

Abstract
We investigated the role of endogenous protein kinase activity on synaptic transmission in the rat nucleus accumbens slice. The isoquinolinesulfonamide H-7 (50 μM), a non-selective serine/threonine protein kinase inhibitor, had no effect on pharmacologically isolated glutamatergic EPSCs. However, it reduced GABA release in a dose-dependent manner. This effect of H-7 was not mimicked by the selective cAMP-dependent protein kinase inhibitor H-89, the PKC inhibitor Bisindolylmaleimide-1, or the cGMP-dependent protein kinase inhibitor KT-5823. However, bath application of the myosin light chain kinase (MLCK) inhibitor, ML-7, significantly reduced IPSC amplitudes and partially occluded the reduction in IPSCs observed following bath application of H-7. These results suggest that endogenous protein kinase activity, specifically MLCK activity, regulates GABA, but not glutamate release, onto medium spiny neurons in the nucleus accumbens.
Keywords: electrophysiology, nucleus accumbens, protein kinase, medium spiny neurons, GABA, myosin light chain kinase
Introduction
Neurotransmitter release from presynaptic nerve terminals is maintained by a precise cycle of vesicle docking, priming and fusion, coupled with endocytosis and refilling (Sudhof, 2004). The functions of many of the components involved in the vesicle cycle are sensitive to protein phosphorylation (Leenders and Sheng, 2005). For example, activation of protein kinase C (PKC) or cyclic-AMP dependent protein kinase A (PKA) has been shown to enhance neurotransmitter release at a number of glutamatergic and GABAergic synapses throughout the brain (Capogna et al., 1995; Carroll et al., 1998; Chavez-Noriega and Stevens, 1994; Chen and Regehr, 1997; Chen and Roper, 2003; Malenka et al., 1986).
While activating serine/threonine protein kinases enhances neurotransmission, to what degree these kinases are endogenously active during synaptic activity and how this endogenous activity controls synaptic strength is less clear. In the hippocampus, the non-selective protein kinase inhibitor, H-7 can inhibit excitatory synaptic transmission at high (e.g. 200-300 μM) concentrations (Malinow et al., 1988; Muller et al., 1990; Waxham et al., 1993). However, at lower concentrations, H-7 selectively reduces GABAergic synaptic transmission leading to a disinhibition of CA1 neurons (Corradetti et al., 1989; Leahy and Vallano, 1991). A similar inhibition of IPSCs has been observed following application of the non-selective kinase inhibitor staurosporine in the VTA (Bonci and Williams, 1997).
Inhibiting protein kinase activity in the nucleus accumbens (NAc) has been shown to interfere with appetitive learning (Baldwin et al., 2002) and to alter cocaine self-administration and relapse to cocaine seeking behavior (Self et al., 1998). Thus, it is thought that the activation of protein kinases is required for instrumental learning. However, given that endogenous protein kinase activity may control synaptic neurotransmission, it is possible that protein kinase inhibitors may be altering basal synaptic activity, as opposed to blocking activity related to a specific learning event. Therefore, we decided to investigate the effect of inhibiting serine/threonine kinase activity on excitatory and inhibitory synaptic responses in the NAc. We find that inhibiting protein kinase activity reduces GABAergic transmission through a presynaptic decrease in neurotransmitter release but has no effect on excitatory glutamatergic transmission. However, we find that neither PKA nor PKC is responsible for this inhibition. Instead, the reduction of GABA release is driven, at least in part, by the inhibition of myosin light chain kinase (MLCK).
Materials and Methods
All animal procedures conformed to NIH and Ernest Gallo Clinic and Research Center animal care policies. Male Sprague Dawley Rats (2 to 4 week old) were anesthetized using isofluorane, decapitated, and their brains were submerged in ice-cold Ringers solution containing (in mM): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 1.0 NaH2PO4, 2.5 CaCl2, 26.2 NaHCO3, and 11 glucose. Coronal slices (350 μm thick) containing the Nac were prepared using a vibratome (Leica, Nusslach, Germany) and allowed to recover in carboxygen (95% O2, 5% CO2)-bubbled Ringer’s solution for > 1 hour. Individual slices were visualized under an Olympus upright microscope with differential interference contrast optics and infrared illumination. A bipolar stimulating electrode was positioned dorsal to the recording area in the medial shell of the NAc. Medium spiny neurons, identified by their size and hyperpolarized resting membrane potentials, were voltage-clamped at -80 mV using patch pipettes (3-5 MOhms) containing (in mM): 107.5 Cs-gluconate, 20 HEPES, 0.2 EGTA, 10 TEA, 8 NaCl, 2 MgATP and 0.3 Na3GTP (pH 7.2 and osmolarity adjusted to 275 mOsm). Recordings were made using an Axopatch 1 D amplifier with a cutoff frequency of 2 kHz and collected at 5 kHz using IgorPro (Wavemetrics, Lake Oswego, OR). Holding current, series and input resistances were monitored throughout the experiment. Recordings were rejected if the series resistance changed by more than 5 MOhms.
