

Abstract
Phosphite dehydrogenase (PTDH)1 is a unique NAD-dependent enzyme that catalyzes the oxidation of inorganic phosphite to phosphate. The enzyme has great potential for cofactor regeneration and mechanistic studies have provided some insight into the residues that are important for catalysis. In this investigation, pre-steady state studies were performed on the His6-tagged wild type (WT) enzyme, several active site mutants, a thermostable mutant (12X-PTDH), and a thermostable mutant with dual cofactor specificity (NADP-12X-PTDH). Stopped-flow kinetic experiments indicate that slow steps after hydride transfer do not significantly limit the rate of reaction for WT, the active site mutants, or the thermostable mutant. Pre-steady state kinetic isotope effects (KIEs) and single turn-over experiments further confirm that slow steps after the chemical step do not significantly limit the rate of reaction for any of these proteins. Collectively, these results suggest that the hydride transfer step is fully rate determining in PTDH and that the observed KIE on kcat is the intrinsic effect in WT PTDH and the mutants examined. In contrast, a slow step after catalysis may partially limit the rate of phosphite oxidation by NADP-12X-PTDH with NADP as cofactor. Finally, site directed mutagenesis of Asp79 indicates that this residue is important in orienting Arg237 for proper interaction with phosphite.
Keywords: stopped-flow, intrinsic isotope effect
Phosphite dehydrogenase (PTDH) is a unique NAD-dependent enzyme that was first isolated from Pseudomonas stutzeri WW88 (1, 2). PTDH catalyzes the oxidation of inorganic phosphite to phosphate (Scheme 1), allowing the organism to use phosphite as its sole phosphorus source. The reaction represents an intriguing phosphoryl transfer reaction in which water or hydroxide is the phosphoryl acceptor. The enzyme has been shown to have great potential for cofactor regeneration (3) and its substrate specificity has been expanded to include the commercially important cofactor NADP (4). Subsequent directed evolution lead to a mutant enzyme with increased activity (5) and substantially improved thermostability (6), thus enhancing the industrial utility of the enzyme.
Scheme 1.
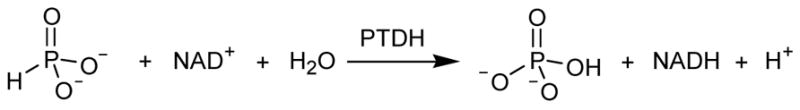
In addition to the engineering work, several studies have probed the mechanistic details of the unusual enzymatic transformation catalyzed by PTDH (7–10). Although the reaction resembles a phosphoryl transfer process, the enzyme shows significant sequence homology (26–35%) to the D-hydroxy acid dehydrogenase family of enzymes (2). Using this homology, important active site residues have been identified in PTDH and site-directed mutagenesis studies have investigated the roles they play in catalysis. Arg237 and Glu266 were shown to be important binding residues whereas His292 mutants demonstrated that this residue is critical for catalysis in PTDH and may act as the active site base to activate the water nucleophile (7). A recently solved X-ray crystallographic structure of the thermostable 12X-PTDH mutant (S. Nair, unpublished results) confirmed the presence of these residues in the active site and provided further support for the roles they play in catalysis (Figure 1). In addition, the X-ray crystallographic structure suggests additional roles for other residues. For example, Asp79 is involved in a hydrogen bond/ionic interaction with Arg237 and possibly acts to orient Arg237 such that it properly positions the substrate phosphite for hydride transfer. Kinetic isotope effects (KIE) studies with deuterium labeled phosphite demonstrated significant isotope effects on Vmax (DV) and Vmax/Km, Pt D(V/K), indicating hydride transfer is at least partially rate determining for the enzyme (8, 10). This is somewhat surprising considering the very favorable energetics of the reaction at pH 7.0, which strongly favors oxidation of phosphite to form phosphate, (E0′ = −0.648 V) while reducing NAD+ to NADH (E0′ = −0.320 V). This leads to an essentially irreversible reaction with an equilibrium constant of 1011. Given the favorable thermodynamics for the reaction, it is tempting to envision an enzyme with a rapid chemical step limited by slow physical steps, such as product release or a slow conformational change. To evaluate the degree to which the hydride transfer is rate limiting in PTDH, pre-steady state studies were performed on the WT enzyme, several active site mutants, the thermostable 12X-PTDH mutant and the thermostable mutant NADP-12X-PTDH that has relaxed cofactor specificity and can utilize both NAD and NADP. These studies indicate that slow steps after hydride transfer do not limit the rate of reaction for wild type PTDH and its mutants. Collectively, the results suggest that the hydride transfer step is fully rate determining for kcat and that the observed KIE is the intrinsic effect in the WT and the mutants examined. A smaller isotope effect observed with kcat/Km compared to kcat suggests that substrate dissociation is somewhat slower than hydride transfer under conditions of low substrate concentration.
Figure 1.
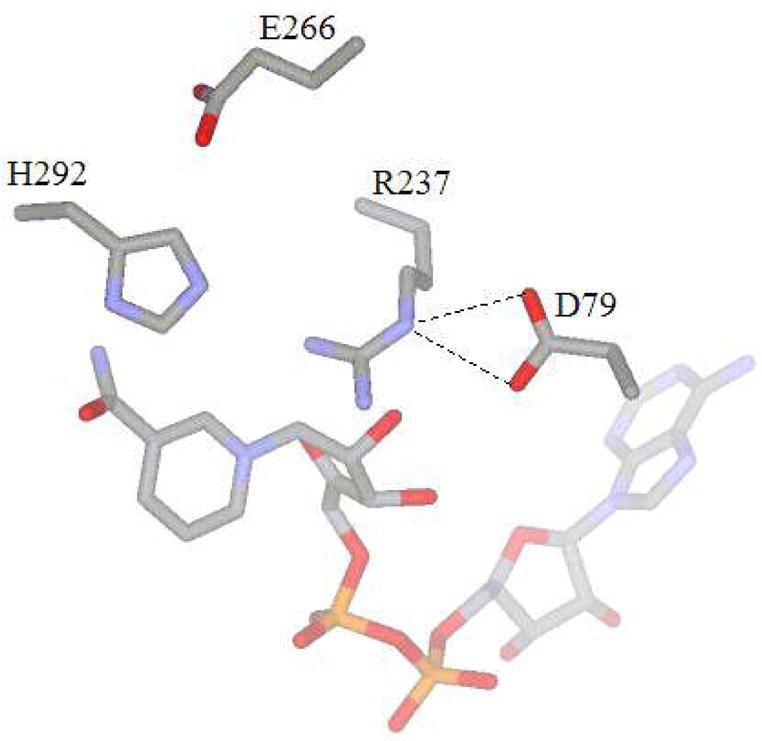
Active site structure of 12X-PTDH PTDH.
Materials and Methods
Materials
Sodium phosphite was purchased from Fluka. All other chemicals were purchased either from Sigma or Aldrich unless otherwise noted.
