

Abstract
γ‐Herpesviruses, Epstein–Barr virus (EBV/HHV‐4) and Kaposi's sarcoma‐associated herpesvirus (KSHV/HHV‐8), are involved in human carcinogenesis, particularly in immunocompromised patients. Virus‐associated malignancies are becoming of significant concern for the mortality of long‐lived immunocompromised patients, and therefore, research of advanced strategies for AIDS‐related malignancies is an important field in cancer chemotherapy. Detailed understanding of the EBV and KSHV lifecycle and related cancers at the molecular level is required for novel strategies of molecular‐targeted cancer chemotherapy. The present review gives a simple outline of the functional interactions between KSHV‐ and EBV‐viral gene products and host cell deregulated signaling pathways as possible targets of chemotherapy against AIDS‐related malignancies. (Cancer Sci 2007; 98: 1288–1296)
Herpesviruses and AIDS‐related malignancy
Herpesviruses are double‐strand DNA viruses and at least eight of them are identified as human pathogens: human herpes simplex virus 1 and 2 (HSV/HHV‐1 and ‐2), varicella‐zoster virus (VZV/HHV‐3), Epstein–Barr virus (EBV/HHV‐4), human cytomegalovirus (HCMV/HHV‐5), human herpesviruses 6 and 7 (HHV‐6 and ‐7), and Kaposi's sarcoma‐associated herpesvirus (KSHV/HHV‐8).( 1 , 2 ) The herpesviruses are divided into three subgroups, α‐ β‐ and γ‐herpesvirus. EBV and KSHV are similar γ‐herpesviruses and associated AIDS‐related malignancies and their genomic DNA encodes various homologous gene products with oncogenic potential.( 1 , 2 )γ‐Herpesviruses are primarily lymphotropic but some show a lytic replication cycle in epithelial and fibroblast cells. EBV and KSHV display two similar alternative phases of infection, latent and lytic. Upon induction of the lytic cycle, lytic viral gene expression initiates lytic viral DNA replication and virion production. In contrast, a restricted set of viral genes is expressed and viral genomes are maintained as circular double‐stranded DNA in latently infected malignant cells (Table 1).( 2 )
Table 1.
Viral genes expressed in latently infected cells
| Malignancies | Viral products | |
|---|---|---|
| Epstein–Barr virus | ||
| Type I latency | Burkitt's lymphoma | EBNA1, EBER, BART |
| Type II latency | Hodgkin's lymphoma | EBNA1, LMP‐1,‐2, |
| Nasopharyngeal carcinoma | EBER, BART | |
| Natural killer/T‐cell lymphoma and AIDS‐related non‐Hodgkin's lymphoma | ||
| Type III latency | AIDS‐related proliferative disorders, post‐transplant lymphoproliferative disorders, and lymphoblastoid cell lines | EBNA1,‐2,‐3 A, ‐3B,‐3C,‐LP, LMP‐1,‐2, EBER, BART |
| Kaposi's sarcoma‐associated herpesvirus | ||
| Kaposi's sarcoma | LANA‐1, k‐Cyclin, vFLIP, vIRF1, Kaposin | |
| Primary effusion lymphomas | LANA‐1, k‐Cyclin, vFLIP, vIRF3, Kaposin, vIL‐6 (~40%) | |
| Multicentric Castleman's disease | LANA‐1, k‐Cyclin, vFLIP, vIL‐6 (~40%) | |
Kaposi's sarcoma and AIDS‐related lymphoma are the most frequent AIDS‐defining malignancies. KSHV was discovered from the Kaposi's sarcoma lesion of an AIDS patient in 1994,( 3 ) and has been linked to this sarcoma and certain lymphoproliferative disorders. Histologically, Kaposi's sarcoma lesions are characterized by neoangiogenesis and proliferative spindle‐shaped cells admixed with aberrant endothelial and inflammatory cells.( 4 ) KSHV is detected in spindle‐shaped cells that are likely the malignant cells in such lesion.( 5 ) In advanced Kaposi's sarcoma lesions, latently expressed KSHV gene products are believed to promote cell growth, to block apoptosis and host immune response, and to induce neoangiogenesis.( 2 ) KSHV‐infection is the only risk factor that is essential for Kaposi's sarcoma development, and the paracrine model is hypothesized for Kaposi's lesion in which KSHV induces a growth‐factor rich microenvironment that supports the proliferation of both neighboring and KSHV‐infected cells.( 6 ) Additionally, stimulated by this virus‐induced microenvironment, the inflammatory cells and cytokines including interleukin (IL)‐6, basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF) and the up‐regulated matrix metalloproteinases (MMP), activity of endothelial cells probably contribute to profound neoangiogenesis. In addition, HIV may itself contribute to Kaposi's sarcoma tumorigenesis.( 7 ) The Tat protein of HIV appears to stimulate endothelial cell migration, protect KSHV‐infected cells from apoptosis, promote spindle‐shaped cell growth, and enhance KSHV transmission.( 8 , 9 )
More than 50% of AIDS‐related lymphoma cases are also associated with γ‐herpesviruses, EBV and KSHV. EBV infects B cells as a persistent infection. EBV latent genes activate resting B cells and induce complete transformation, but the host immune system suppresses EBV‐induced malignancies.( 10 ) In healthy individuals, immunosurveillance systems including cytotoxic T‐cell responses against EBV latent antigen suppress the expansion of these lymphoblasts.( 11 ) However, EBV has strategies to escape from an immunosurveillance system in a host.( 12 , 13 ) Moreover, HIV‐infection increases the incidence of lymphoma, because such virus‐induced lymphoblasts have a good opportunity to expand under an immunocompromised condition. KSHV is also associated with another B‐cell lymphoproliferative disorder called multicentric Castleman's disease, in which lymphadenopathy with polyclonal hyperimmunoglobulinemia, and high levels of IL‐6 in serum are characteristics and there is often HIV co‐infection.( 14 )
Many cellular oncogenic and anti‐oncogenic molecules are involved in the mechanisms for malignant cell proliferation and cell survival.( 15 ) Deregulated cell growth signaling, deregulated cell cycle control, dysfunction of tumor suppressor genes, and resistance to apoptotic cell death contribute to malignant phenotypes.( 16 ) Intriguingly, infection of KSHV and EBV has been found to manipulate host cell mechanism, and pathogenic viral gene products also deregulate cellular signaling to promote cell growth and survival. The molecular and epidemiological linkage between AIDS‐related malignancies and KSHV‐ and EBV‐infection suggests that these viral gene products are potential targets for molecular‐targeted chemotherapy for AIDS‐related malignancies. This review describes an outline of the molecular aspects for KSHV‐ and EBV‐associated cell signaling, latency, and malignancy as targets for molecular‐targeted chemotherapy of AIDS‐associated malignancies.
Latent replication and maintenance of viral genomes
EBV‐ and KSHV‐associated malignancies involve the viruses’ latent cycle. During latency, the viral genome DNA is maintained as an episome, and behaves like a host cellular chromosome. The episomal viral genomes of EBV (~165 kbp) and KSHV (~165 kbp) (Fig. 1) are replicated semiconservatively once per cell cycle and partitioned faithfully to daughter cells during mitosis, and viral factors (trans‐ and cis‐) and host cell mechanisms are employed to keep these viral latencies in infected cells.( 17 , 18 ) All EBV‐ and KSHV‐associated malignancies express EBNA1 and LANA‐1 proteins, respectively, and these two viral antigens are required for latent replication and maintenance of viral genome.
