3-K, RhoA-GTPase, and Dia-2 (47). Since hyperacetylation is a quantitative indication of changes in MT stabilization, we next quantified the levels of MT acetylation by Western blot analysis. Moderate levels of acetylated MTs were observed in uninfected cells (Fig. 9, upper panel, lane 13). In contrast, MTs were heavily acetylated in infected cells, with 8-fold induction as early as 5 min p.i. (Fig. 9, upper panel, lane 1); this increased to 10-fold between 15 and 60 min p.i. and persisted at about 7- to 8-fold during the 120 min of observation (Fig. 9, lanes 2 to 5). The levels of total tubulin remained unaltered, thus demonstrating the specificity of acetylation (Fig. 9, lower panel). KSHV-induced hyperacetylation was comparable to the induction by lysophosphatidic acid, a known inducer (Fig. 9A, upper panel, lane 12). Treatment with 5 mM MβCD for 1 h alone moderately induced MT acetylation by ∼2-fold (Fig. 9A, upper panel, lane 11). Infection of cells treated with MβCD inhibited the virus-induced MT acetylation by ca. 50% at 15 min p.i. (Fig. 9A, upper panel, lane 7).
The observed acetylation was also confirmed by determining the relative amounts of acetylated tubulin in KSHV-infected cells by IFA. Serum-starved uninfected HMVEC-d cells exhibited the classical pattern of finely spread out loose bundles of MTs radiating from the perinuclear nucleation center of MTs, the microtubular organizing center, toward the edges of the cell (Fig. 9Ba). As early as 5 min p.i., KSHV induced the aggregation of MTs, leading to an increased thickening of MT bundles (Fig. 9Bb), which were sustained for the 30 min of observation (Fig. 9Bb to d and inset panel c). These results demonstrated the modulation of MT dynamics early during infection by KSHV, resulting in transient reorganization of the MT network into bundles, replacing the normal MT cytoskeletal morphology. Cells pretreated with 5 mM MβCD alone exhibited less-organized disrupted MTs and thickened rounded plasma membranes (Fig. 9Be, inset). When lipid raft-disrupted cells were infected, the robust bundling of acetylated tubulin observed during KSHV infection alone was not observed. Instead, as seen in Fig. 4D, the bundles were disorganized, MT thickening was absent, and the less-organized disrupted MTs and thickened rounded plasma membranes were prominently visible in these cells (Fig. 9Bf to h and Fig. 9Bg, inset). Taken together, these results suggested that KSHV profoundly influences MT dynamics during the early stages of endothelial cell infection and that lipid rafts play important roles in KSHV-induced MT acetylation and aggregation that are essential for the effective delivery of viral DNA to the infected cell nuclei.
DISCUSSION
Viruses, bacteria, and parasites utilize lipid rafts, probably due to their capacity to concentrate receptors and signal transduction molecules from which multiple signaling pathways arise (6, 58, 63). Lipid rafts have been implicated not only in the entry of many enveloped and nonenveloped viruses but also in viral replication, assembly, budding, and release (16). We presented here comprehensive evidence to demonstrate that HMVEC-d cell lipid rafts are essential for KSHV infection and gene expression, and this is due to the potential role of lipid rafts in the proper functioning of KSHV-induced signal molecules critical for postbinding and entry stages of infection such as movement in the cytoplasm and nuclear delivery of viral DNA (Fig. 10). Our studies differ from other reports since this is the first demonstration of a direct role for lipid rafts in virus-induced signal pathways in the migration of viral capsid in the cytoplasm, MT dynamics, and nuclear delivery.
FIG. 10.