All drugs were obtained from either Sigma or Tocris. Stock solutions were prepared either in DMSO or H2O and diluted immediately before use. For preincubation experiments, the drug was dissolved in 50 ml of Ringer’s solution, prewarmed to 35° C and slices were immersed in this solution for one hour.
The coefficient of variation (CV) was calculated as the SD/mean amplitude for 20 consecutive IPSCs in each condition. Statistical analysis was performed using Student’s t-test or the appropriate ANOVA and significance was defined as p < 0.05. All results are presented as means ± SEM.
Results
The general protein kinase inhibitor, H-7 inhibits IPSC, but not EPSC, amplitudes
We initially investigated the effects of the non-selective protein kinase inhibitor, H-7, on synaptic transmission in the nucleus accumbens shell. Medium spiny neurons (MSNs) were voltage clamped at -80 mV. Application of 50 μM H-7 had no significant effect on the amplitude of pharmacologically isolated EPSCs, recorded in the presence of 100 μM picrotoxin (Fig. 1A; 93.2 ± 6.5 % of control, n=5). On the other hand, the amplitude of pharmacologically isolated IPSCs, recorded in the presence of 10 μM DNQX, were significantly reduced following the bath application of 50 μM H-7 (Fig. 1B, reduced to 24.2 ± 4.8 % of control, p < 0.001, n=5). This effect was rapid and the peak reduction in amplitudes occurred within 3 min. The magnitude of reduction in IPSC amplitude was dose-dependent with an IC50 of 0.8 μM (Fig. 1C). This effect on IPSCs could be mimicked by a second non-selective protein kinase inhibitor, staurosporine (1 μM, Fig. 1D; 32.1 ± 8.4% of control, n=4, p < .01). EPSCs were unaffected by staurosporine application (98.6 ± 5% of control, n=3). The above results suggest that endogenous protein kinase activity controls GABAergic but not glutamatergic transmission on MSNs in the NAc.