Figure 1.
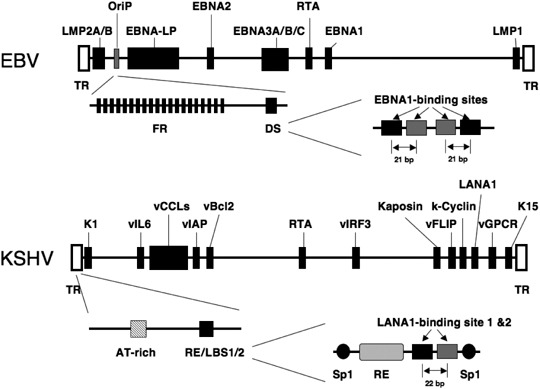
Schematic drawing of the organization of Epstein–Barr virus (EBV) and Kaposi's sarcoma‐associated herpesvirus (KSHV) linear genomes. Each viral gene‐coding region is flanked by multiple terminal repeat (TR) units. Different viral transcription patterns are observed in each latency types I, II, and III, and different EBNA are encoded by individual mRNA generated by different splicing of the long transcript. The open reading frames and latent replication origins discussed here are indicated., LMP, latent membrane protein.
Latent replication of the EBV genome requires only two viral factors, EBNA1 antigen acting in trans, and the latent DNA replication origin, the OriP region in cis.( 19 ) OriP consists of two major cis elements, namely the family of repeats (FR) and the dyad symmetry (DS) region (Fig. 1). The EBNA1 protein (641 amino acids) consists of three parts, the N‐terminus (1–391), the joining region (392–458), and the C‐terminus (459–641). The N‐terminus contains two regions that can link two independent EBNA1‐binding DNA elements, DS and FR, and can form a loop chromatin structure in a viral episome.( 20 ) The two linking regions are separated by Gly‐Ala rich repeats, and these repeats contribute to the escape from the host cell immune surveillance system by preventing EBNA1‐peptide presentation on the major histocompatibility complex (MHC) class I complex.( 12 , 13 ) The C‐terminus of EBNA1 functions as a DNA‐binding and EBNA1‐dimerization domain, which is essential for OriP‐dependent DNA replication.( 21 ) EBNA1 does not have helicase or ATPase activity, but EBNA1‐binding to the DS region appears to bend DNA at binding sites,( 22 ) which may stimulate a locally unique chromatin structure and an assembly of components for DNA replication.
The DS region contains four EBNA1‐binding sites consisting of two pairs of 21 bp‐spacing, and EBNA1‐binding on DS induces DNA replication licensed by host cell machineries. EBNA1‐binding on DS appears to initiate an assembly of host cell pre‐replication complexes, including the Origin recognition complex (ORC) and the Minichromosome maintenance (MCM) complex.( 23 ) Functions of ORC1 and ORC2 are important in facilitating OriP‐plasmid DNA replication,( 24 , 25 ) and, conversely, the licensing factor Geminin negatively controls OriP‐plasmid replication during cell cycle.( 24 ) Interestingly, viral OriP‐mediated plasmid replication is more severely affected by ORC1/2 activity changes than host cell DNA replication and proliferation. ORC1 mutations or ORC2 reduction significantly impairs OriP‐dependent plasmid replication and maintenance without inhibiting host cell proliferation. Within the DS region, other host cell factors also assemble to help OriP‐plasmid replication and maintenance. The DS region contains three nonamer sites (TTAGGGTTA), which resemble telomeric repeats. TRF2 and TRF2‐interacting tankyrase assemble on the DS region cooperatively with EBNA1, and these factors contribute to EBNA1‐dependent OriP‐plasmid replication/maintenance.( 26 )
The FR consists of a 20‐member family of 30‐bp repeats, and each member has one 18‐bp element with high affinity to EBNA1. EBNA1‐binding to FR has various effects. Multiple EBNA1‐binding to FR is critical for OriP‐mediated maintenance/partitioning of EBV episomal chromosome.( 27 ) Moreover, the EBNA1‐binding at FR enhances transcriptional activity of the latency promoters, called BamHIC and LMP1 promoter, and contributes to five latent gene expressions during latent proliferation of infected cells.( 28 )
Another γ‐herpesvirus, KSHV, shows latent infection on some tumors, and most KSHV‐infected cells in PEL and Kaposi's sarcoma lesions express latent genes for LANA‐1, k‐cyclin, vFLIP, and vIRF3.( 2 ) The KSHV genome also has viral latent replication origin for persistent infection. The trans‐acting factor is KSHV latency antigen LANA‐1, and the cis‐acting is the terminal repeat (TR) region consisting of two LANA‐1‐binding sites (LBS1/2) and a replication element (RE)( 18 , 29 , 30 , 31 ) (Fig. 1). LANA‐1 is a functional counterpart of EBNA1, and the minimum replication origin in the TR structurally related to EBV DS confers LANA‐1‐dependent replication origin activity on episomal plasmid DNA. LANA‐1 protein (1162 amino acids) comprises three regions, and is required for stable KSHV episomal maintenance in infected cells. The N‐terminal region of LANA‐1 tethers LANA‐1 to chromatin and mitotic chromosomes during cell cycle by binding with nucleosomal histone H2A‐H2B.( 32 ) Specifically, a peptide of LANA‐1 residues at positions 5–13 binds to an acidic patch of nucleosomal H2A‐H2B. The central region of LANA‐1 contains three different acidic blocks, and the C‐terminus (996–1139) encodes a DNA‐binding domain that binds to an 18‐bp imperfect palindrome in the LBS1/2.( 18 ) LANA‐1 binds cooperatively to LBS1 and LBS2 with high and low affinity, respectively,( 30 ) and this structural organization is similar to half of the DS element in OriP. Functions of the LANA‐1 N‐ and C‐terminal regions are involved in LANA‐1‐mediated bridging between plasmid DNA and mitotic chromosome and are required for efficient TR‐containing plasmid DNA replication and faithful segregation of episomes during cell division.( 33 )
As in the case of EBNA1‐OriP, LANA‐1‐dependent KSHV genome replication depends on host cell machineries, and LANA‐1‐bound TR is associated with host cell DNA replication components. ORC and MCM complexes assemble at or near the LBS of the TR in the KSHV‐latently infected cells.( 29 , 34 , 35 ) ORC2 is able to interact with the LANA‐1 C‐terminus at least in vitro, and the LANA‐1‐DNA complex is likely to facilitate ORC recruitment at TR in vivo. Furthermore, various cellular proteins are assembled on the TR region in KSHV‐infected cells.( 36 )
LANA‐1 also contributes to persistent latency by repressing the lytic replication cycle. The immediately early gene product RTA, KSHV ORF50, plays a critical role in switching viral replication from the latent to the lytic phase by transactivating lytic viral replication gene expression through interaction with RBP‐Jκ.( 37 ) LANA‐1 physically interacts with RTA and inhibits RTA's promoter activity. Such effects by LANA‐1 decrease RTA transactivation activity and contribute to the maintenance of viral latency.
Overall, latently expressed major antigens EBNA1 and LANA‐1 have pivotal roles in persistent infection and malignancy development caused by EBV and KSHV. Thus, functions of these antigens are attractive targets for molecular therapy/chemoprevention of latently infected cell malignancy.