Overexpression and Purification of WT and Mutant PTDH
His6-tagged PTDH was overexpressed as reported previously (10). The His-tag was shown in a prior study not to have a significant effect on the kinetics of the enzyme (11). Briefly, 3 L of LB was inoculated with ~20 mL overnight culture of Escherichia coli BL21(DE3) harboring a pET15b vector containing the PTDH gene and grown at 37 °C until OD600 = 0.4. The culture was cooled on ice for ~20 min and IPTG was added to a concentration of 0.3 mM. The culture was grown at 25 °C for 8–10 h following induction. The cells were pelleted by centrifugation (5000g, 20 min), resuspended in 20 mM Tris pH 7.6, 500 mM NaCl, 10% (v/v) glycerol and stored at –80 °C. The resuspended cell pellet was thawed on ice, incubated with 0.3 mg/mL lysozyme for 20 min and then sonicated for 15 min with a pulse sequence of 5 s on, 9.9 s off. After removal of cell debris by centrifugation (15,000g, 30 min), the cell free extract was filtered (0.44 μm) and loaded onto a 8 mL POROS (PerSeptive Biosystems) metal chelate affinity column loaded with Ni2+ and equilibrated in buffer A. The column was washed with 10–20 column volumes buffer A followed by a linear gradient of 20 column volumes from 100% buffer A to 100% buffer B (Buffer A: 20 mM Tris pH 7.6, 100 mM NaCl, 10 mM imidazole, 10% (v/v) glycerol, Buffer B: 20 mM Tris pH 7.6, 100 mM NaCl, 500 mM imidazole, 10% (v/v) glycerol.) The fractions containing PTDH, as determined by absorbance at 280 nm, SDS-PAGE and enzyme activity, were pooled, concentrated, dialyzed into 50 mM MOPS pH 7.25 and flash frozen. The protein concentration was determined using a calculated extinction coefficient for PTDH, 28,000 M−1cm−1.
Preparation of D79A PTDH
The mutant was made using the Quik Change protocol (Stratagene) with some modifications. The primer pair used to introduce the desired mutation was D79A: 5'-GCG CTC AAG GGC TTC GCA AAT TTC GAT GTG GAC GCC-3' and 5'-GGC GTC CAC ATC GAA ATT TGC GAA GCC CTT GAG CGC-3'; (with the mutated codon underlined). The primers were desalted and the PCR reaction used Pfx polymerase. Once the mutant genes were constructed they were sequenced in their entirety to ensure that the desired mutation was incorporated and no other mutations were generated.
Steady State Kinetic Assays
Initial rates were obtained at 25 °C using a Cary 100 Bio UV-vis spectrophotometer (Varian). The concentration of stock NAD solutions were determined using the extinction coefficients (NAD ε260 = 18,000 M−1cm−1) (12). The concentration of phosphite stock solutions was determined by adding PTDH and excess NAD, incubating the reaction at 25 °C until complete and determining the concentration of NADH produced using its extinction coefficient. Reactions were monitored by the formation of NADH by absorbance at 340 nm. Typical reactions were performed in 100 mM MOPS pH 7.25, with 0.2–0.5 μM PTDH, and varying concentrations of NAD and phosphite.
Steady State Kinetic Isotope Effects
KIEs were determined by holding the NAD concentration at 10 x Km and varying the concentration of either 1H or 2H phosphite. It has been shown previously that PTDH observes an ordered mechanism in which NAD binds first (2), resulting in no observed KIE on V/Km, NAD (10). The data in this study were fitted to the Michaelis-Menten equation to obtain Vmax and Km, Pt. Direct comparison of the kinetic parameters determined with 1H and 2H phosphite gave DV and D(V/Km, Pt).
Pre-Steady State Kinetics and KIEs of WT and Active Site Mutants
Pre-steady state kinetics experiments were performed using an Olis RSM 1000 stopped flow spectrophotometer. The dead time of the instrument under experimental conditions was determined to be ~4 ms using ascorbate and 2,6-dichloroindophenol (13). Data was collected between 320–420 nm at a rate of 1000 scans/s for a total time of 1–2 s for traces with WT and all mutants except R237K and D79A. For the latter, less active mutants data was collected in the same wavelength range but at a rate of 62 scans/s for a total time of 30 s. For all experiments, one syringe contained PTDH in 100 mM MOPS pH 7.25, the other syringe contained NAD and either 1H or 2H phosphite such that the final concentration of both substrates was saturating (~10 x Km) after mixing, also in 100 mM MOPS pH 7.25. The final concentration of enzyme was 15 μM for WT and all of the mutants tested, except R237K and D79A for which the final concentration were 33 μM and 28 μM, respectively. Aggregation and precipitation of the enzymes did not allow the use of higher concentrations of enzyme. The traces shown here are the average of four shots. The kobs was calculated by fitting the data to a zero order equation; the errors given for kobs are the standard deviation of four individual shots. Controls showed that none of the t = 0 absorbance is due to product formation within the dead time of the stopped-flow instrument.
Pre-Steady State Kinetics and KIEs of Thermostable 12X-PTDH and NADP-12X-PTDH
Pre-steady state kinetic studies were performed as described above except higher concentrations of enzyme could be used because of the higher solubility of these proteins. For 12X-PTDH the final concentration of enzyme was 150 μM and the concentrations of all substrates were 5 mM. The data were fit to a zero order equation as described above. The traces are the average of four shots. For NADP-12X-PTDH the final concentration of enzyme was 120 μM and the concentration of all substrates was 2.5 mM. When NAD was used the traces were fit to a zero order equation. The curvature in the traces when NADP was used as a substrate prevented fitting to a zero order equation. The data were not well fit by the burst equation (eq 1) (14).
| eq 1 |
Single Turnover Experiments with 12X-PTDH
For single turnover experiments data were collected between 320–420 nm at a rate of 1000 scans/s for a total time of 2 s for 1H phosphite and at a rate of 62 scans/s for a total time of 4 s for 2H phosphite. These experiments used 2.5 mM NAD, 150 μM 1H or 2H phosphite and 150 μM 12X-PTDH PTDH. The data were fit to a single exponential equation.
Results
Steady State Kinetics of Asp79 Mutants
The D79A mutant showed significant differences in its kinetic constants compared to WT (Table 1). Whereas Km,NAD remained relatively unaffected, the value of kcat decreased approximately 80-fold and Km,Pt increased about 20-fold, leading to a 2600-fold decrease in catalytic efficiency. On the other hand, the D79N mutant had kinetic parameters more similar to WT. The mutant displayed only a 10-fold decrease in kcat whereas the Km values for phosphite and NAD were essentially unaffected (Table 1).
Table 1.
Steady State Kinetic Constants for Asp79 and Arg237 Mutantsa
| kcat (s−1) | Km, Pt (μM) | Km, NAD (μM) | kcat/Km, Pt (M−1s−1) | |
|---|---|---|---|---|
| WT | 2.42 (0.03) | 55 (5) | 35 (5) | 6.5 (0.7) x 104 |
| D79A | 0.0305 (0.0008) | 1200 (100) | 62 (3) | 25 (2) |
| D79N | 0.23 (0.01) | 35 (5) | 12 (1) | 6.60 (0.90) x 103 |
| R237K | 0.0126 (0.0006) | 1.00 (0.10) x 104 | 1000 (100) | 1.25 (0.14) |
All assays were performed at 25 °C, pH 7.25 in 100 mM MOPS. The steady state parameters kcat and Km,Pt were measured at saturating concentrations of NAD and the phosphite concentration was varied; Km,NAD was determined by holding the phosphite concentration at a saturating level and varying the concentration of NAD. The errors given in parentheses are obtained from fitting to the Michaelis Menten equation.
Pre-Steady State Kinetics of WT PTDH
The pre-steady state behavior of PTDH was investigated to determine if step(s) after catalysis were partially rate determining. WT PTDH showed no clear pre-steady state burst of activity (Figure 2A). The trace does not show any obvious curvature and at time t = 0 the absorbance is ~0.05 absorbance units (AU) when uncorrected for the absorbance of enzyme and substrates; when corrected, the t = 0 absorbance is approximately zero. The amplitude of a full stoichiometric burst would be expected to be ~ 0.1 AU based on the amount of enzyme used in the experiment, suggesting that a stoichiometric burst does not take place in the dead time of the instrument. Because it was not possible to increase the enzyme concentration to enhance any burst amplitude due to aggregation and precipitation of the enzyme, we cannot definitely rule out the possibility that a small substoichiometric burst occurs within the dead time of the instrument. The similarity of the pre-steady state and steady state rates provides additional support that no rapid reaction occurs upon the initial mixing of enzyme and substrates.