Model depicting the overlapping dynamic phases of KSHV-induced preexisting signal pathways and viral infection in HMVEC-d cells with intact lipid raft (A) and in lipid raft-disrupted cells (B). In phase 1, KSHV binds to adherent target cell HS molecules via its envelope glycoproteins gpK8.1A and gB, followed by interaction with α3β1 integrin via gB, interaction to CD98-xCT molecules, and possibly to other yet-to-be-identified molecules. Interactions with cell surface receptors trigger the cascades of host cell preexisting signal pathways (phases 2 to 4), which overlaps with virus entry by endocytosis. Activation of FAK and Src leads to the activation of PI3-K and Rho-GTPases, and RhoA activates Dia-2. Activated Src, PI3-K, Rho-GTPases, and Dia-2 colocalize with lipid raft domains. These signal cascades lead to the formation of endocytic vesicles, their movement in the cytoplasm, MT acetylation, and modulation of MT dynamics. In phase 5, the endosome moves in the cytoplasm, and the capsid is released, probably facilitated by the induced signaling pathways such as PKC-ζ. The endocytic vesicles with virus or released capsid or tegument complexes bind to dynein motor components, transported along the RhoA GTPase modulated MT to reach the nuclear vicinity, and deliver the viral DNA into the nucleus for viral gene expression (phase 6). When lipid rafts are disrupted (B), FAK and ERK activation is not altered. Increased Src phosphorylation occurs, which facilitates more viral internalization. Decreased activation of PI3-K and decreased association with lipid raft occurs, leading to reduced RhoA-GTPase and Dia-2 activation, reduced association with lipid raft domains, and reduced MT stabilization. The reduction in the activation of lipid raft-associated signal molecules such as PI3-K, RhoA-GTPase, and Dia-2 molecules essential for postbinding and entry stages of infection such as modulation of microtubular dynamics, movement of virus in the cytoplasm, and nuclear delivery of viral DNA, together with reduced NF-κB activation, probably account for the inhibition in the viral gene expression after lipid raft disruption. The present study thus demonstrates that lipid rafts of endothelial cells play critical roles in the postentry stage of KSHV infection.
The association of initial contact receptor(s) of a particular virus with lipid rafts probably dictates the role of lipid rafts in virus attachment, since lipid raft disruption affected the attachment of certain viruses but not others. Our studies demonstrate that the lipid raft does not play a direct role in KSHV binding to endothelial cells, suggesting that the initial contact receptors, such as HS, are probably not associated with lipid rafts. The association of other receptors, such as integrins and CD98-xCT, with lipid rafts is under study. Similar to our results, the disruption of rafts with MβCD significantly reduced the binding of human immunodeficiency virus type 1 (HIV-1) to T cells (Jurkat-1), affected HIV-1 envelope fusion in T cells, and did not affect binding of several strains of HIV-1 virions to brain microvascular endothelial cells at 4°C and HIV-1 infectivity at 37°C (8). These results demonstrate the cell type specificity of lipid raft requirements for HIV and suggest that the HS molecule that is believed to be involved in HIV-1 binding to endothelial cells may not be associated with lipid rafts and, in contrast, that CD4 and chemokine receptors mediating HIV-1 binding and entry into T cells must be intimately associated with lipid rafts in T cells and/or require an intact lipid raft.
The role of the lipid raft in virus entry appears to be a complex phenomenon and varies according to the virus and cell type, probably reflecting several independent variables such as the association of entry receptor(s) with the lipid raft, the mode of viral entry, the role of the lipid raft in fusion of viral and cellular membranes, lipid raft-associated signal molecules, their activation, downstream effector molecules, and the consequences of such activations, etc. Ongoing studies suggest that, similar to its entry into BJAB (B cell line), 293 (human epithelial), and HFF (fibroblast) cells, KSHV also enters HMVEC-d cells by endocytosis via macropinocytic vesicles (data not shown). Endothelial cell lipid rafts also do not appear to play a direct role in KSHV entry. This is similar to many enveloped viruses that use endocytosis as a mode of entry into the target cells, where the lipid raft is not required for entry. For example, Semliki Forest virus and Sindbis virus enter the target cells by endocytosis, and lipid rafts are not required for binding and for fusion with endosomal membranes (77). Similarly, vesicular stomatitis virus and coronavirus entry by endocytosis is not affected by MβCD (11, 70). Treatment of influenza virus by envelope cholesterol depletion did not affect infectious virus morphology, binding, and internalization (69). However, depletion of influenza virus envelope-associated cholesterol inhibited influenza virus fusion in the endosomes (69).
In contrast, lipid raft disruption by MβCD inhibited the entry of many viruses such as poliovirus, HIV-1 (in T cells), rotavirus, herpes simplex virus type 1 (HSV-1), and Ebola virus (9, 11, 23, 52, 57, 69). MβCD and nystatin efficiently inhibited EBV infection of Burkitt's lymphoma B-cell lines, as determined by the measurement of eGFP expression from the viral genome (38). Since EBV has been shown to enter these cells by fusion at the cell membrane, in contrast to endocytosis in primary B cells (44), and since infection of vesicular stomatitis virus G protein-pseudotyped retrovirus expressing GFP entering cells by endocytosis was not affected, it was concluded that lipid raft disruption affected the entry of EBV, presumably by affecting EBV receptors such as major histocompatibility complex class II proteins that are known to be associated with lipid rafts (38). However, whether EBV binds to the lipid raft-disrupted target cells was not examined.