Cell cycle deregulation by viral gene products
Some KSHV encoding genes have the potential ability to modulate the cell cycle control mechanism (Fig. 2). k‐Cyclin (ORF72) is a homolog of cyclin D2 (32% identity and 54% similarity).( 38 ) The predominant partner for k‐cyclin is CDK6, and this viral CDK complex phosphorylates RB protein like cellular CDK complexes.( 38 ) Moreover, k‐cyclin/CDK6 shows broader substrate specificity than cellular cyclinD/CDK6. k‐Cyclin/CDK6 can phosphorylate CDK2 substrates, including ORC1, CDC6, p27Kip1, histone H1, and others.( 39 ) In contrast to their cellular homolog, viral CDK complexes are constitutively active in KSHV‐infected cells.( 40 ) k‐Cyclin lacking degron is more stable than other cellular cyclins, and viral CDK complexes do not require activation by the upstream kinase, CAK.( 41 ) Moreover, viral CDK complexes antagonize the cellular CDK inhibitor, p27Kip1 and p21Cip1. Viral CDK complexes appear to inactivate p27Kip1 by phosphorylation‐mediated degradation and mislocalization.( 42 , 43 , 44 ) Viral CDK6 complex associates with and phosphorylates p21Cip1 on serine 130, and this phosphorylated p21Cip1 is not able to inhibit CDK2.( 45 ) Thus, owing to the unusual dual substrate preference of both CDK2 and CDK6, and escaping from functional CDK‐inhibitors,( 46 ) viral CDK complexes promote S‐phase entry efficiently. Moreover, LANA‐1 also has roles in G1/S transition. LANA‐1 expression is co‐regulated with k‐cyclin expression at latency locus by the major latency promoter.( 47 ) LANA‐1 is reported to inactivate RB and p53 like other oncogenic viral proteins,( 48 , 49 ) and to enhance E2F activity for S‐phase entry. These LANA‐1 functions during latency modulate host cell growth machinery in multiple ways and contribute to B‐cell hyperplasia.( 50 , 51 ) Another viral protein, KSHV vIRF1, is able to interact with both p53 and ATM to inhibit p53 transcriptional activity.( 52 )
Figure 2.
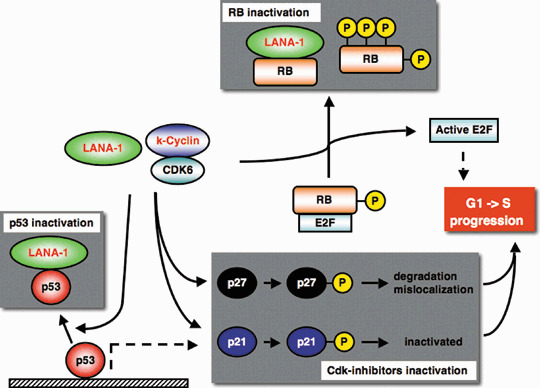
Cell cycle regulatory proteins and viral gene products. Kaposi's sarcoma‐associated herpesvirus (KSHV) latent antigen LANA‐1 inactivates RB and p53, and KSHV k‐cyclin/CDK complex inactivates Cdk‐inhibitors and RB by phosphorylation (P). These viral product expressions promote G1‐S progression of the host cell. Viral products are indicated by red font.
Modulation of cellular signaling pathway (PI3K‐Akt, NF‐κB, MAPK, Wnt, Notch)
During EBV‐ and KSHV‐associated malignancies, different types of latent gene expression have been described (Table 1). Some EBV and KSHV viral products have the ability to manipulate the host cellular signaling pathway during latent and lytic phases (Fig. 3). Such deregulated signaling by viral products contributes to malignant cell proliferation, survival and viral expansion. These manipulated cell signalings are as follows.
Figure 3.
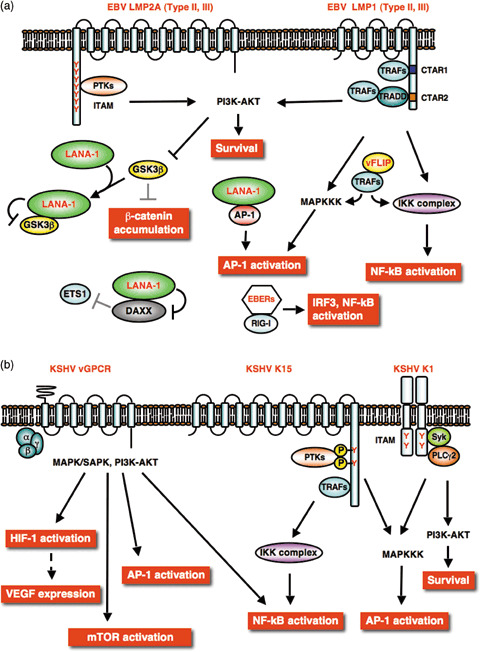
Cell signaling manipulated by Epstein–Barr virus (EBV) and Kaposi's sarcoma‐associated herpesvirus (KSHV) viral products. (a) Latent phase. KSHV latent antigen LANA‐1, vFLIP and EBV non‐coding‐RNA EBER are expressed in latently infected cells. EBV latent membrane protein (LMP)2A and LMP1 are expressed in type II and III latency. (b) Lytic phase. The KSHV G‐protein coupled receptor (vGPCR), K15, and K1 induce multiple signals. The tyrosine residues (Y) in the cytoplasmic tails of K15 and K1 mediate signal initiation. Complex cell signaling manipulated by viral products is an advantage to virus‐infected cell survival. Viral products are indicated by red font.
NF‐κB and MAPK. KSHV vFLIP encoded by ORF71, related to cellular FLIP, was initially considered to be an apoptosis inhibitor like its cellular homolog, but its anti‐apoptotic activity has recently been shown to be primarily associated with activation of the nuclear factor‐κB (NF‐κB) pathway (Fig. 3a). vFLIP is expressed in latently infected Kaposi's sarcoma spindle‐shaped and PEL cells, and TRAF2‐mediated association of vFLIP with IKK‐γ causes constitutive activation of the NF‐κB pathway.( 53 , 54 ) EBV latent membrane protein 1 (LMP1) also activates the NF‐κB pathway (Fig. 3a).( 55 ) The LMP1 C‐terminal cytoplasmic tail contains two distinct functional domains, C‐terminal activation regions 1 and 2 (CTAR1 and 2). Each CTAR1‐TRAF2 and CTAR2‐TRADD complex links to the IKK–NIK–NF‐κB pathway.( 56 , 57 ) These unscheduled activations of the NF‐κB pathway by viral products are required for virus‐induced cellular transformation and cell survival. KSHV vFLIP‐TRAF and EBV LMP1‐TRAF complexes also activate another signaling pathway, the JNK‐pathway that induces AP‐1 activation to support cell survival.( 58 , 59 ) In addition, LMP1‐mediated activations of NF‐κB and/or mitogen‐activated protein kinase (MAPK) pathways contribute to VEGF production.( 60 ) KSHV LANA‐1 also has an ability to function as a co‐activator of c‐Jun to activate an AP‐1 responsive element (Fig. 3a).( 61 )
EBV latent membrane protein 2 A (LMP2A) has an N‐terminal cytoplasmic region containing eight tyrosine residues, two of which (Tyr‐74 and ‐85) function as the immunoreceptor tyrosine‐based activation domain (ITAM) motif (Fig. 3a). The phosphorylated ITAM region recruits Src‐type tyrosine kinases, and this ITAM signalsome‐mediated ERK‐activation also stimulates the c‐Jun/AP‐1 pathway.( 62 ) Analogously, KSHV K1 has a C‐terminal cytoplasmic tail bearing an ITAM motif (Fig. 3b),( 63 ) which is highly conserved among different K1 subtypes and is similar to that of EBV LMP2A. K1 is expressed in Kaposi's sarcoma lesions, but is not correlated with latency. However, K1 has the ability to immortalize and extend the life span of primary human umbilical vein endothelial cells in culture.( 64 ) K1 homodimerization leads to the formation of signalsome, including Syk and phospholipase C, that activates downstream AP‐1 and NF‐AT.( 65 ) KSHV ORF K15, a lytic gene product, is a positional and functional homolog of EBV LMP2A. K15 analogously interacts with cellular proteins, TRAF and Src kinases, and activates AP‐1, NF‐κB, JNK and ERK (Fig. 3b).( 66 )
In addition, non‐protein products from EBV, EBER1 and 2 are shown to affect host cell signaling.( 67 ) EBER1 and 2 are non‐translated viral small RNA abundantly expressed in EBV latently infected cells. EBER activate the NF‐κB and IRF3 pathways through association with RIG‐I (Fig. 3a),( 68 ) and EBER‐mediated signaling is thought to affect cytokine expressions in EBV‐infected cells.( 69 ) These activations of NF‐κB and AP‐1 by viral products lead to expression of cellular IL‐6, IL‐8, bFGF, VEGF and others, the expression of which contributes to host cell survival and malignancy.