Figure 2.
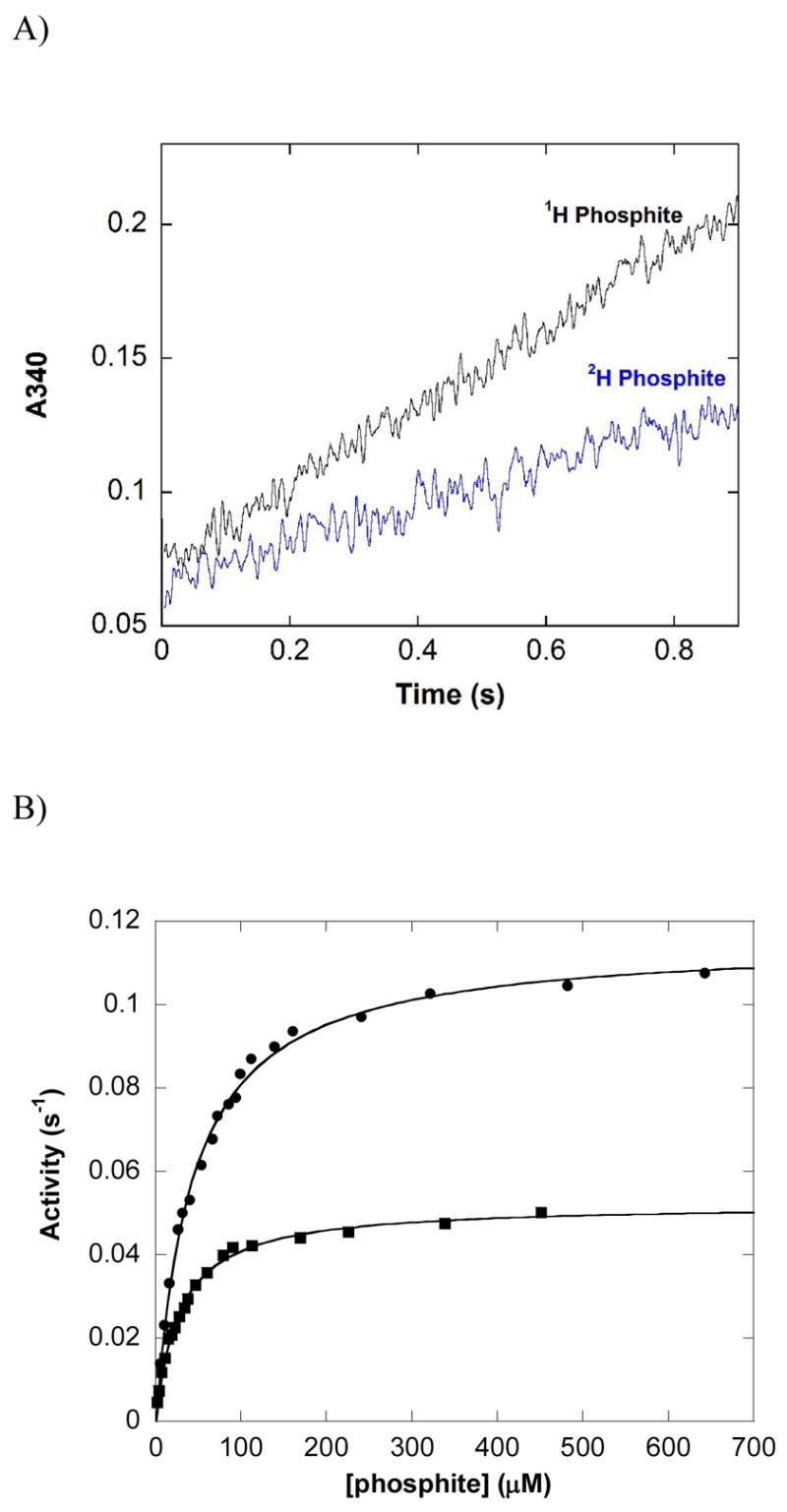
Reaction of wild type PTDH with protiated and deuterated phosphite in the steady state and pre-steady state. (A) Average of four pre-steady state traces in which a solution of 15 μM WT PTDH was mixed with an equal volume of a solution of 500 μM 1H or 2H phosphite and 500 μM NAD+ in 50 mM MOPS pH 7.25. (B) Phosphite concentration dependence of the steady state activity of 0.13 μM PTDH in the presence of 1 mM NAD+ in 100 mM MOPS pH 7.25. The lines represent fits to the Michaelis-Menten equation for protiated (circles) and deuterated phosphite (squares).
Pre-Steady State KIEs with WT PTDH
Comparison of the rates in the pre-steady state using 1H and 2H phosphite allows determination of the observed isotope effect in the pre-steady state, Dkobs (Figure 2A and Table 2). The primary substrate KIE in the pre-steady state was determined to be Dkobs = 2.0 (0.2) (Table 2) in good agreement with the steady state KIE on Vmax, DV = 2.22 (0.03) (Figure 2B).
Table 2.
Steady State Kinetic Parameters for NADP-12X-PTDHa
| kcat (s−1) | kcat/Km, Pt (M−1s−1) | DV | D(V/K) | |
|---|---|---|---|---|
| 12X | 3.26 (0.15) | 7.6 (1.1) x 104 | 2.5 (0.15) | 1.0 (0.2) |
| NADP-12X NAD | 2.04 (0.04) | 4.3 (0.2) x 104 | 2.17 (0.08) | 1.48 (0.18) |
| NADP-12X NADP | 0.58 (0.02) | 2.5 (0.3) x 104 | 1.66 (0.07) | 1.1 (0.2) |
All assays were performed at 25 °C, pH 7.25 in 100 mM MOPS. The steady state parameters kcat and Km,Pt were measured at saturating concentrations of NAD or NADP and the phosphite concentration was varied. The errors given in parentheses are obtained from fitting to the Michaelis-Menten equation.
Pre-Steady State Kinetics of 12X-PTDH
The pre-steady state behavior of the thermostable mutant, 12X-PTDH, was investigated next because of its current use in commercial settings. This enzyme does not aggregate at high enzyme concentrations allowing pre-steady state studies that used ~10-fold more enzyme than the experiments with WT PTDH. Pre-steady state traces did not show a burst of activity nor any curvature during the period of data collection (Figure 3A). A stoichiometric burst within the dead time of the stopped-flow instrument can be definitively ruled out; based on the amount of enzyme used, the burst amplitude would have been ~0.9 AU. It is clear from the trace that no such burst occurs within the dead time of the stopped-flow instrument. A significant substoichiometric burst can also be ruled out for 12X-PTDH; if the burst amplitude was reduced by 90% the expected burst would be ~0.09 AU. Clearly, such a burst does not occur within the dead time of the instrument or during the course of the experiment. In addition, the rate observed in the pre-steady state is within error of the steady state kcat (Table 2). Collectively, these observations indicate that a step after catalysis is not partially rate limiting for 12X-PTDH. As found for WT-PTDH, the isotope effect for 12X-PTDH observed in the pre-steady state, Dkobs = 2.23 (0.15), is in good agreement with the observed isotope effect on Vmax (DV =2.50 (0.15), Table 2).
Figure 3.