Figure 1.
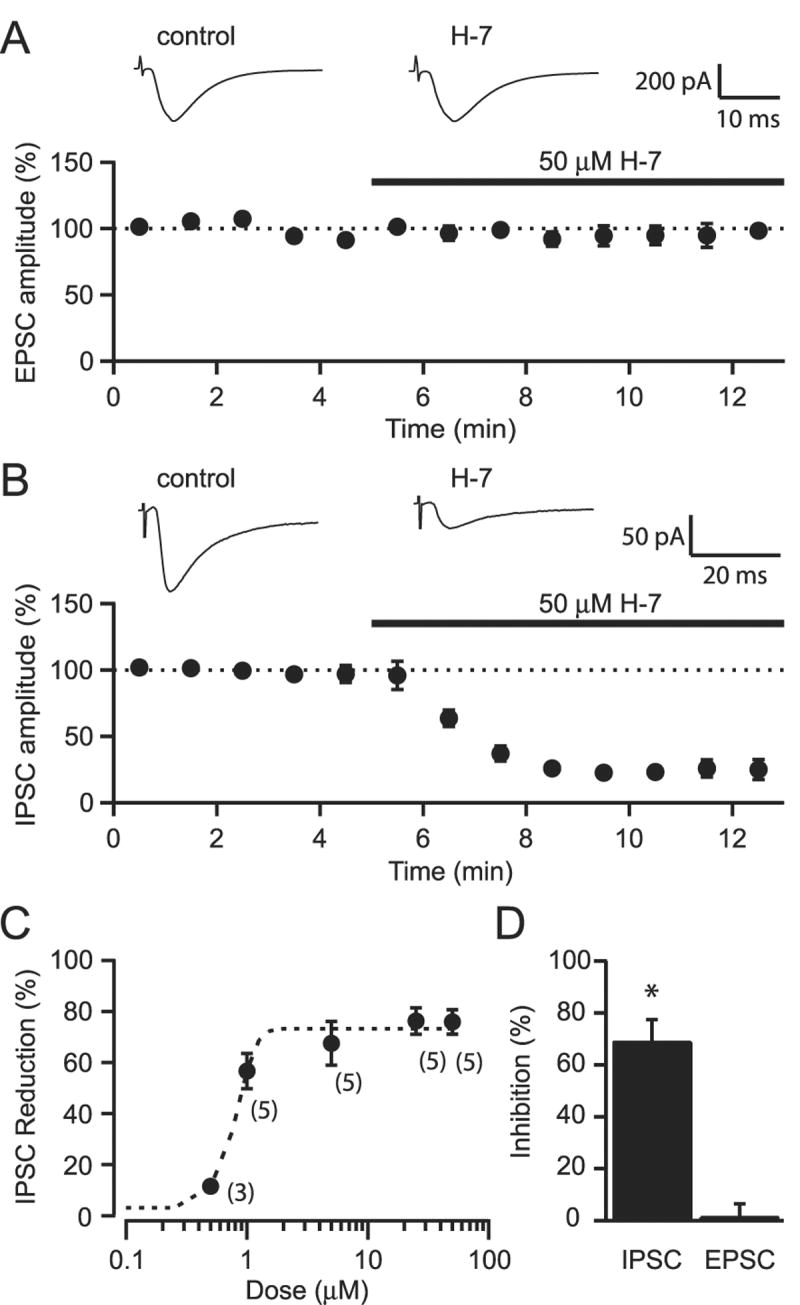
Inhibiting protein kinase activity reduces IPSC but not EPSC amplitudes. A. Bath application of 50 μM H-7 has no effect on EPSC amplitudes. Averaged EPSCs from a representative experiment for control and H-7 conditions are displayed above. B. Bath application of 50 μM H-7 produces a rapid reduction in IPSC amplitudes. Averaged EPSCs from a representative experiment are shown. C. Dose response curve of the H-7 effect on IPSCs. The dotted line shows a sigmoid fit to the data. The experimental n for each dose are provided in parenthesis. D. Bath application of 1 μM staurosporine inhibited IPSC amplitudes but had no effect on EPSC amplitudes * indicates p < 0.01
H-7 reduces IPSC amplitudes by inhibiting GABA release
To determine whether the observed inhibition of IPSCs was due to a decrease in GABA release or a postsynaptic change in GABAA receptor function, we looked at the effects of H-7 on miniature IPSCs (mIPSCs) recorded at 0 mV in the presence of 1 μM TTX, 10 μM DNQX and 100 μM d-AP5. Bath application of 50 μM H-7 decreased the mean mIPSC frequency to 11.9 ± 2.9% of control values (p < .01; n=4). There was also a reduction in mIPSC amplitude that did not reach statistical significance (Fig. 2 D, 76.1 ± 7.9% of control, p< .06).
Figure 2.
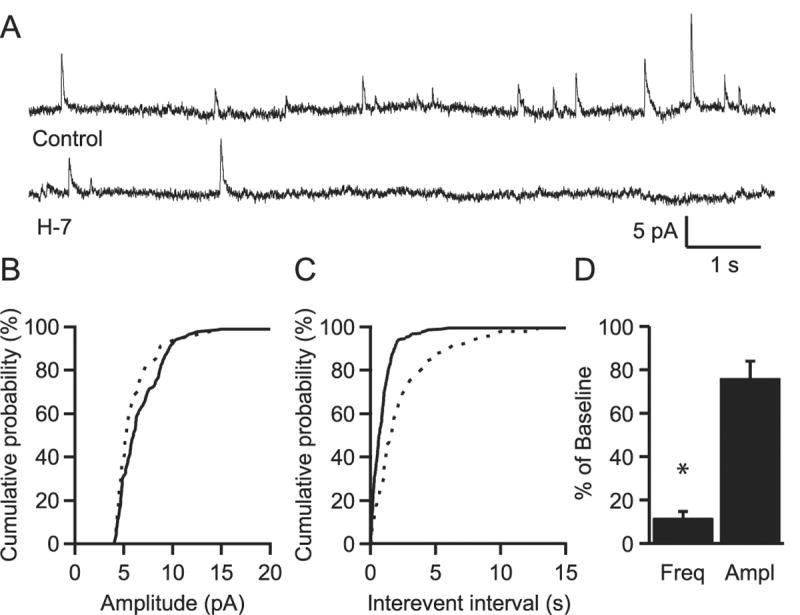
H-7 reduces the frequency of mIPSCs. A. Representative traces of mIPSCs recorded either before (Control) or after bath application of H-7 from an individual experiment. B. Cumulative probability distribution of mini amplitudes in control (solid line) and H-7 (dashed line) for the same experiment. C. Cumulative distribution of the inter-event intervals in control and H-7 for the same experiment. D. Average of four experiments shows a significant reduction in frequency but not in the mean amplitude of mIPSCs. * indicates p < 0.005.