PI3K‐Akt. EBV LMP1 is able to activate the PI3K /Akt pathway in rodent fibroblast through the CTAR1 domain (Fig. 3a), and PI3K inhibitor suppresses CTAR1‐induced focus formation and anchorage‐independent growth of rodent fibroblast cells.( 70 ) EBV LMP2A activates the RAS/PI3K /Akt pathway (Fig. 3a), and LMP2A‐ITAM‐dependent Akt activation contributes to suppression of pro‐apoptotic activity of forkhead transcription factor (FHKR) and cell survival of B cells from LMP2A‐transgenic mice.( 71 , 72 ) LMP2A‐induced activation of the PI3K‐Akt axis leads to mTOR‐activation.( 73 ) KSHV LANA‐1 functions to promote VEGF/KDR signaling. LANA‐1 binds to Daxx, and this interaction interferes with Daxx‐mediated repression of Ets‐dependent VEGF receptor expression (Fig. 3a).( 74 )
During the lytic cycle, the cytoplasmic tail of KSHV K1 has the ability to activate the PI3K /Akt pathway (Fig. 3b), and K1 lytic expression and activation of PI3K /Akt in B‐cell appears to inactivate the FHKR family members and promote cell survival by preventing apoptosis.( 75 ) The KSHV G‐protein coupled receptor (vGPCR) encoded by ORF74 has been revealed to be an attractive therapeutic target for KSHV, although only a very small fraction of cells in advanced Kaposi's sarcoma lesions express it.( 76 , 77 ) The expression of vGPCR, which appears to activate multiple pathways including those of MAPK, NF‐κB, and Akt (Fig. 3b), leads to endothelial cell immortalization and tumorigenicity, and induces an angiogenic phenotype mediated by VEGF/KDR autocrine/paracrine signaling.( 78 , 79 ) vGPCR in PEL cells activates the PI3K/Akt pathway via the heterotrimeric GTP‐binding protein family.( 80 ) VEGF/ KDR autocrine/paracrine activation also promotes PI3K/Akt‐mTOR signaling, and pharmacological inhibitions against this signaling axis effectively block vGPCR‐induced endothelial cell proliferation.( 77 , 79 ) EBV also encodes a lytic G‐protein‐coupled receptor protein, BILF1, that stimulates endogenous GTP‐binding protein,( 81 ) although the biological significance of BILF1 on EBV pathogenesis remains to be determined.
Wnt. A recent advance in γ‐herpesvirus studies has uncovered another important oncogenic pathway manipulated by viral gene products. Both EBV and KSHV modulate the Wnt‐signaling pathway during development of malignancy.( 82 ) Deregulated Wnt‐signaling (accumulation of β‐catenin and activation of TCF) is often responsible for many types of human cancer development.( 83 ) EBV LMP2A‐mediated PI3K /Akt activation leads to phosphorylation and suppression of GSK3β in epithelial cells (Fig. 3a).( 72 ) GSK3β is a negative regulator of Wnt signaling, and its inactivation by LMP2A results in stabilization of β‐catenin and activation of TCF transcriptional factors. ITAM and phospho‐tyrosine motifs of LMP2A are required for accumulation and nuclear translocation of β‐catenin. In addition, EBV LMP1 can up‐regulate β‐catenin in B lymphoma cells by inhibiting the Siah‐1‐dependent ubiquitination and degradation of β‐catenin.( 84 ) Stable expression of β‐catenin in Kaposi's sarcoma lesion and PEL cell lines is also observed,( 85 ) and β‐catenin deregulation is suspected to play a role in KSHV‐tumorigenesis. Fujimuro et al. found that LANA‐1 causes GSK3β nuclear localization through interaction via C‐terminal axin‐like and N‐terminal domains of LANA‐1.( 85 ) LANA‐1‐binding depletes cytoplasmic GSK3β, and thus subsequently allows accumulation of β‐catenin to promote S‐phase entry (Fig. 3a).( 86 , 87 )
Notch. Notch receptor and its neighboring cell ligand pathway regulate cell proliferation, differentiation, and apoptosis during development. Notch signaling is involved in physiological angiogenesis, and disruption of the Notch pathway has been shown in multiple types of tumors.( 88 ) Ligand‐binding activates receptor proteolysis and induces nuclear translocation of the cleaved intracellular domain, Notch intracellular domain (NICD), which binds to DNA‐binding protein CSL /CBF‐1/ RBP‐Jκ to convert CSL‐complex to a positive transcriptional complex from a co‐repressor complex. EBV EBNA2 has been shown to be partially interchangeable with NICD for transactivation of CSL /CBF‐1/RBP‐Jκ target genes such as anti‐apoptotic bfl‐1, and this modulation may contribute to EBV‐mediated‐transformation (Fig. 4).( 89 , 90 , 91 )
Figure 4.
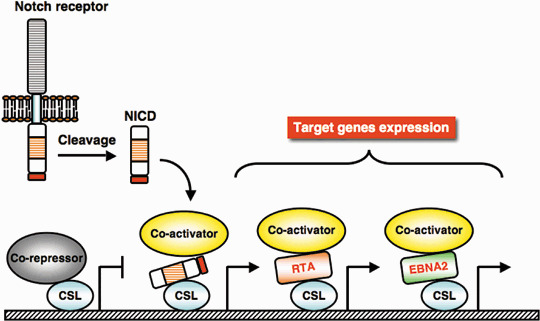
Epstein–Barr virus (EBV) EBNA2 and Kaposi's sarcoma‐associated herpesvirus (KSHV) RTA promote Notch‐signaling. Ligand‐receptor interaction initiate Notch signaling, and Notch receptor cleaved product, Notch intracellular domain (NICD) activates CSL‐initiated transcription of target genes by recruiting transcriptional co‐activators. Expression of EBNA2 induces ligand‐independent CSL activation, and RTA expression activates Notch‐signaling by stimulation of CSL DNA‐binding. Although RTA causes reactivation of the KSHV lytic cycle, and both EBNA2 and RTA promote Notch‐signaling, EBNA2 does not reactivate the KSHV lytic cycle. Viral products are indicated by red font.