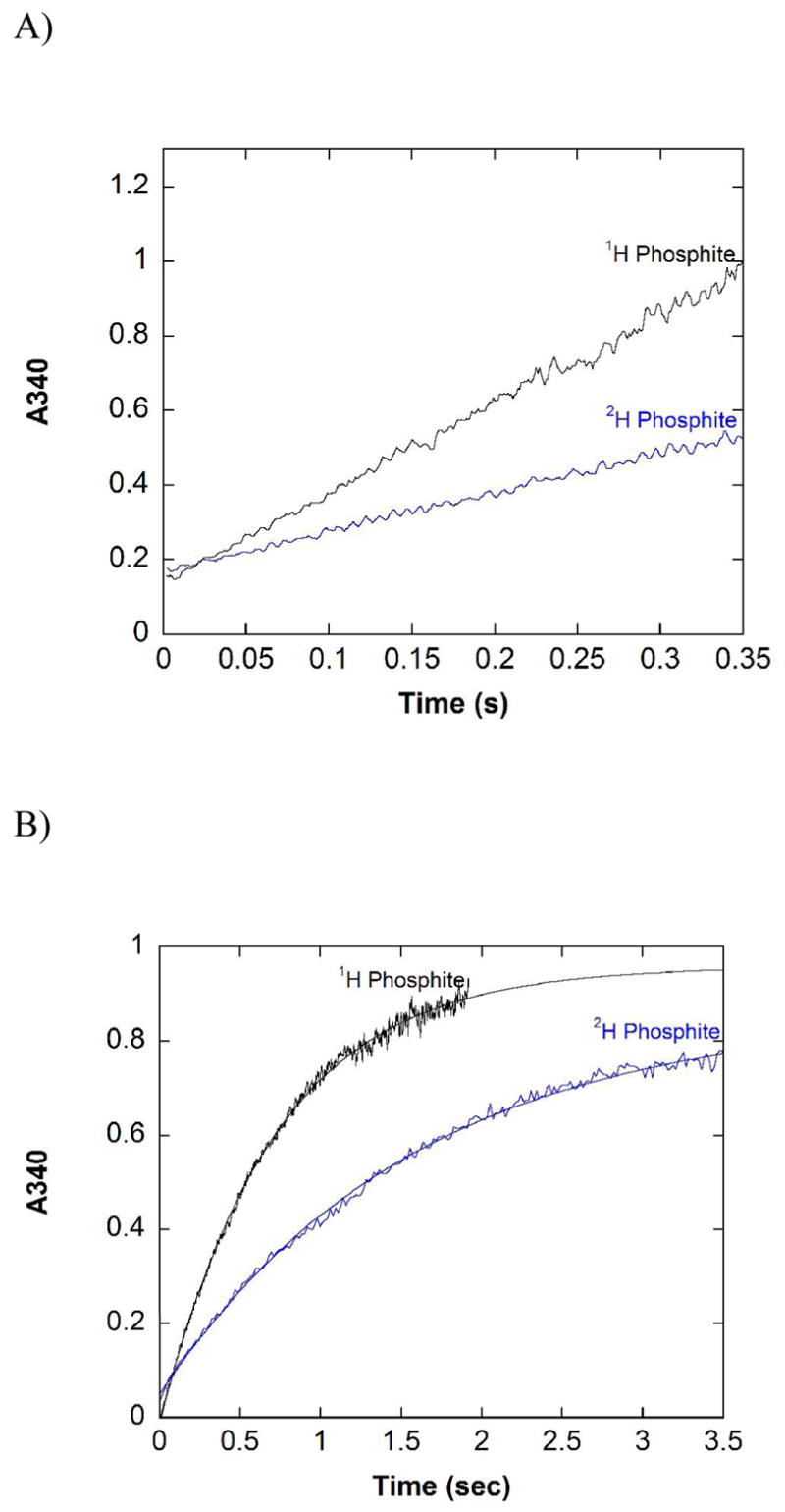
Pre-steady state traces for 12X-PTDH. A) Traces with saturating NAD+, 1H or 2H phosphite. Reaction conditions: 100 mM MOPS pH 7.25, 5 mM 1H or 2H phosphite, 5 mM NAD, 150 μM 12X-PTDH. Each trace is the average of four separate shots. B) Single turn over experiments with 1H or 2H phosphite. Reaction conditions: 100 mM MOPS pH 7.25, 150 μM 1H or 2H phosphite, 2.5 mM NAD+, 150 μM 12X-PTDH. Each trace is the average of four separate shots. The solid line represents the fit to a single exponential equation.
Single Turnover Experiments with 12X-PTDH
The higher solubility of 12X-PTDH allowed investigation of the pre-steady state behavior under single turnover conditions using 150 μM of enzyme. These single turnover experiments were performed to probe whether a step involving phosphite binding or a conformational change associated with binding could be partially rate limiting. At phosphite concentrations of 50–150 μM, the experimental traces for 1H and 2H phosphite were well fit by a single exponential equation without any evidence for a lag phase (Figure 3B). The observed rates under single turnover conditions for 1H phosphite were similar to the steady state rates under the same conditions, providing further evidence that steps after hydride transfer are not rate determining. Unfortunately, attempts to separate binding steps from catalysis in these experiments were unsuccessful. The observed rates were independent of phosphite concentrations under conditions that still provided reliable absorbance changes (30–150 μM) suggesting that the KD,Pt is below 25 μM. The isotope effect under single turnover conditions was determined to be 2.2 (0.16), close to the isotope effect observed in the pre-steady state multiple turnover studies determined under saturating concentrations of substrates, Dkobs = 2.23 (0.15). Furthermore, the isotope effect determined under single turnover conditions was not significantly reduced compared to the steady state effect, DV = 2.5 (0.15).
Steady State KIEs of NADP-12X-PTDH
The steady state KIEs were determined next for the engineered PTDH mutant with dual cofactor specificity using both NAD and NADP as substrates (Table 3). With NAD as a substrate DV was 2.17 (0.08) but when NADP was used this value decreased significantly to 1.66 (0.07). This same trend was also observed previously with the dual cofactor specificity mutant in the WT background (i.e. without the mutations conferring the increased thermostability) (15). The kcat value with NADP as a substrate also decreased ~6-fold compared to when NAD was used as the cofactor (Table 3).
Table 3.
Summary of Steady State and Pre-steady State Kinetic Constants for WT-PTDH and Mutantsa
| Steady Stateb |
Pre-Steady Statec |
|||||
|---|---|---|---|---|---|---|
| kcat (s−1) | kcat/Km, Pt (M−1s−1) | D(V/Km,Pt) | DV | kobs (s−1) | Dkobs | |
| WT | 2.42 (0.03) | 6.5 (0.7) x 104 | 1.44 (0.08) | 2.22 (0.03) | 1.56 (0.02) | 2.0 (0.1) |
| E266Q | 7.5 (0.1) | 250 (10) | 1.78 (0.15) | 3.3 (0.1) | 5.3 (0.5) | 3.0 (0.3) |
| K76A | 0.62 (0.01) | 295 (15) | 1.9 (0.1) | 2.34 (0.04) | 0.42 (0.07) | 2.1 (0.4) |
| R237K | 0.0126 (0.0006) | 1.25 (0.14) | 2.1 (0.3) | 2.36 (0.06) | 0.0152 (0.002) | 2.37 (0.35) |
| D79A | 0.0305 (0.0008) | 25 (2) | 1.5 (0.1) | 1.97 (0.08) | 0.0188 (0.0007) | 2.01 (0.16) |
| 12X- PTDH | 3.25 (0.15) | 7.6 (1.1) x 104 | 1.0 (0.2) | 2.50 (0.15) | 2.5 (0.1) | 2.23 (0.15) |
All assays were performed at 25 °C, pH 7.25 in 100 mM MOPS.