Although MβCD treatment did not block the attachment of intracellular mature vaccinia virus to mammalian cells, it blocked a postbinding step of entry, possibly membrane fusion (21). Similarly, lipid rafts are not required for murine coronavirus binding, but are essential for entry following virus binding, suggesting that murine coronavirus entry requires specific interactions between the spike protein and lipid rafts during the internalization step (20). Lipid raft disruption has been shown to affect the entry of other nonenveloped virus such as Echo virus, echovirus II, and simian virus 40 (18, 20, 68). The receptors for these viruses, α2β1 integrin, CD55, and major histocompatibility complex class I, respectively, localize to cholesterol-rich domains and, as the result of virus attachment, allow the viruses to access the caveolin-dependent pathways that occur from these domains.
To define the role of lipid raft in HSV-1 cells, lipid raft-disrupted Vero cells were infected with HSV-1 and assayed for β-galactosidase (β-Gal) activity under the control of HSV-1 early gene (11). A dose-dependent inhibition of β-Gal activity (HSV-1 early gene expression) was observed that was taken as evidence for the role of lipid rafts in HSV-1 entry into target cells. However, the observed inhibition of β-Gal gene expression from HSV-1 genome in lipid raft-disrupted cells could be due to the inhibition of virus binding to the target cells, the inhibition of internalization of viral DNA, the inhibition of viral capsid transport in the cytoplasm, or the inhibition of viral gene expression after nuclear entry.
Lipid rafts serve as foci for the recruitment of transmembrane proteins and intracellular signaling molecules at the plasma membrane (7, 60, 64). Several membrane proteins, such as caveolin and glycosyl phosphotidylinositol (GPI)-anchored proteins, as well as cytoplasmic proteins such as Src family kinases, are enriched in rafts, and recruitment of transmembrane receptors such as integrins into rafts promotes their interaction with local kinases and other signal molecules, resulting in downstream signaling (7, 60, 64). Various models have been proposed for the mechanism by which the above-mentioned recruitment occurs, and many aspects of the recruitment and activation phases are not clearly understood (7, 60, 64). Cholesterol depletion has been shown to induce Src and ERK1/2 activation and reduce PI3-K activation (17, 31, 49, 50). One of the surprising findings in our studies is the increased KSHV DNA internalization in MβCD- and nystatin-treated cells compared to untreated infected cells (Fig. 4C and D), and the reason for this increase is not clear. Since KSHV-induced FAK and Src phosphorylation is essential for viral entry and since no change in FAK activation and increase in Src activation by lipid raft disruption was observed, we speculate that increased Src activation could be attributed to the increased KSHV entry. This is probably a valid speculation since Src activation has been shown to be essential for endocytosis (1) (Fig. 10).
After ligand interaction with receptors, associated tyrosine kinases are activated by autophosphorylation on tyrosine residues and recruit signaling complexes to the plasma membrane, which then rapidly translocate to clathrin, caveolae, and other vesicles (80). Src-mediated tyrosine phosphorylation of clathrin regulates clathrin translocation to the plasma membrane, and clathrin subsequently interacts with a number of other essential proteins, such as AP2 and Eps15 (1, 80). Src-dependent phosphorylation also regulates dynamin self-assembly and ligand-induced endocytosis by releasing the internalized endocytic vesicles from the plasma membrane (1). Protein components of signal transduction cascades assemble at endocytic vesicles and remain associated with endocytic vesicles after their dynamin-dependent or independent release from the plasma membrane (15, 41). Src-dependent phosphorylation initiates the assembly of a plasma membrane-associated Ras activation complex. Rho- and Rab-GTPases activated by PI3-K and Ras are critical for the formation of various types of endocytic vesicles and their movement, as well as for MT and microfilament reorganization (15, 41, 43, 66). Increased Src activation and increased KSHV entry by lipid raft disruption suggests that Src activation is probably tightly regulated by lipid raft-associated factor(s), which probably regulates the amount of KSHV entry into HMVEC-d cells. Further work is in progress to define the association of Src kinases in lipid rafts during infection, their regulation, and their role in KSHV entry.