Modulation of the Notch pathway is involved in KSHV viral reactivation from latency. KSHV lytic replication may contribute to the spread of virus and progression of Kaposi's sarcoma pathogenesis, and RTA, a transactivator of the lytic gene expression program, is a switch molecule from latency to lytic replication cycle.( 92 ) RTA binds to and transactivates CSL‐complex, and RTA‐targeted gene activation requires CSL (Fig. 4).( 93 ) In addition, RTA‐CSL interaction contributes to KSHV latency establishment. During the early stage of infection, RTA activates LANA‐1 expression by using CSL,( 37 , 94 ) and LANA‐1 expression conversely represses RTA expression by down‐regulation of its promoter.( 95 ) This feedback mechanism seems to function during KSHV latency establishment. Recent analysis has also showed that NICD expression was elevated in latent Kaposi's sarcoma lesions and PEL cells.( 96 ) Increased NICD is potentially able to reactivate KSHV from latency by activating the CSL complex on the RTA promoter. However, the above studies suggest that LANA‐1, predominantly expressed in latently infected cells, represses NICD‐mediated transactivation on the RTA promoter. Therefore, Notch signaling is regulating the balance between latency and the lytic cycle of KSHV.
Apoptotic pathway deregulated by viral gene products
Viral infection sometimes causes host cell apoptosis by activating endogenous machinery during the lytic cycle. Therefore, escaping from apoptosis is advantageous for viral expansion and survival. γ‐Herpesviruses, KSHV and EBV, encode anti‐apoptotic genes to guard the host cell during the lytic cycle( 97 ) (Fig. 5). The KSHV gene ORF16‐encoded protein (vBcl‐2) is highly homologous to Bcl‐2, particularly at the BH1 and BH2 domains, and shows functionally anti‐apoptotic activity.( 98 ) vBcl‐2 is expressed in spindle‐shaped cells at late stages of the Kaposi's sarcoma lesions, and the anti‐apoptotic effect of KSHV vBcl‐2 seems to contribute to Kaposi's sarcoma progression. Similarly, EBV also encodes a Bcl‐2 homolog, BHRF1, an early lytic cycle protein.( 99 ) The function of BHRF1 resembles anti‐apoptotic Bcl‐2 in some types of cells, and contributes to both the initial evasion of apoptosis during early infection and the establishment of latency for cellular transformation. Another type of anti‐apoptotic mode is provided by viral products related to IAP (vIAP), encoded by KSHV ORF K7.( 100 ) ORF K7 is structurally related to survivin ΔEX3, a splice variant of survivin that inhibits apoptosis. The vIAP BIR domain interacts with active caspase‐3, and the vIAP BH2 domain binds to Bcl‐2. Thus, vIAP promotes anti‐apoptotic activity of Bcl‐2 against active caspase‐3.
Figure 5.
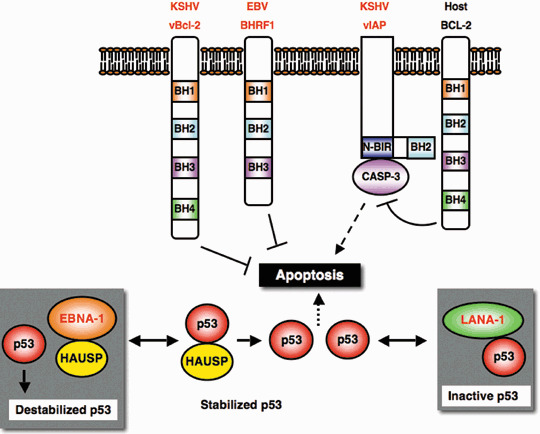
Anti‐apoptotic effects by viral products. Epstein–Barr virus (EBV) BHRF1 and Kaposi's sarcoma‐associated herpesvirus (KSHV) vBcl‐2 are functionally related to host cell anti‐apoptotic Bcl‐2 family containing Bcl‐2‐homology region 1–4 (BH1‐4). KSHV vIAP bridges active caspase‐3 (CASP‐3) and host cell Bcl‐2 through baculovirus IAP repeat domain (N‐BIR) to inhibit apoptosis‐execution by caspase‐3. Latent antigens, EBV EBNA1 and KSHV LANA‐1 suppress p53 activity and prevent p53‐induced apoptosis execution. Viral products are indicated by red font.
In addition, LANA‐1 interaction with p53 leads to inactivation of p53‐induced cell death during latency, which is considered to increase the chance for viral persistence and KSHV‐tumorigenesis.( 48 ) p53‐induced apoptosis is also antagonized by a latent antigen of EBV, EBNA1. USP7/ HAUSP is a key de‐ubiquitination enzyme regulating the p53–Mdm2 pathway, and stabilizes p53 by preventing p53 protein lowering.( 101 ) Competitive binding of EBNA1 to USP7/HAUSP disrupts p53 stabilization control balance, and protects cells from p53‐induced apoptosis by destabilizing p53 protein.( 102 )
Concluding remarks
Numerous studies on cancer research have shown that multiple genetic alterations cause malignancies, and here γ‐herpesviruses, EBV and KSHV, are considered to be strong causes of AIDS‐related malignancies. Although some viral gene products do not themselves have oncogenic activity, they contribute to virus survival and ensure persistent infection of the host cells. Molecular studies on these viral lifecycles have provided us knowledge of molecular bases for viral‐associated malignancies. At present, these viral gene products from γ‐herpesviruses are found to manipulate host cell signaling to initiate cell transformation and contribute to the progression and development of some types of cancer. Therefore, all these viral proteins modulating cell signaling are recognized as selective targets for chemotherapy/chemoprevention of EBV and KSHV‐associated malignancies. Admittedly, there are many unsolved questions in AIDS‐related malignancies, and such questions must be investigated to understand virus‐associated malignancies. Further extensive research is crucial but holds promise for the development of selective molecular‐targeted therapy for AIDS‐associated malignancies.