To confirm that the action of H-7 on IPSCs is presynaptic, we analyzed the coefficient of variation (CV) of the amplitudes of evoked IPSCs collected for a period of 5 minutes during control and following H-7 application. Application of H-7 (1 – 50 μM) produced an overall increase in the CV ratio (2.45 ± 0.6, p < .05, n=16 data not shown). Finally, we included 50 μM H-7 in the internal solution and looked for changes in IPSC amplitudes after whole-cell break-in. IPSC amplitudes showed a small, non-significant reduction 10 minutes following break-in (90.9 ± 11.3% compared to the mean amplitude during the initial two minutes, n=3). Subsequent bath application of 50 μM H-7 inhibited the IPSCs, supporting a presynaptic locus.
Reduction is partially mediated by Myosin Light Chain Kinase (MLCK)
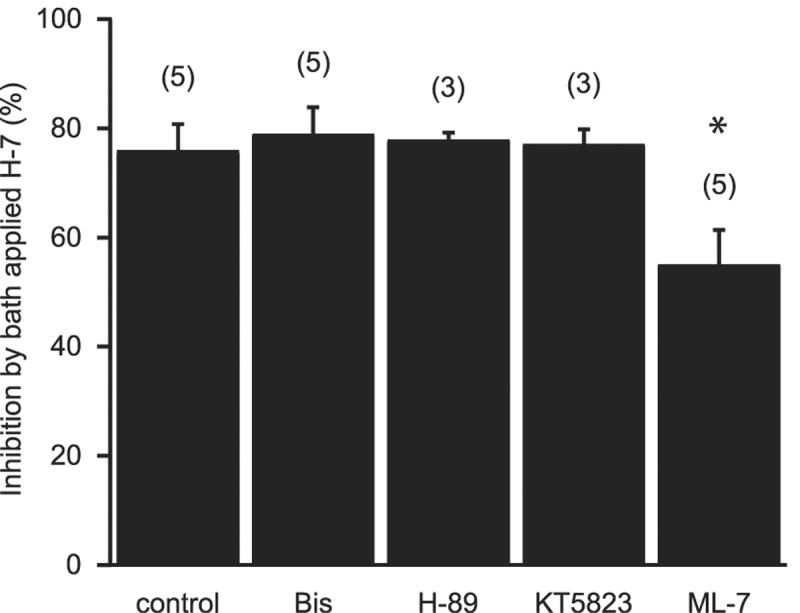
We next investigated which serine-threonine kinase might account for the reduction in GABA release. Bath application of the PKC inhibitor, bisindolylmaleimide I (Bis; 1 μM) for 10 min had no significant effect on evoked IPSC amplitudes (Fig. 4A, 92.4 ± 4.8 % of control, n=5). Similarly, application of the selective PKA inhibitor, N-[2-(p-bromocinnamylamino)ethyl]-5-soquinolinesulfonamide (H-89), at 5 μM did not significantly inhibit IPSC amplitudes (Fig. 4B, 106.0 ±13.3 % of control, n=3). This lack of inhibition was not observed even when 1 μM Bis or 5 μM H-89 were continuously applied for a period of 1 hr (data not shown), suggesting that neither PKA nor PKC accounts for the H-7 induced reduction in GABA transmission.
Figure 4.