KSHV induction of Src required for the formation of endocytic vesicles and their movement and the activation of PI3-K, RhoA, and the associated Dia-2 required for microtubular stabilization suggest that KSHV has evolved to manipulate signal pathways to aid in its entry and in the movement of its capsid and/or tegument in the cytoplasm (47). Extraction of cholesterol by MβCD has been shown to perturb host cell signal cascades. Association of T-cell receptor with lipid rafts promotes aggregation, which facilitates localization of signaling proteins, which includes Lck, LAT, and T-cell receptor while excluding CD45, thereby initiating protein tyrosine phosphorylation (28), and lipid raft disruption disrupts these signal cascades. Similarly, our studies demonstrate that lipid rafts play an active role in the modulation of KSHV-induced signal cascades, especially PI3-K activation. PI3-K plays a significant role(s) in the postentry stages of KSHV infection by modulating the downstream host cell signaling effector molecules such as RhoA-GTPases and Dia-2 molecules and the associated cascades. Downregulation of PI3-K upon lipid raft disruption has been shown in a number of studies (31, 50). Although studies have shown that MβCD affected the nucleocytoplasmic trafficking of RNPs and export (14), further studies are essential to define the role of lipid rafts in the minus end transport of cargo to the nucleus via dynein motors (47).
The role of the lipid raft in MT dynamics is not known. Increased viral entry in lipid raft-disrupted cells did not result in a concomitant increase in nuclear delivery and infection, and instead we observed the dramatic decrease in infected nucleus-associated viral DNA. This can be directly attributed to the reduced association of viral capsid to MTs and MT disruption in lipid raft-disrupted cells, which in turn could be attributed to defects in lipid raft-associated PI3-K, RhoA-GTPase, and Dia-2 activation and function that are critical for MT dynamics, movement of virus containing vesicles, and movement of viral capsid via dynein motor along the MTs toward the nucleus (47). At present, we cannot also rule out the potential possibility of a defect in KSHV escape from endosomes in lipid raft-disrupted cells, as shown for adenovirus type 2 entry in HeLa cells during cholesterol depletion by MβCD (35). Further studies are essential to decipher the fate of KSHV entering lipid raft-disrupted cells to determine whether viral particles get degraded in lysosome compartments due to a defect in lipid raft-mediated signaling and to determine whether viral capsids migrate toward the nucleus after lipid raft restoration.
Similar to other viruses, the initiation of target cell infection by herpesviruses is a complex multistep event, which includes the initial attachment to the target cells involving multiple receptors and multiple viral proteins, followed by penetration or internalization of viral DNA into the cytosol by fusion of the viral envelope with the host cell membranes either by fusion of viral envelope with the plasma membrane or with the endosome membranes, transport to the nuclear periphery, and release of the viral genome to the nucleus. Even though several host cell surface receptors have been identified for human and animal herpesviruses, how these interactions with cell surfaces facilitate the infection is not fully understood. Within minutes of its binding to adherent target cells, KSHV induces a variety of host cell preexisting signal pathway molecules, such as FAK, Src, PI3-K, Rho-GTPases, PKC-ζ, MEK, and ERK1/2 (46, 47, 61) (Fig. 10). Our studies are the initial attempts to understand the role of lipid rafts in KSHV infection, which reveals several complex interplays between lipid rafts and KSHV. Additional studies, including biochemical, proteomic, and virological approaches, are essential to decipher the association of KSHV entry receptors such as integrins, CD98/xCT, etc., and induced signal molecules with lipid rafts, the association of viral envelope glycoproteins with lipid rafts, and the nature of lipid raft-mediated signal cascades and their role in infection, including dynein motor-mediated transport via MTs. Such studies would provide a better understanding of KSHV biology and an insight into whether lipid rafts could be targeted to block KSHV infection of endothelial cells.
Acknowledgments
This study was supported in part by Public Health Service grant AI 057349 and the Rosalind Franklin University of Medicine and Science-H. M. Bligh Cancer Research Fund to B.C.
We thank Keith Philibert for critically reading the manuscript.
Footnotes
Published ahead of print on 16 May 2007.