Acknowledgments
We are grateful to Drs Harutaka Katano and Shizuko Harada (National Institute of Infectious Diseases), and Masahiro Fujimuro (Hokkaido University) for valuable comments on this manuscript. We apologize that we could not cite the excellent work of many investigators owing to space limitations. This work was supported by a grant‐in‐aid for Cancer Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Boshoff C, Weiss R. AIDS‐related malignancies. Nat Rev Cancer 2002; 2: 373–82. [DOI] [PubMed] [Google Scholar]
- 2. Damania B. Oncogenic gamma‐herpesviruses: comparison of viral proteins involved in tumorigenesis. Nat Rev Microbiol 2004; 2: 656–68. [DOI] [PubMed] [Google Scholar]
- 3. Chang Y, Cesarman E, Pessin MS et al . Identification of herpesvirus‐like DNA sequences in AIDS‐associated Kaposi's sarcoma. Science 1994; 266: 1865–9. [DOI] [PubMed] [Google Scholar]
- 4. Katano H, Sato Y, Kurata T et al . High expression of HHV‐8‐encoded ORF73 protein in spindle‐shaped cells of Kaposi's sarcoma. Am J Pathol 1999; 155: 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boshoff C, Schulz TF, Kennedy MM et al . Kaposi's sarcoma‐associated herpesvirus infects endothelial and spindle cells. Nat Med 1995; 1: 1274–8. [DOI] [PubMed] [Google Scholar]
- 6. Hayward GS. Initiation of angiogenic Kaposi's sarcoma lesions. Cancer Cell 2003; 3: 1–3. [DOI] [PubMed] [Google Scholar]
- 7. Pati S, Foulke JS Jr, Barabitskaya O et al . Human herpesvirus 8‐encoded vGPCR activates nuclear factor of activated T cells and collaborates with human immunodeficiency virus type 1 Tat. J Virol 2003; 77: 5759–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barillari G, Sgadari C, Palladino C et al . Inflammatory cytokines synergize with the HIV‐1 Tat protein to promote angiogenesis and Kaposi's sarcoma via induction of basic fibroblast growth factor and the alpha v beta 3 integrin. J Immunol 1999; 163: 1929–35. [PubMed] [Google Scholar]
- 9. Aoki Y, Tosato G. HIV‐1 Tat enhances Kaposi sarcoma‐associated herpesvirus (KSHV) infectivity. Blood 2004; 104: 810–14. [DOI] [PubMed] [Google Scholar]
- 10. Thorley‐Lawson DA. Epstein‐Barr virus: exploiting the immune system. Nat Rev Immunol 2001; 1: 75–82. [DOI] [PubMed] [Google Scholar]
- 11. Voo KS, Fu T, Wang HY et al . Evidence for the presentation of major histocompatibility complex class I‐restricted Epstein‐Barr virus nuclear antigen 1 peptides to CD8 + T lymphocytes. J Exp Med 2004; 199: 459–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Levitskaya J, Coram M, Levitsky V et al . Inhibition of antigen processing by the internal repeat region of the Epstein‐Barr virus nuclear antigen‐1. Nature 1995; 375: 685–8. [DOI] [PubMed] [Google Scholar]
- 13. Yin Y, Manoury B, Fahraeus R. Self‐inhibition of synthesis and antigen presentation by Epstein‐Barr virus‐encoded EBNA1. Science 2003; 301: 1371–4. [DOI] [PubMed] [Google Scholar]
- 14. Soulier J, Grollet L, Oksenhendler E et al . Kaposi's sarcoma‐associated herpesvirus‐like DNA sequences in multicentric Castleman's disease. Blood 1995; 86: 1276–80. [PubMed] [Google Scholar]
- 15. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10: 789–99. [DOI] [PubMed] [Google Scholar]
- 16. Tsuruo T, Naito M, Tomida A et al . Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci 2003; 94: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yates JL, Warren N, Sugden B. Stable replication of plasmids derived from Epstein‐Barr virus in various mammalian cells. Nature 1985; 313: 812–15. [DOI] [PubMed] [Google Scholar]
- 18. Hu J, Garber AC, Renne R. The latency‐associated nuclear antigen of Kaposi's sarcoma‐associated herpesvirus supports latent DNA replication in dividing cells. J Virol 2002; 76: 11 677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J, Sugden B. Origins of bidirectional replication of Epstein‐Barr virus: models for understanding mammalian origins of DNA synthesis. J Cell Biochem 2005; 94: 247–56. [DOI] [PubMed] [Google Scholar]
- 20. Goldsmith K, Bendell L, Frappier L. Identification of EBNA1 amino acid sequences required for the interaction of the functional elements of the Epstein‐Barr virus latent origin of DNA replication. J Virol 1993; 67: 3418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bochkarev A, Barwell JA, Pfuetzner RA et al . Crystal structure of the DNA‐binding domain of the Epstein‐Barr virus origin‐binding protein, EBNA1, bound to DNA. Cell 1996; 84: 791–800. [DOI] [PubMed] [Google Scholar]
- 22. Bashaw JM, Yates JL. Replication from oriP of Epstein‐Barr virus requires exact spacing of two bound dimers of EBNA1 which bend DNA. J Virol 2001; 75: 10 603–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schepers A, Ritzi M, Bousset K et al . Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein‐Barr virus. EMBO J 2001; 20: 4588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dhar SK, Yoshida K, Machida Y et al . Replication from oriP of Epstein‐Barr virus requires human ORC and is inhibited by geminin. Cell 2001; 106: 287–96. [DOI] [PubMed] [Google Scholar]
- 25. Noguchi K, Vassilev A, Ghosh S et al . The BAH domain facilitates the ability of human Orc1 protein to activate replication origins in vivo. EMBO J 2006; 25: 5372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deng Z, Lezina L, Chen CJ et al . Telomeric proteins regulate episomal maintenance of Epstein‐Barr virus origin of plasmid replication. Mol Cell 2002; 9: 493–503. [DOI] [PubMed] [Google Scholar]
- 27. Rawlins DR, Milman G, Hayward SD et al . Sequence‐specific DNA binding of the Epstein‐Barr virus nuclear antigen (EBNA‐1) to clustered sites in the plasmid maintenance region. Cell 1985; 42: 859–68. [DOI] [PubMed] [Google Scholar]
- 28. Reisman D, Sugden B. trans activation of an Epstein‐Barr viral transcriptional enhancer by the Epstein‐Barr viral nuclear antigen 1. Mol Cell Biol 1986; 6: 3838–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lim C, Sohn H, Lee D et al . Functional dissection of latency‐associated nuclear antigen 1 of Kaposi's sarcoma‐associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J Virol 2002; 76: 10 320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu J, Renne R. Characterization of the minimal replicator of Kaposi's sarcoma‐associated herpesvirus latent origin. J Virol 2005; 79: 2637–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Verma SC, Choudhuri T, Robertson ES. The minimal replicator element of KSHV terminal repeat supports replication in a semi‐conservative and cell cycle dependent manner. J Virol 2007; 81: 3402–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barbera AJ, Chodaparambil JV, Kelley‐Clarke B et al . The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 2006; 311: 856–61. [DOI] [PubMed] [Google Scholar]
- 33. Komatsu T, Ballestas ME, Barbera AJ et al . KSHV LANA1 binds DNA as an oligomer and residues N‐terminal to the oligomerization domain are essential for DNA binding, replication, and episome persistence. Virology 2004; 319: 225–36. [DOI] [PubMed] [Google Scholar]