The steady state parameters were measured at saturating NAD concentration for each enzyme tested and phosphite concentration was varied. The errors given in parentheses were obtained from fitting to the Michaelis-Menten equation.
The pre-steady state studies were performed at saturating phosphite and NAD concentrations for each mutant studied. kobs was obtained by fitting the absorbance change observed in the pre-steady state to a zero order equation; the errors given in parentheses are standard deviations.
Pre-Steady State Behavior of NADP-12X-PTDH with NAD as Substrate
The pre-steady state behavior of NADP-12X-PTDH with NAD (Figure 4A) is very similar to that observed with 12X-PTDH and the wild type enzyme. The trace is linear, no significant burst of activity in the pre-steady state is observed, and the rate in the pre-steady state, kobs = 2.1 (0.2) s−1, is within error of the steady state rate, kcat = 2.04 (0.04) s−1. The isotope effect observed in the pre-steady state, Dkobs = 2.26 (0.23), is also within error of DV = 2.17 (0.08), as observed with the other enzymes tested.
Figure 4.
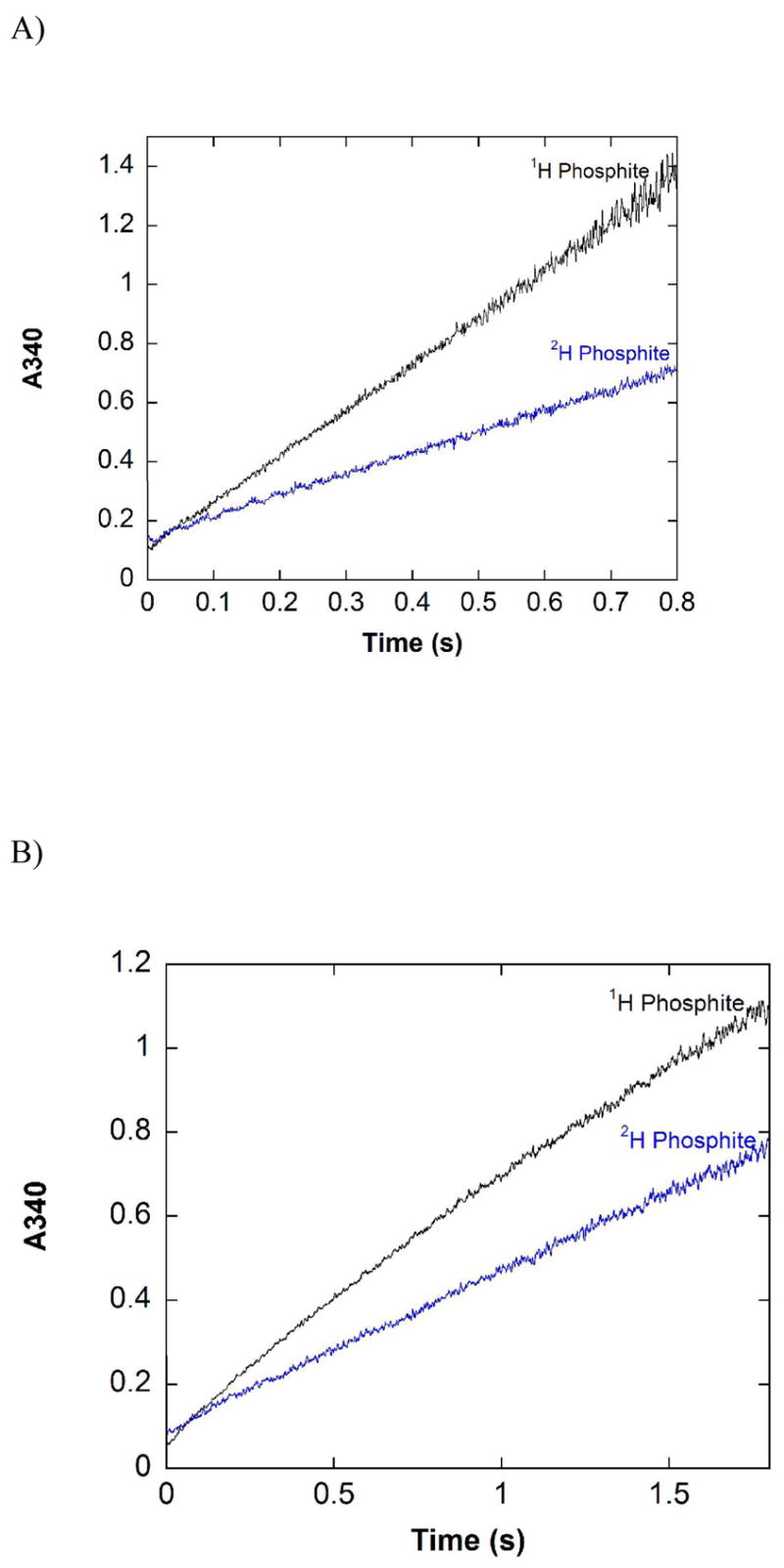
Pre-steady state traces for NADP-12X-PTDH. A) Traces with saturating NAD+, 1H or 2H phosphite. Reaction conditions: 100 mM MOPS pH 7.25, 5 mM 1H or 2H phosphite, 5 mM NAD+, 123 μM NADP-12X-PTDH. Each trace is the average of four separate shots. B) Traces with saturating NADP, 1H or 2H phosphite. Reaction conditions: 100 mM MOPS pH 7.25, 5 mM 1H or 2H phosphite, 5 mM NADP, 123 μM NADP-12X-PTDH. Each trace is the average of four separate shots.
Pre-Steady State Behavior of NADP-12X-PTDH with NADP as Substrate
The pre-steady state kinetics of NADP-12X-PTDH with NADP as cofactor were investigated to evaluate the possibility that a step after catalysis becomes partially rate determining, providing a possible explanation for the observed decreases in kcat and DV in the steady state experiments discussed above (Table 3). In contrast to what was observed when NAD was used as cofactor, the pre-steady state traces showed curvature with NADP, particularly with protiated phosphite (Figure 4B). The curvature is not due to consumption of substrates; only ~3% of the substrates were consumed during the period of data collection. The curvature is most likely due to burst kinetics in the pre-steady state. Analysis of the data finds that the rate immediately after mixing of enzyme and substrates is ~2-fold larger than kcat and then decreases to nearly kcat at the latest time points collected. We were unable to accurately fit the data to the generic burst equation (14).
Pre-Steady State Kinetics of PTDH Active Site Mutants
The pre-steady state behavior of the active site mutants K76A, R237K, E266Q, and D79A was investigated as they have steady-state rates spanning more than three orders of magnitude. Pre-steady state traces for R237K and E266Q are shown in Figure 5. The trace for R237K has relatively low signal-to-noise because the low activity of the mutant leads to a relatively small absorbance change. As for the WT enzyme, aggregation of the protein occurs at higher concentrations for all of the mutants tested, precluding studies using higher enzyme concentrations that would generate a larger absorbance change and improve signal-to-noise. Although noisy, the R237K trace (Figure 5A) clearly shows the absence of a significant burst in the pre-steady state for this mutant. The data shown in Figure 5 correspond to less than one half-life, but the observed rate (1.52 x 10−2 s−1) is in very good agreement with the steady state rate for this mutant under saturating conditions (1.26 x 10−2 s−1), again indicating that no significant burst occurs. Similarly, no observable burst of activity was observed in the pre-steady state for K76A or D79A (data not shown), and their pre-steady state rates are approximately the same as the steady state rates (Table 2). The signal-to-noise is much improved for E266Q, which has higher activity, but for this mutant as well a significant burst of product formation was not observed in the first two half-lives, and the steady state and pre-steady state rates are comparable (Figure 5B). The lack of a clear burst in the pre-steady state suggests a step after catalysis does not significantly limit the rate of reaction for all of the active site mutants tested.