REFERENCES
- 1.Ahn, S., J. Kim, C. L. Lucaveche, M. C. Reedy, L. M. Luttrell, R. J. Lefkowitz, and Y. Daaka. 2002. Src-dependent tyrosine phosphorylation regulates dynamin self-assembly and ligand-induced endocytosis of the epidermal growth factor receptor. J. Biol. Chem. 277:26642-26651. [DOI] [PubMed] [Google Scholar]
- 2.Akula, S. M., P. P. Naranatt, N. S. Walia, F. Z. Wang, B. Fegley, and B. Chandran. 2003. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J. Virol. 77:7978-7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akula, S. M., N. P. Pramod, F. Z. Wang, and B. Chandran. 2001. Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 284:235-249. [DOI] [PubMed] [Google Scholar]
- 4.Akula, S. M., N. P. Pramod, F. Z. Wang, and B. Chandran. 2002. Integrin α3β1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407-419. [DOI] [PubMed] [Google Scholar]
- 5.Akula, S. M., F. Z. Wang, J. Vieira, and B. Chandran. 2001. Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 282:245-255. [DOI] [PubMed] [Google Scholar]
- 6.Anderson, H. A., E. M. Hiltbold, and P. A. Roche. 2000. Concentration of MHC class II molecules in lipid rafts facilitates antigen presentation. Nat. Immunol. 1:156-162. [DOI] [PubMed] [Google Scholar]
- 7.Anderson, R. G., and K. Jacobson. 2002. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science 296:1821-1825. [DOI] [PubMed] [Google Scholar]
- 8.Argyris, E. G., E. Acheampong, G. Nunnari, M. Mukhtar, K. J. Williams, and R. J. Pomerantz. 2003. Human immunodeficiency virus type 1 enters primary human brain microvascular endothelial cells by a mechanism involving cell surface proteoglycans independent of lipid rafts. J. Virol. 77:12140-12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bavari, S., C. M. Bosio, E. Wiegand, G. Ruthel, A. B. Will, T. W. Geisbert, M. Hevey, C. Schmaljohn, A. Schmaljohn, and M. J. Aman. 2002. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 195:593-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bechtel, J. T., Y. Liang, J. Hvidding, and D. Ganem. 2003. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 77:6474-6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bender, F. C., J. C. Whitbeck, M. Ponce de Leon, H. Lou, R. J. Eisenberg, and G. H. Cohen. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 77:9542-9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borza, C. M., and L. M. Hutt-Fletcher. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8:594-599. [DOI] [PubMed] [Google Scholar]
- 13.Brown, D. 2002. Structure and function of membrane rafts. Int. J. Med. Microbiol. 291:433-437. [DOI] [PubMed] [Google Scholar]
- 14.Carrasco, M., M. J. Amorim, and P. Digard. 2004. Lipid raft-dependent targeting of the influenza A virus nucleoprotein to the apical plasma membrane. Traffic 5:979-992. [DOI] [PubMed] [Google Scholar]
- 15.Cavalli, V., M. Corti, and J. Gruenberg. 2001. Endocytosis and signaling cascades: a close encounter. FEBS Lett. 498:190-196. [DOI] [PubMed] [Google Scholar]
- 16.Chazal, N., and D. Gerlier. 2003. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. 67:226-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen, X., and M. D. Resh. 2002. Cholesterol depletion from the plasma membrane triggers ligand-independent activation of the epidermal growth factor receptor. J. Biol. Chem. 277:49631-49637. [DOI] [PubMed] [Google Scholar]
- 18.Chen, Y., and L. C. Norkin. 1999. Extracellular simian virus 40 transmits a signal that promotes virus enclosure within caveolae. Exp. Cell Res. 246:83-90. [DOI] [PubMed] [Google Scholar]
- 19.Chillaron, J., R. Roca, A. Valencia, A. Zorzano, and M. Palacin. 2001. Heteromeric amino acid transporters: biochemistry, genetics, and physiology. Am. J. Physiol. Renal Physiol. 281:F995-F1018. [DOI] [PubMed] [Google Scholar]