- 34. Stedman W, Deng Z, Lu F et al . ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma‐associated herpesvirus latent replication origin. J Virol 2004; 78: 12 566–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Verma SC, Choudhuri T, Kaul R et al . Latency‐associated nuclear antigen (LANA) of Kaposi's sarcoma‐associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J Virol 2006; 80: 2243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohsaki E, Ueda K, Sakakibara S et al . Poly (ADP‐ribose) polymerase 1 binds to Kaposi's sarcoma‐associated herpesvirus (KSHV) terminal repeat sequence and modulates KSHV replication in latency. J Virol 2004; 78: 9936–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lan K, Kuppers DA, Verma SC et al . Induction of Kaposi's sarcoma‐associated herpesvirus latency‐associated nuclear antigen by the lytic transactivator RTA. a novel mechanism for establishment of latency. J Virol 2005; 79: 7453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li M, Lee H, Yoon DW et al . Kaposi's sarcoma‐associated herpesvirus encodes a functional cyclin. J Virol 1997; 71: 1984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Verschuren EW, Jones N, Evan GI. The cell cycle and how it is steered by Kaposi's sarcoma‐associated herpesvirus cyclin. J General Virol 2004; 85: 1347–61. [DOI] [PubMed] [Google Scholar]
- 40. Van Dross R, Yao S, Asad S et al . Constitutively active K‐cyclin/cdk6 kinase in Kaposi sarcoma‐associated herpesvirus‐infected cells. J Natl Cancer Inst 2005; 97: 656–66. [DOI] [PubMed] [Google Scholar]
- 41. Kaldis P, Ojala PM, Tong L et al . CAK‐independent activation of CDK6 by a viral cyclin. Mol Biol Cell 2001; 12: 3987–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ellis M, Chew YP, Fallis L et al . Degradation of p27 (Kip) cdk inhibitor triggered by Kaposi's sarcoma virus cyclin‐cdk6 complex. EMBO J 1999; 18: 644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mann DJ, Child ES, Swanton C et al . Modulation of p27 (Kip1) levels by the cyclin encoded by Kaposi's sarcoma‐associated herpesvirus. EMBO J 1999; 18: 654–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sarek G, Jarviluoma A, Ojala PM. KSHV viral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood 2006; 107: 725–32. [DOI] [PubMed] [Google Scholar]
- 45. Jarviluoma A, Child ES, Sarek G et al . Phosphorylation of the cyclin‐dependent kinase inhibitor p21Cip1 on serine 130 is essential for viral cyclin‐mediated bypass of a p21Cip1‐imposed G1 arrest. Mol Cell Biol 2006; 26: 2430–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Swanton C, Mann DJ, Fleckenstein B et al . Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature 1997; 390: 184–7. [DOI] [PubMed] [Google Scholar]
- 47. Talbot SJ, Weiss RA, Kellam P et al . Transcriptional analysis of human herpesvirus‐8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 1999; 257: 84–94. [DOI] [PubMed] [Google Scholar]
- 48. Friborg J Jr, Kong W, Hottiger MO et al . p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999; 402: 889–94. [DOI] [PubMed] [Google Scholar]
- 49. Radkov SA, Kellam P, Boshoff C. The latent nuclear antigen of Kaposi sarcoma‐associated herpesvirus targets the retinoblastoma‐E2F pathway and with the oncogene Hras transforms primary rat cells. Nat Med 2000; 6: 1121–7. [DOI] [PubMed] [Google Scholar]
- 50. Katano H, Sato Y, Kurata T et al . Expression and localization of human herpesvirus 8‐encoded proteins in primary effusion lymphoma, Kaposi's sarcoma, and multicentric Castleman's disease. Virology 2000; 269: 335–44. [DOI] [PubMed] [Google Scholar]
- 51. Fakhari FD, Jeong JH, Kanan Y et al . The latency‐associated nuclear antigen of Kaposi sarcoma‐associated herpesvirus induces B cell hyperplasia and lymphoma. J Clin Invest 2006; 116: 735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shin YC, Nakamura H, Liang X et al . Inhibition of the ATM/p53 signal transduction pathway by Kaposi's sarcoma‐associated herpesvirus interferon regulatory factor 1. J Virol 2006; 80: 2257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu L, Eby MT, Rathore N et al . The human herpes virus 8‐encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J Biol Chem 2002; 277: 13 745–51. [DOI] [PubMed] [Google Scholar]
- 54. Field N, Low W, Daniels M et al . KSHV vFLIP binds to IKK‐gamma to activate IKK. J Cell Sci 2003; 116: 3721–8. [DOI] [PubMed] [Google Scholar]
- 55. Mosialos G, Birkenbach M, Yalamanchili R et al . The Epstein‐Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell 1995; 80: 389–99. [DOI] [PubMed] [Google Scholar]
- 56. Izumi KM, Kieff ED. The Epstein‐Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor‐associated death domain protein to mediate B lymphocyte growth transformation and activate NF‐kappaB. Proc Natl Acad Sci USA 1997; 94: 12 592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sylla BS, Hung SC, Davidson DM et al . Epstein‐Barr virus‐transforming protein latent infection membrane protein 1 activates transcription factor NF‐kappaB through a pathway that includes the NF‐kappaB‐inducing kinase and the IkappaB kinases IKKalpha and IKKbeta. Proc Natl Acad Sci USA 1998; 95: 10 106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kieser A, Kilger E, Gires O et al . Epstein‐Barr virus latent membrane protein‐1 triggers AP‐1 activity via the c‐Jun N‐terminal kinase cascade. EMBO J 1997; 16: 6478–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. An J, Sun Y, Sun R et al . Kaposi's sarcoma‐associated herpesvirus encoded vFLIP induces cellular IL‐6 expression: the role of the NF‐kappaB and JNK/AP1 pathways. Oncogene 2003; 22: 3371–85. [DOI] [PubMed] [Google Scholar]
- 60. Murono S, Inoue H, Tanabe T et al . Induction of cyclooxygenase‐2 by Epstein‐Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc Natl Acad Sci USA 2001; 98: 6905–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. An J, Sun Y, Rettig MB. Transcriptional coactivation of c‐Jun by the KSHV‐encoded LANA. Blood 2004; 103: 222–8. [DOI] [PubMed] [Google Scholar]
- 62. Chen SY, Lu J, Shih YC et al . Epstein‐Barr virus latent membrane protein 2A regulates c‐Jun protein through extracellular signal‐regulated kinase. J Virol 2002; 76: 9556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lanier LL. Viral immunoreceptor tyrosine‐based activation motif (ITAM) ‐mediated signaling in cell transformation and cancer. Trends Cell Biol 2006; 16: 388–90. [DOI] [PubMed] [Google Scholar]
- 64. Wang L, Dittmer DP, Tomlinson CC et al . Immortalization of primary endothelial cells by the K1 protein of Kaposi's sarcoma‐associated herpesvirus. Cancer Res 2006; 66: 3658–66. [DOI] [PubMed] [Google Scholar]
- 65. Lagunoff M, Majeti R, Weiss A et al . Deregulated signal transduction by the K1 gene product of Kaposi's sarcoma‐associated herpesvirus. Proc Natl Acad Sci USA 1999; 96: 5704–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brinkmann MM, Glenn M, Rainbow L et al . Activation of mitogen‐activated protein kinase and NF‐kappaB pathways by a Kaposi's sarcoma‐associated herpesvirus K15 membrane protein. J Virol 2003; 77: 9346–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Komano J, Maruo S, Kurozumi K et al . Oncogenic role of Epstein‐Barr virus‐encoded RNAs in Burkitt's lymphoma cell line Akata. J Virol 1999; 73: 9827–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Samanta M, Iwakiri D, Kanda T et al . EB virus‐encoded RNAs are recognized by RIG‐I and activate signaling to induce type I IFN. EMBO J 2006; 25: 4207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Iwakiri D, Eizuru Y, Tokunaga M et al . Autocrine growth of Epstein‐Barr virus‐positive gastric carcinoma cells mediated by an Epstein‐Barr virus‐encoded small RNA. Cancer Res 2003; 63: 7062–7. [PubMed] [Google Scholar]