Figure 5.
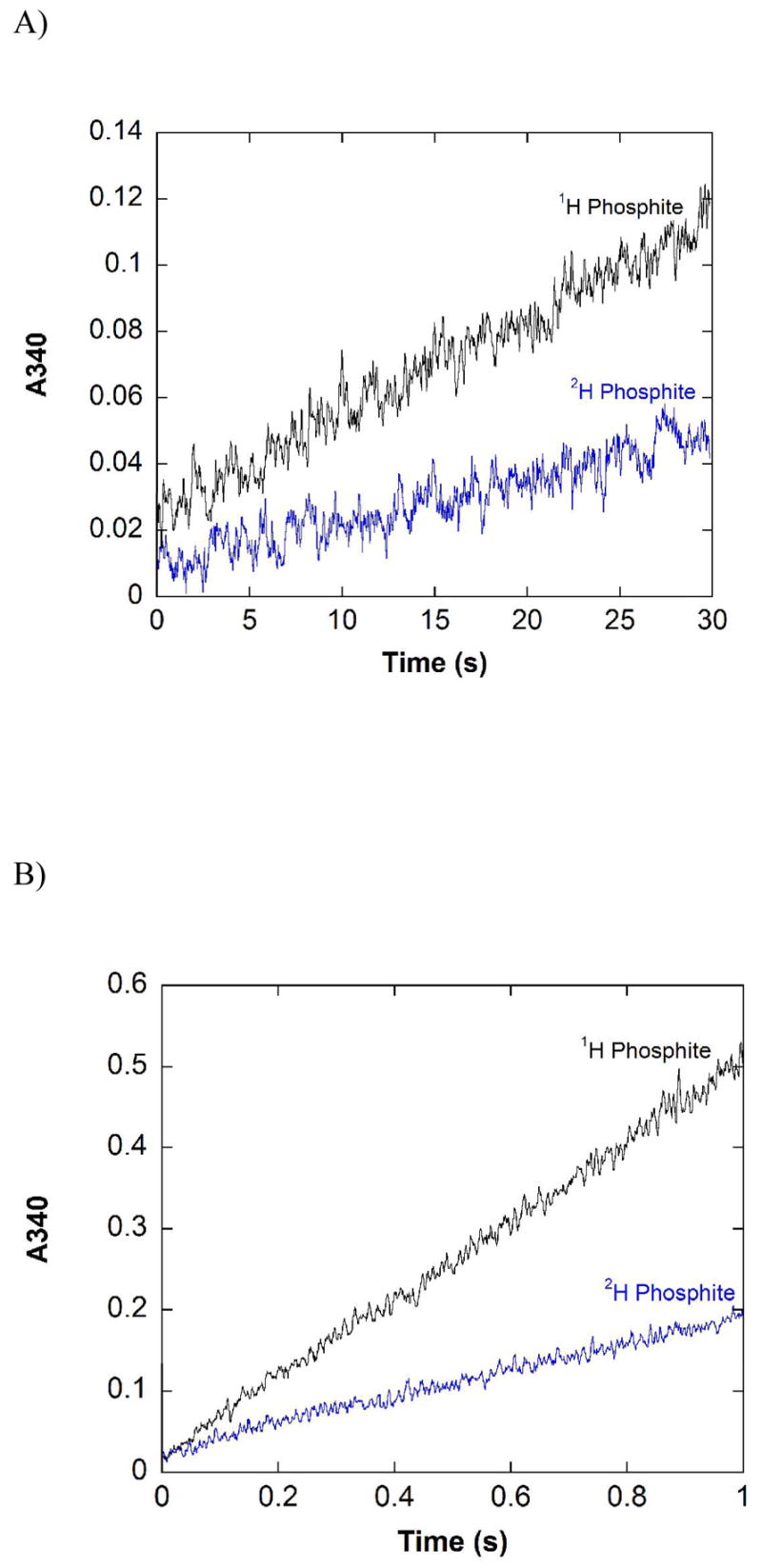
Reaction of representative active site mutants with protiated and deuterated phosphite in the pre-steady state. A) Reaction of R237K with protiated and deuterated phosphite in the pre-steady state. Reaction conditions: 100 mM MOPS pH 7.25, 100 mM 1H or 2H phosphite, 10 mM NAD+, 33 μM R237K. B) Reaction of E266Q with protiated and deuterated phosphite in the pre-steady state. Reaction conditions: 100 mM MOPS pH 7.25, 100 mM 1H or 2H phosphite, 5 mM NAD+, 15 μM E266Q. Each trace is the average of four separate shots. Background due to absorbance of NAD+ at 340 nm has been subtracted.
For completeness, the isotope effects in the pre-steady state were also determined for these mutants. As expected based on the lack of burst kinetics, the isotope effect in the pre-steady state for R237K, Dkobs = 2.37 (0.35), was within error of DV = 2.36 (0.06), whereas for E266Q Dkobs was 3.3 (0.3), also within error of the steady state KIE, DV = 3.3 (0.1). In fact, Dkobs was within error of the steady state DV for all of the mutants tested (Table 2).
Discussion
The reaction catalyzed by PTDH, the oxidation of inorganic phosphite to phosphate (Scheme 1), represents an intriguing phosphoryl transfer reaction in which water or hydroxide is the phosphoryl acceptor. Previous site-directed mutagenesis studies (7) have investigated the roles played by several active site residues in catalysis. These studies demonstrated the importance of Arg237 and Glu266 for phosphite binding and the critical importance of His292 for catalysis. The recently solved X-ray crystallographic structure (unpublished data) has confirmed the presence of these residues in the enzyme active site and has suggested other residues that might be important to enzyme function (Figure 1). In the structure, the conserved Asp79 interacts with Arg237, but is not expected to make direct contacts with phosphite. The site directed mutagenesis studies presented here suggest that Asp79 acts to orient Arg237. The D79A mutation, which eliminates any functionality that could hold Arg237 in place, led to significantly decreased values of both kcat and kcat/Km, Pt and a large increase in Km, Pt compared to the WT enzyme (Table 1). In fact, the kinetic constants for D79A are similar to those found for R237K, a mutant in which the arginine that directly interacts with phosphite is mutated to lysine. In contrast, the D79N mutant has nearly WT activity and Km, Pt has actually decreased compared to WT (Table 1). Presumably the hydrogen bonding interactions between Asn79 and Arg237 are sufficient to position the arginine for efficient binding of phosphite.
The extremely favorable energetics of the reaction catalyzed by PTDH, Keq = 1011, makes it tempting to envision a mechanism in which the chemical step of hydride transfer is rapid and the steady state rate is determined by physical steps such as product release or a slow conformational change. However, previous studies with PTDH showed significant KIEs on both Vmax and V/Km, Pt, indicating that the chemical step of hydride transfer is at least partially rate determining (8). The isotope effect on V/Km, NAD was unity, consistent with an ordered mechanism with NAD binding first. It should be noted that the values found for the isotope effects are larger than they may first appear because the theoretical maximal classical isotope effect has been calculated to be 5.0 at 25 °C based on infrared stretching frequencies of P-H and P-D bonds (10). Unlike other dehydrogenases (16), the possibility of quantum mechanical tunneling has not yet been investigated for PTDH.