The MLCK inhibitor, ML-7 inhibits IPSC amplitudes. A. Bath application of the PKA inhibitor H-89 (5 μM) had no effect on IPSC amplitudes. B. Bath application of the PKC inhibitor Bis (1 μM) had no effect on IPSC amplitudes. C. Bath application of the PKG inhibitor KT5823 (1 μM) had no effect on IPSC amplitudes. D. Bath application of the MLCK inhibitor ML-7 (10 μM) significantly reduced IPSC amplitudes.
Other serine/threonine kinases that are sensitive to H-7 include protein kinase G (PKG) and myosin light chain kinase I (MLCK) (Hidaka et al., 1984). Bath application of the PKG antagonist, KT5823, had no significant effect on the amplitude of IPSCs (Fig. 4C, 86.5 ± 8.1 % of control, n=4). The MLCK inhibitor, (5-iodonaphthalene-1-sulfonyl) homopiperazine (ML-7, 10 μM), on the other hand, did significantly reduce IPSC amplitudes (Fig. 4D, 61.2 ± 13.6 % of control, p < .05, n=5) implicating MLCK as a potential kinase that regulates GABA release at MSN synapses.
The lack of effects seen with PKC, PKA and the PKG inhibitors could be due to incomplete equilibration of these drugs and an inability to reach the site of action during the period of application. To account for this possibility, we incubated slices in the selective protein kinase inhibitors for 1 hr prior to recording and investigated whether this preincubation occludes the effects of bath applied H-7. Under control conditions, 50 μM H-7 reduced IPSC amplitudes by 75.9 ± 4.8 % (n=5; Fig. 1). Preincubation with Bis, H-89 or KT5823 had no significant effect on the subsequent bath application of H-7 (Fig. 5, IPSC amplitudes were reduced by 79.0 ± 4.8%, n=5; 77.9 ± 1.2%, n=3; and 77.2 ± 2.6 %, n=3 respectively). Pretreatment with the MLCK inhibitor, ML-7 partially occluded the reduction seen in H-7 (Fig. 5, H-7 inhibited IPSC amplitudes by 55.1 ± 6.4%, n=5, p<.001 compared to control group, one-way ANOVA w/ Holm-Sidak posthoc test). The above results suggest that MLCK, but not PKC, PKA or PKG, plays an important role in controlling GABA release at MSN synapses and that an effect on MLCK at least partially explains IPSC amplitude reduction seen in H-7.
Figure 5.
Preincubation with ML-7 partially occludes the H-7 mediated inhibition of IPSCs. Slices were incubated for 1 hour in control, 1 μM Bis, 5 μM H-89, 1 μM KT5823 or 10 μM ML-7. IPSC amplitudes before and after bath application of 50uM H-7 are compared and % reduction for each pretreatment is shown. * indicates p < 0.01 compared to control (one way ANOVA with Holms-Sidak posthoc test).
Discussion
GABAergic synaptic transmission in the NAc shell is selectively inhibited by the protein kinase inhibitor H-7. This effect is similar to that observed in the hippocampus, where H-7 produces a disinhibition of CA1 neurons at concentrations similar to those used in this study (Corradetti et al., 1989; Leahy and Vallano, 1991). These previous studies postulated that the disinhibition by H-7 was mediated through PKC. However, in the NAc, our data indicates that the effects of H-7 on GABA release are independent of either PKC or PKA, as more selective inhibitors for the two are unable to mimic or occlude the effects of H-7. Likewise, inhibition of PKG also had no effect on GABAergic transmission. Instead, we found that the MLCK inhibitor ML-7 was able to significantly inhibit GABA release. Moreover, incubating slices in the inhibitor partially occluded the effects of H-7.