- 70. Mainou BA, Everly DN Jr, Raab‐Traub N. Epstein‐Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K. Oncogene 2005; 24: 6917–24. [DOI] [PubMed] [Google Scholar]
- 71. Swart R, Ruf IK, Sample J et al . Latent membrane protein 2A‐mediated effects on the phosphatidylinositol 3‐Kinase/Akt pathway. J Virol 2000; 74: 10 838–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morrison JA, Klingelhutz AJ, Raab‐Traub N. Epstein‐Barr virus latent membrane protein 2A activates beta‐catenin signaling in epithelial cells. J Virol 2003; 77: 12 276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Moody CA, Scott RS, Amirghahari N et al . Modulation of the cell growth regulator mTOR by Epstein‐Barr virus‐encoded LMP2A. J Virol 2005; 79: 5499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Murakami Y, Yamagoe S, Noguchi K et al . Ets‐1‐dependent expression of vascular endothelial growth factor receptors is activated by latency‐associated nuclear antigen of Kaposi's sarcoma‐associated herpesvirus through interaction with Daxx. J Biol Chem 2006; 281: 28 113–21. [DOI] [PubMed] [Google Scholar]
- 75. Tomlinson CC, Damania B. The K1 protein of Kaposi's sarcoma‐associated herpesvirus activates the Akt signaling pathway. J Virol 2004; 78: 1918–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Montaner S, Sodhi A, Ramsdell AK et al . The Kaposi's sarcoma‐associated herpesvirus G protein‐coupled receptor as a therapeutic target for the treatment of Kaposi's sarcoma. Cancer Res 2006; 66: 168–74. [DOI] [PubMed] [Google Scholar]
- 77. Sodhi A, Chaisuparat R, Hu J et al . The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi's sarcoma‐associated herpesvirus G protein‐coupled receptor. Cancer Cell 2006; 10: 133–43. [DOI] [PubMed] [Google Scholar]
- 78. Montaner S, Sodhi A, Molinolo A et al . Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell 2003; 3: 23–36. [DOI] [PubMed] [Google Scholar]
- 79. Bais C, Van Geelen A, Eroles P et al . Kaposi's sarcoma associated herpesvirus G protein‐coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor‐2/KDR. Cancer Cell 2003; 3: 131–43. [DOI] [PubMed] [Google Scholar]
- 80. Cannon M, Philpott NJ, Cesarman E. The Kaposi's sarcoma‐associated herpesvirus G protein‐coupled receptor has broad signaling effects in primary effusion lymphoma cells. J Virol 2003; 77: 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Paulsen SJ, Rosenkilde MM, Eugen‐Olsen J et al . Epstein‐Barr virus‐encoded BILF1 is a constitutively active G protein‐coupled receptor. J Virol 2005; 79: 536–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fujimuro M, Hayward SD. Manipulation of glycogen‐synthase kinase‐3 activity in KSHV‐associated cancers. J Mol Med 2004; 82: 223–31. [DOI] [PubMed] [Google Scholar]
- 83. Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci 2003; 94: 225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jang KL, Shackelford J, Seo SY et al . Up‐regulation of beta‐catenin by a viral oncogene correlates with inhibition of the seven in absentia homolog 1 in B lymphoma cells. Proc Natl Acad Sci USA 2005; 102: 18 431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fujimuro M, Wu FY, ApRhys C et al . A novel viral mechanism for dysregulation of beta‐catenin in Kaposi's sarcoma‐associated herpesvirus latency. Nat Med 2003; 9: 300–6. [DOI] [PubMed] [Google Scholar]
- 86. Fujimuro M, Liu J, Zhu J et al . Regulation of the interaction between glycogen synthase kinase 3 and the Kaposi's sarcoma‐associated herpesvirus latency‐associated nuclear antigen. J Virol 2005; 79: 10 429–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fujimuro M, Hayward SD. The latency‐associated nuclear antigen of Kaposi's sarcoma‐associated herpesvirus manipulates the activity of glycogen synthase kinase‐3beta. J Virol 2003; 77: 8019–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rehman AO, Wang CY. Notch signaling in the regulation of tumor angiogenesis. Trends Cell Biol 2006; 16: 293–300. [DOI] [PubMed] [Google Scholar]
- 89. Hsieh JJ, Hayward SD. Masking of the CBF1/RBPJ kappa transcriptional repression domain by Epstein‐Barr virus EBNA2. Science 1995; 268: 560–3. [DOI] [PubMed] [Google Scholar]
- 90. Henkel T, Ling PD, Hayward SD et al . Mediation of Epstein‐Barr virus EBNA2 transactivation by recombination signal‐binding protein J kappa. Science 1994; 265: 92–5. [DOI] [PubMed] [Google Scholar]
- 91. Pegman PM, Smith SM, D'Souza BN et al . Epstein‐Barr virus nuclear antigen 2 trans‐activates the cellular antiapoptotic bfl‐1 gene by a CBF1/RBPJ kappa‐dependent pathway. J Virol 2006; 80: 8133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lukac DM, Renne R, Kirshner JR et al . Reactivation of Kaposi's sarcoma‐associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998; 252: 304–12. [DOI] [PubMed] [Google Scholar]
- 93. Liang Y, Chang J, Lynch SJ et al . The lytic switch protein of KSHV activates gene expression via functional interaction with RBP‐Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev 2002; 16: 1977–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lan K, Kuppers DA, Robertson ES. Kaposi's sarcoma‐associated herpesvirus reactivation is regulated by interaction of latency‐associated nuclear antigen with recombination signal sequence‐binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J Virol 2005; 79: 3468–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lan K, Kuppers DA, Verma SC et al . Kaposi's sarcoma‐associated herpesvirus‐encoded latency‐associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus‐mediated control of latency. J Virol 2004; 78: 6585–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lan K, Murakami M, Choudhuri T et al . Intracellular‐activated Notch1 can reactivate Kaposi's sarcoma‐associated herpesvirus from latency. Virology 2006; 351: 393–403. [DOI] [PubMed] [Google Scholar]
- 97. Cuconati A, White E. Viral homologs of BCL‐2: role of apoptosis in the regulation of virus infection. Genes Dev 2002; 16: 2465–78. [DOI] [PubMed] [Google Scholar]
- 98. Sarid R, Sato T, Bohenzky RA et al . Kaposi's sarcoma‐associated herpesvirus encodes a functional bcl‐2 homologue. Nat Med 1997; 3: 293–8. [DOI] [PubMed] [Google Scholar]
- 99. Henderson S, Huen D, Rowe M et al . Epstein‐Barr virus‐coded BHRF1 protein, a viral homologue of Bcl‐2, protects human B cells from programmed cell death. Proc Natl Acad Sci USA 1993; 90: 8479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang HW, Sharp TV, Koumi A et al . Characterization of an anti‐apoptotic glycoprotein encoded by Kaposi's sarcoma‐associated herpesvirus which resembles a spliced variant of human survivin. EMBO J 2002; 21: 2602–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Li M, Brooks CL, Kon N et al . A dynamic role of HAUSP in the p53‐Mdm2 pathway. Mol Cell 2004; 13: 879–86. [DOI] [PubMed] [Google Scholar]
- 102. Saridakis V, Sheng Y, Sarkari F et al . Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein‐Barr nuclear antigen 1 implications for EBV‐mediated immortalization. Mol Cell 2005; 18: 25–36. [DOI] [PubMed] [Google Scholar]