- 20.Choi, K. S., H. Aizaki, and M. M. Lai. 2005. Murine coronavirus requires lipid rafts for virus entry and cell-cell fusion but not for virus release. J. Virol. 79:9862-9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung, C. S., C. Y. Huang, and W. Chang. 2005. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J. Virol. 79:1623-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark, E. A., W. G. King, J. S. Brugge, M. Symons, and R. O. Hynes. 1998. Integrin-mediated signals regulated by members of the Rho family of GTPases. J. Cell Biol. 142:573-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danthi, P., and M. Chow. 2004. Cholesterol removal by methyl-beta-cyclodextrin inhibits poliovirus entry. J. Virol. 78:33-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.del Pozo, M. A., N. Balasubramanian, N. B. Alderson, W. B. Kiosses, A. Grande-Garcia, R. G. Anderson, and M. A. Schwartz. 2005. Phospho-caveolin-1 mediates integrin-regulated membrane domain internalization. Nat. Cell. Biol. 7:901-908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dourmishev, L. A., A. L. Dourmishev, D. Palmeri, R. A. Schwartz, and D. M. Lukac. 2003. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 67:175-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fenczik, C. A., R. Zent, M. Dellos, D. A. Calderwood, J. Satriano, C. Kelly, and M. H. Ginsberg. 2001. Distinct domains of CD98hc regulate integrins and amino acid transport. J. Biol. Chem. 276:8746-8752. [DOI] [PubMed] [Google Scholar]
- 27.Feral, C. C., N. Nishiya, C. A. Fenczik, H. Stuhlmann, M. Slepak, and M. H. Ginsberg. 2005. CD98hc (SLC3A2) mediates integrin signaling. Proc. Natl. Acad. Sci. USA 102:355-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galbiati, F., B. Razani, and M. P. Lisanti. 2001. Emerging themes in lipid rafts and caveolae. Cell 106:403-411. [DOI] [PubMed] [Google Scholar]
- 29.Ganem, D. 1997. KSHV and Kaposi's sarcoma: the end of the beginning? Cell 91:157-160. [DOI] [PubMed] [Google Scholar]
- 30.Giancotti, F. G., and E. Ruoslahti. 1999. Integrin signaling. Science 285:1028-1032. [DOI] [PubMed] [Google Scholar]
- 31.Gomez-Mouton, C., R. A. Lacalle, E. Mira, S. Jimenez-Baranda, D. F. Barber, A. C. Carrera, A. C. Martinez, and S. Manes. 2004. Dynamic redistribution of raft domains as an organizing platform for signaling during cell chemotaxis. J. Cell Biol. 164:759-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grundhoff, A., and D. Ganem. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Investig. 113:124-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris, T. J., and C. H. Siu. 2002. Reciprocal raft-receptor interactions and the assembly of adhesion complexes. Bioessays 24:996-1003. [DOI] [PubMed] [Google Scholar]
- 34.Herndier, B., and D. Ganem. 2001. The biology of Kaposi's sarcoma. Cancer Treat. Res. 104:89-126. [DOI] [PubMed] [Google Scholar]
- 35.Imelli, N., O. Meier, K. Boucke, S. Hemmi, and U. F. Greber. 2004. Cholesterol is required for endocytosis and endosomal escape of adenovirus type 2. J. Virol. 78:3089-3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoue, N., J. Winter, R. B. Lal, M. K. Offermann, and S. Koyano. 2003. Characterization of entry mechanisms of human herpesvirus 8 by using an Rta-dependent reporter cell line. J. Virol. 77:8147-8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaleeba, J. A., and E. A. Berger. 2006. Kaposi's sarcoma-associated herpesvirus fusion-entry receptor: cystine transporter xCT. Science 311:1921-1924. [DOI] [PubMed] [Google Scholar]
- 38.Katzman, R. B., and R. Longnecker. 2003. Cholesterol-dependent infection of Burkitt's lymphoma cell lines by Epstein-Barr virus. J. Gen. Virol. 84:2987-2992. [DOI] [PubMed] [Google Scholar]
- 39.Krishnan, H. H., P. P. Naranatt, M. S. Smith, L. Zeng, C. Bloomer, and B. Chandran. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishnan, H. H., N. Sharma-Walia, D. N. Streblow, P. P. Naranatt, and B. Chandran. 2006. Focal adhesion kinase is critical for entry of Kaposi's sarcoma-associated herpesvirus into target cells. J. Virol. 80:1167-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lanzetti, L., P. P. Di Fiore, and G. Scita. 2001. Pathways linking endocytosis and actin cytoskeleton in mammalian cells. Exp. Cell Res. 271:45-56. [DOI] [PubMed] [Google Scholar]