When KIEs were determined for active site mutants such as K76A and R237K the isotope effect observed on Vmax was within error of the effect on WT. Considering that the catalytic efficiency of these mutants is diminished by up to four orders of magnitude compared to the WT enzyme and that kcat is decreased by more than 100-fold, identical Vmax isotope effects in the mutants and WT presents the possibility that the observed isotope effect may be the intrinsic effect, i.e. the observed KIE on Vmax is not masked by other steps. To evaluate the degree to which the hydride transfer step is rate limiting in PTDH, pre-steady state studies were performed on the WT enzyme, several active site mutants, and the thermostable 12X-PTDH mutant. Studies with the WT enzyme did not show a significant burst of activity nor clear curvature during the course of the measurement or in the dead time of the stopped-flow instrument. The same behavior was observed for several active site mutants including E266Q, K76A, R237K, D79A, and the thermostable mutants 12X-PTDH and NADP-12X-PTDH with NAD as a substrate. These results indicate that slow steps after hydride transfer, such as product release or a slow conformational change, do not significantly limit the steady state rate in the WT enzyme and mutants tested. Attempts to confirm this conclusion by determining both the deuterium and tritium KIEs using the method of Northrop (17) were inconclusive due to the relatively large error in the tritium isotope effects. Single turnover experiments with the 12X-PTDH mutant also did not provide any evidence that steps preceding catalysis limit the rate of reaction for 12X-PTDH.
The pre-steady state behavior of NADP-12X-PTDH using the alternate cofactor, NADP, differs from that observed with WT and other PTDH mutants. The rate immediately after mixing enzyme and substrates is ~2-fold faster than the steady state rate, suggesting that a step after catalysis is partially rate determining for this mutant under these conditions. This model is consistent with the decrease in both kcat and the steady state KIE in this mutant when NADP is used as the cofactor (Table 4). Together with the small dissociation constant previously determined for NADPH for the NADP specific mutations in the WT background (15), the data suggest that product release may be partially rate determining for NADP-12X-PTDH using NADP as a cofactor.
In several other enzymes where chemistry has been shown to be fully rate limiting (18–20) it was observed that the isotope effects on Vmax and V/K were the same as a result of a very small or no commitment to catalysis. In PTDH DV is greater than D(V/K) for WT and all the active site mutants tested (Table 2). This finding suggests that PTDH may have a relatively large forward commitment to catalysis. Examination of the expressions for DV (eq. 2) and D(V/K) (eq. 3), simplified for an irreversible reaction as occurs in PTDH, shows that DV and D(V/K) depend on different terms in addition to the intrinsic effect Dk. Using Northrop’s terminology (21), D(V/K) depends also on commitment to catalysis, Cf, whereas DV depends on the ratio of Rf, the ratio of catalysis, to Ef, the equilibrium preceding catalysis (the ratio Rf/Ef has also been termed cVf in the nomenclature of Cleland (22)).
| eq 2 |
| eq 3 |
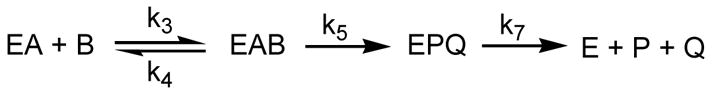
The terms Cf, Rf, and Ef, in turn depend on different ratios of microscopic rate constants. The pre-steady state data demonstrate that there are no kinetically important steps after catalysis and the single turnover experiments did not provide clear evidence for a slow conformational change prior to hydride transfer. Therefore, the minimal mechanism given in Scheme 2 can be used to represent the initial reaction rates catalyzed by PTDH in the presence of saturating concentrations of NAD. In this Scheme, no equilibria preceed catalysis under conditions of substrate saturation (DV) and thus Ef is unity. Cf and Rf are described by the expressions in equations 4 and 5 (21). The stopped-flow data clearly show that product release (k7) must be much faster than hydride transfer (k5), resulting in a value for Rf approaching zero and observed isotope effects on V that are equal to the intrinsic KIE (eq 3). Phosphite release from the ternary complex (k4) on the other hand cannot be much faster than the hydride transfer step to account for D(V/K) values that are smaller than those for DV. In the case of PTDH, the observed data are consistent with a rate of Michaelis complex dissociation (k4) that is approximately 2-fold slower than the chemical step (k5). In other words, the commitment to catalysis is about 2 as a result of a “sticky” substrate (22).
Scheme 2.
| eq 4 |
| eq 5 |
A free energy diagram describing the minimal kinetic mechanism is given in Figure 6A. The energy of the ES complex was determined from the Kd for phosphite calculated from D(V/K), DV and Km,Pt using the method of Klinman and Matthews (23). The dissociation constant for phosphite so determined is 16 μM which corresponds to an ES complex ~6.5 kcal/mol lower in energy than the NAD-enzyme complex and free phosphite. The total free energy change for the reaction catalyzed by PTDH at pH 7.0 (15 kcal/mol) was determined from the redox potentials of phosphite and NAD as reported previously (10). As discussed above, the experimental data suggest that kcat is fully limited by hydride transfer, thus the barrier height for hydride transfer (17 kcal/mol) was calculated using the Eyring equation, assuming k5 = kcat. Using the experimentally determined isotope effect on V/K, the assumption of k5 = kcat, and the expression for D(V/K) given in equation 2, the value of k4 and the barrier to the formation of the ES complex was determined. The free energy diagram depicted in Figure 6A accounts for the experimental observations of nearly identical Vmax isotope effects in mutants with a wide range of catalytic efficiency and activity, the lack of a significant burst in the pre-steady state, and the identity between the steady state DV and the isotope effects observed in the pre-steady state. Such a kinetic mechanism predicts that as commitment to catalysis becomes smaller, i.e. the substrate becomes less sticky or k5 becomes smaller than k4, the isotope effect will become more fully expressed in D(V/K) to the limit where D(V/K) = DV. Interestingly, such a trend is observed in the active site mutants. Although D(V/K) never reaches the limit of DV, as kcat decreases D(V/K) approaches DV in the active site mutants (Table 2). Free energy profiles for the K76A and R237K mutants, determined as described above for WT PTDH, show graphically the (small) decrease in commitment to catalysis that occurs in these mutants compared to WT (Figure 6B).
Figure 6.

Proposed free energy diagram of the reaction catalyzed by PTDH (A) and PTDH mutants (B).
It has been noted that the classic picture of an enzyme catalyzed reaction in which physical steps such as substrate binding and product release are rapid and a single chemical transformation step is slow and completely rate determining is often incorrect (24). More often enzymes have multiple rate determining steps and in some cases the overall rate of reaction is not determined by chemistry at all and is entirely limited by physical steps (25). In fact, it has been suggested that during evolution the step with the largest barrier is being optimized ultimately resulting in several partially rate determining steps (26). The observation that chemistry is fully rate determining in PTDH may suggest that the enzyme has only relatively recently evolved the ability to oxidize phosphite. However, the low sequence conservation between PTDH and another confirmed phosphite dehydrogenase (27) as well as putative phosphite dehydrogenases in the protein databases suggests that these enzymes may have had a long time to evolve and diverge. It is intriguing that the two members of the D-hydroxy acid dehydrogenase family catalyzing the thermodynamically most favorable reactions, formate dehydrogenase and phosphite dehydrogenase, are amongst the slowest enzymes in the family and that for both chemistry is fully rate limiting (18). The fact that we have not been able to improve the activity of PTDH significantly through in vitro evolution techniques (5) suggests that perhaps the nucleophilic displacement reaction catalyzed by PTDH has a relatively large intrinsic barrier.
Footnotes
This work was supported by the National Institutes of Health (GM63003).