- 42.Lo, P., X. Yu, I. Atanasov, B. Chandran, and Z. H. Zhou. 2003. Three-dimensional localization of pORF65 in Kaposi's sarcoma-associated herpesvirus capsid. J. Virol. 77:4291-4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McPherson, P. S., B. K. Kay, and N. K. Hussain. 2001. Signaling on the endocytic pathway. Traffic 2:375-384. [DOI] [PubMed] [Google Scholar]
- 44.Miller, N., and L. M. Hutt-Fletcher. 1992. Epstein-Barr virus enters B cells and epithelial cells by different routes. J. Virol. 66:3409-3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore, P. S., and Y. Chang. 2003. Kaposi's sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu. Rev. Microbiol. 57:609-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naranatt, P. P., S. M. Akula, C. A. Zien, H. H. Krishnan, and B. Chandran. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J. Virol. 77:1524-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Naranatt, P. P., H. H. Krishnan, M. S. Smith, and B. Chandran. 2005. Kaposi's sarcoma-associated herpesvirus modulates microtubule dynamics via RhoA-GTP-diaphanous 2 signaling and utilizes the dynein motors to deliver its DNA to the nucleus. J. Virol. 79:1191-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palazzo, A. F., C. H. Eng, D. D. Schlaepfer, E. E. Marcantonio, and G. G. Gundersen. 2004. Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science 303:836-839. [DOI] [PubMed] [Google Scholar]
- 49.Parpal, S., M. Karlsson, H. Thorn, and P. Stralfors. 2001. Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control. J. Biol. Chem. 276:9670-9678. [DOI] [PubMed] [Google Scholar]
- 50.Peres, C., A. Yart, B. Perret, J. P. Salles, and P. Raynal. 2003. Modulation of phosphoinositide 3-kinase activation by cholesterol level suggests a novel positive role for lipid rafts in lysophosphatidic acid signalling. FEBS Lett. 534:164-168. [DOI] [PubMed] [Google Scholar]
- 51.Piperno, G., M. LeDizet, and X. J. Chang. 1987. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J. Cell Biol. 104:289-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Popik, W., and T. M. Alce. 2004. CD4 receptor localized to non-raft membrane microdomains supports HIV-1 entry. Identification of a novel raft localization marker in CD4. J. Biol. Chem. 279:704-712. [DOI] [PubMed] [Google Scholar]
- 53.Rappocciolo, G., F. J. Jenkins, H. R. Hensler, P. Piazza, M. Jais, L. Borowski, S. C. Watkins, and C. R. Rinaldo, Jr. 2006. DC-SIGN is a receptor for human herpesvirus 8 on dendritic cells and macrophages. J. Immunol. 176:1741-1749. [DOI] [PubMed] [Google Scholar]
- 54.Renne, R., D. Blackbourn, D. Whitby, J. Levy, and D. Ganem. 1998. Limited transmission of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 72:5182-5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ridley, A. J. 2001. Rho GTPases and cell migration. J. Cell Sci. 114:2713-2722. [DOI] [PubMed] [Google Scholar]
- 56.Sadagopan, S., N. Sharma-Walia, M. V. Veettil, H. Raghu, R. Sivakumar, V. Bottero, and B. Chandran. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-κB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 81:3949-3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanchez-San Martin, C., T. Lopez, C. F. Arias, and S. Lopez. 2004. Characterization of rotavirus cell entry. J. Virol. 78:2310-2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sargiacomo, M., M. Sudol, Z. Tang, and M. P. Lisanti. 1993. Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J. Cell Biol. 122:789-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sato, H., M. Tamba, K. Kuriyama-Matsumura, S. Okuno, and S. Bannai. 2000. Molecular cloning and expression of human xCT, the light chain of amino acid transport system xc. Antioxid. Redox Signal 2:665-671. [DOI] [PubMed] [Google Scholar]
- 60.Schmid, S. L. 1997. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annu. Rev. Biochem. 66:511-548. [DOI] [PubMed] [Google Scholar]
- 61.Sharma-Walia, N., H. H. Krishnan, P. P. Naranatt, L. Zeng, M. S. Smith, and B. Chandran. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 79:10308-10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma-Walia, N., P. P. Naranatt, H. H. Krishnan, L. Zeng, and B. Chandran. 2004. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 78:4207-4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature 387:569-572. [DOI] [PubMed] [Google Scholar]
- 64.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell. Biol. 1:31-39. [DOI] [PubMed] [Google Scholar]
- 65.Simons, K., and A. Wandinger-Ness. 1990. Polarized sorting in epithelia. Cell 62:207-210. [DOI] [PubMed] [Google Scholar]
- 66.Somsel Rodman, J., and A. Wandinger-Ness. 2000. Rab GTPases coordinate endocytosis. J. Cell Sci. 113(Pt. 2):183-192. [DOI] [PubMed] [Google Scholar]
- 67.Song, K. S., S. Li, T. Okamoto, L. A. Quilliam, M. Sargiacomo, and M. P. Lisanti. 1996. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains: detergent-free purification of caveolae microdomains. J. Biol. Chem. 271:9690-9697. [DOI] [PubMed] [Google Scholar]
- 68.Stuart, A. D., H. E. Eustace, T. A. McKee, and T. D. Brown. 2002. A novel cell entry pathway for a DAF-using human enterovirus is dependent on lipid rafts. J. Virol. 76:9307-9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun, X., and G. R. Whittaker. 2003. Role for influenza virus envelope cholesterol in virus entry and infection. J. Virol. 77:12543-12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thorp, E. B., and T. M. Gallagher. 2004. Requirements for CEACAMs and cholesterol during murine coronavirus cell entry. J. Virol. 78:2682-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsui-Pierchala, B. A., M. Encinas, J. Milbrandt, and E. M. Johnson, Jr. 2002. Lipid rafts in neuronal signaling and function. Trends Neurosci. 25:412-417. [DOI] [PubMed] [Google Scholar]
- 72.Tugizov, S. M., J. W. Berline, and J. M. Palefsky. 2003. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 9:307-314. [DOI] [PubMed] [Google Scholar]
- 73.Veettil, M. V., N. Sharma-Walia, S. Sadagopan, H. Raghu, R. Sivakumar, P. P. Naranatt, and B. Chandran. 2006. RhoA-GTPase facilitates entry of Kaposi's sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J. Virol. 80:11432-11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vieira, J., P. O'Hearn, L. Kimball, B. Chandran, and L. Corey. 2001. Activation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) lytic replication by human cytomegalovirus. J. Virol. 75:1378-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Viejo-Borbolla, A., and T. F. Schulz. 2003. Kaposi's sarcoma-associated herpesvirus (KSHV/HHV8): key aspects of epidemiology and pathogenesis. AIDS Rev. 5:222-229. [PubMed] [Google Scholar]
- 76.Vivanco, I., and C. L. Sawyers. 2002. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2:489-501. [DOI] [PubMed] [Google Scholar]
- 77.Waarts, B. L., R. Bittman, and J. Wilschut. 2002. Sphingolipid and cholesterol dependence of alphavirus membrane fusion. Lack of correlation with lipid raft formation in target liposomes. J. Biol. Chem. 277:38141-38147. [DOI] [PubMed] [Google Scholar]
- 78.Wang, F. Z., S. M. Akula, N. P. Pramod, L. Zeng, and B. Chandran. 2001. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J. Virol. 75:7517-7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang, X., W. J. Kenyon, Q. Li, J. Mullberg, and L. M. Hutt-Fletcher. 1998. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J. Virol. 72:5552-5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilde, A., E. C. Beattie, L. Lem, D. A. Riethof, S. H. Liu, W. C. Mobley, P. Soriano, and F. M. Brodsky. 1999. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell 96:677-687. [DOI] [PubMed] [Google Scholar]
- 81.Yamada, K. M., and S. Even-Ram. 2002. Integrin regulation of growth factor receptors. Nat. Cell. Biol. 4:E75-E76. [DOI] [PubMed] [Google Scholar]
- 82.Young, R. M., D. Holowka, and B. Baird. 2003. A lipid raft environment enhances Lyn kinase activity by protecting the active site tyrosine from dephosphorylation. J. Biol. Chem. 278:20746-20752. [DOI] [PubMed] [Google Scholar]
- 83.Zhong, W., H. Wang, B. Herndier, and D. Ganem. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 93:6641-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]