Abbreviations: PTDH, phosphite dehydrogenase; NAD, β-nicotinamide adenine dinucleotide; WT, wild type; 12X-PTDH, thermostable PTDH generated by random mutagenesis; NADP-12X-PTDH, NADP specific 12X PTDH; NADP, β-nicotinamide adenine dinucleotide phosphate; KIE, kinetic isotope effect; DV, primary kinetic isotope effect on Vmax; D(V/K), primary kinetic isotope effect of V/Km, Pt; LB, Luria Bertani broth; OD600, optical density at 600 nm; Tris, tris(hydroxymethyl)aminomethane; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; MOPS, 4-morpholinepropanesulfonic acid; PCR, polymerase chain reaction; UV-vis, ultraviolet and visible; 1H phosphite, protiated phosphite; 2H phosphite, deuterated phosphite; AU, absorbance units; Dkobs, primary kinetic isotope effect observed in the pre-steady state; Cf, commitment to catalysis; Rf, the ratio of catalysis; Ef, the equilibrium preceding catalysis; Dk, intrinsic isotope effect.
References
- 1.White AK, Metcalf WW. The htx and ptx operons of Pseudomonas stutzeri WM88 are new members of the pho regulon. J Bacteriol. 2004;186:5876–5882. doi: 10.1128/JB.186.17.5876-5882.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costas AM, White AK, Metcalf WW. Purification and characterization of a novel phosphorus-oxidizing enzyme from Pseudomonas stutzeri WM88. J Biol Chem. 2001;276:17429–17436. doi: 10.1074/jbc.M011764200. [DOI] [PubMed] [Google Scholar]
- 3.Vrtis JM, White AK, Metcalf WW, van der Donk WA. Phosphite dehydrogenase: a versatile cofactor-regeneration enzyme. Angew Chem Int Ed Engl. 2002;41:3257–3259. doi: 10.1002/1521-3773(20020902)41:17<3257::AID-ANIE3257>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 4.Woodyer R, van der Donk WA, Zhao H. Relaxing the nicotinamide cofactor specificity of phosphite dehydrogenase by rational design. Biochemistry. 2003;42:11604–11614. doi: 10.1021/bi035018b. [DOI] [PubMed] [Google Scholar]
- 5.Woodyer R, van der Donk WA, Zhao H. Optimizing a biocatalyst for improved NAD(P)H regeneration: directed evolution of phosphite dehydrogenase. Comb Chem High Throughput Screen. 2006;9:237–245. doi: 10.2174/138620706776843246. [DOI] [PubMed] [Google Scholar]
- 6.Johannes TW, Woodyer RD, Zhao H. Directed evolution of a thermostable phosphite dehydrogenase for NAD(P)H regeneration. Appl Environ Microbiol. 2005;71:5728–5734. doi: 10.1128/AEM.71.10.5728-5734.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woodyer R, Wheatley JL, Relyea HA, Rimkus S, van der Donk WA. Site-directed mutagenesis of active site residues of phosphite dehydrogenase. Biochemistry. 2005;44:4765–4774. doi: 10.1021/bi047868c. [DOI] [PubMed] [Google Scholar]
- 8.Relyea HA, Vrtis JM, Woodyer R, Rimkus SA, van der Donk WA. Inhibition and pH dependence of phosphite dehydrogenase. Biochemistry. 2005;44:6640–6649. doi: 10.1021/bi047640p. [DOI] [PubMed] [Google Scholar]
- 9.Relyea HA, van der Donk WA. Mechanism and applications of phosphite dehydrogenase. Bioorg Chem. 2005;33:171–189. doi: 10.1016/j.bioorg.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Vrtis JM, White AK, Metcalf WW, van der Donk WA. Phosphite dehydrogenase: an unusual phosphoryl transfer reaction. J Am Chem Soc. 2001;123:2672–2673. doi: 10.1021/ja004301k. [DOI] [PubMed] [Google Scholar]
- 11.Woodyer R, van der Donk WA, Zhao H. Relaxing the Nicotinamide Cofactor Specificity of Phosphite Dehydrogenase by Rational Design. Biochemistry. 2003;42:11604–11614. doi: 10.1021/bi035018b. [DOI] [PubMed] [Google Scholar]
- 12.Trimboli AJ, Barber MJ. Assimilatory nitrate reductase: reduction and inhibition by NADH/NAD+ analogs. Arch Biochem Biophys. 1994;315:48–53. doi: 10.1006/abbi.1994.1469. [DOI] [PubMed] [Google Scholar]
- 13.Hoa GH, Douzou P. Stopped flow method at subzero temperatures. Anal Biochem. 1973;51:127–136. doi: 10.1016/0003-2697(73)90460-0. [DOI] [PubMed] [Google Scholar]
- 14.Johnson KA. In: The Enzymes. Sigman DS, editor. Academic Press; San Diego: 1992. pp. 1–61. [Google Scholar]
- 15.Woodyer R, Zhao H, van der Donk WA. Mechanistic investigation of a highly active phosphite dehydrogenase mutant and its application for NADPH regeneration. Febs J. 2005;272:3816–3827. doi: 10.1111/j.1742-4658.2005.04788.x. [DOI] [PubMed] [Google Scholar]
- 16.Nagel ZD, Klinman JP. Tunneling and dynamics in enzymatic hydride transfer. Chem Rev. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- 17.Northrop DB. In: Isotope Effects on Enzyme-Catalyzed Reaction. Cleland WW, O'Leary MH, Northrop DB, editors. University Park Press; Baltimore, MD: 1977. pp. 122–152. [Google Scholar]
- 18.Blanchard JS, Cleland WW. Kinetic and chemical mechanisms of yeast formate dehydrogenase. Biochemistry. 1980;19:3543–3550. doi: 10.1021/bi00556a020. [DOI] [PubMed] [Google Scholar]
- 19.Patel MP, Blanchard JS. Mycobacterium tuberculosis mycothione reductase: pH dependence of the kinetic parameters and kinetic isotope effects. Biochemistry. 2001;40:5119–5126. doi: 10.1021/bi0029144. [DOI] [PubMed] [Google Scholar]
- 20.Sobrado P, Daubner SC, Fitzpatrick PF. Probing the relative timing of hydrogen abstraction steps in the flavocytochrome b2 reaction with primary and solvent deuterium isotope effects and mutant enzymes. Biochemistry. 2001;40:994–1001. doi: 10.1021/bi002283d. [DOI] [PubMed] [Google Scholar]
- 21.Northrop DB. In: Enzyme Mechanism from Isotope Effects. Cook PF, editor. CRC Press; Boca Raton: 1991. pp. 181–196. [Google Scholar]
- 22.Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. Garland Science; New York: 2007. [Google Scholar]
- 23.Klinman JP, Matthews RG. Calculation of substrate dissociation constants from steady-state isotope effects in enzyme-catalyzed reactions. J Am Chem Soc. 1985;107:1058–1060. [Google Scholar]
- 24.Northrop DB. Steady-state analysis of kinetic isotope effects in enzymic reactions. Biochemistry. 1975;14:2644–2651. doi: 10.1021/bi00683a013. [DOI] [PubMed] [Google Scholar]
- 25.Cleland WW, Northrop DB. Energetics of substrate binding, catalysis, and product release. Methods Enzymol. 1999;308:3–27. doi: 10.1016/s0076-6879(99)08003-9. [DOI] [PubMed] [Google Scholar]
- 26.Burbaum JJ, Raines RT, Albery WJ, Knowles JR. Evolutionary optimization of the catalytic effectiveness of an enzyme. Biochemistry. 1989;28:9293–9305. doi: 10.1021/bi00450a009. [DOI] [PubMed] [Google Scholar]
- 27.Wilson MM, Metcalf WW. Genetic diversity and horizontal transfer of genes involved in the oxidation of reduced P compounds by Alcaligenes faecalis WM2072. Appl Environ Microbiol. 2005;71:290–296. doi: 10.1128/AEM.71.1.290-296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]