Previous studies have implicated MLCK in synaptic transmission in cultured cervical ganglion neurons (Mochida et al., 1994), chromaffin cells (Kumakura et al., 1994) as well as in hippocampal cultures (Ryan, 1999). Specifically, inhibiting MLCK reduces the size of the recycling vesicle pool by blocking the recruitment of vesicles to the readily releasable pool (Ryan, 1999; but see Tokuoka and Goda, 2006). While our data indicate that the actions of H-7 at GABA synapses in the NAc are mediated through the inhibition of MLCK, the reported Ki value for the inhibition MLCK by H-7 is closer to 100 μM (Hidaka et al., 1984), considerably larger than the doses required to inhibit GABA release. This, combined with the lack of complete occlusion between ML-7 and H-7, makes it likely that additional serine/threonine protein kinases are involved. It also raises the possibility that ML-7 may be acting on a protein kinase besides MLCK. It has been proposed that ML-7 has non-selective effects on action potentials and/or voltage gated calcium channels (Tokuoka and Goda, 2006), although this explanation does not explain the effects of H-7 on mIPSC frequency, which are Ca2+ channel independent (Nicola and Malenka, 1997). Nor can it explain the selective reduction of IPSCs with no effect on EPSCs.
These data add to the differences between the control of excitatory and inhibitory synaptic transmission in the NAc. For example, while dopamine inhibits the release of glutamate and GABA in the NAc, it does so via distinct mechanisms (Nicola and Malenka, 1997). A similar situation exists for the inhibition of glutamate and GABA release by kappa opioid agonists (Hjelmstad and Fields, 2003). In each case, the inhibition of GABA release is calcium dependent and is sensitive to TTX, whereas the inhibition of glutamate release is downstream of calcium entry. Since H-7 inhibits mIPSC frequency in the presence of TTX, it is unlikely that the presynaptic inhibition produced by either dopamine or opioids is mediated through this pathway.
Protein kinase activation in the NAc has been hypothesized to drive the long lasting changes associated with instrumental learning and the development of behaviors associated with drug addiction (Baldwin et al., 2002; Self et al., 1998). For example, microinjection of H-7 or the specific PKA inhibitor, Rp-cAMPs into the NAc delays the acquisition of lever-pressing for a food reward (Baldwin et al., 2002). While the data in our study do not speak to the role of protein kinase activation during learning events, they raise the caveat that protein kinase inhibitors can have effects on basal synaptic transmission in the NAc.
Figure 3.
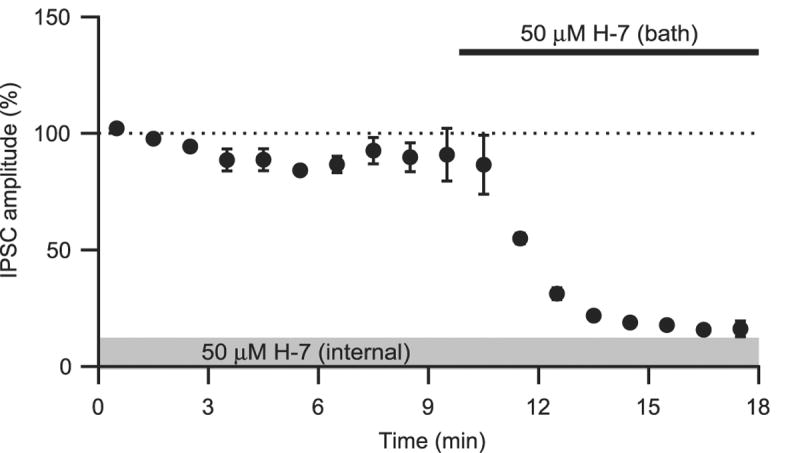
Intracellular application of H-7 does not inhibit IPSCs. IPSC amplitude (normalized to the first two minutes) recorded for 10 min with 50 μM H-7 present in the internal solution. Subsequent bath application of 50 μM H-7 (solid bar) inhibited the IPSCs.
Acknowledgments
The authors thank Dr. Y.F. Xia for comments on the manuscript. This work was supported by NIH grant DA-15686 to GOH and by funds provided by the state of California for medical research on alcohol and substance abuse through the University of California, San Francisco.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baldwin AE, Sadeghian K, Holahan MR, Kelley AE. Appetitive instrumental learning is impaired by inhibition of cAMP-dependent protein kinase within the nucleus accumbens. Neurobiol Learn Mem. 2002;77:44–62. doi: 10.1006/nlme.2000.4002. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, Gahwiler BH, Thompson SM. Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. J Neurosci. 1995;15:1249–1260. doi: 10.1523/JNEUROSCI.15-02-01249.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Nicoll RA, Malenka RC. Effects of PKA and PKC on miniature excitatory postsynaptic currents in CA1 pyramidal cells. J Neurophysiol. 1998;80:2797–2800. doi: 10.1152/jn.1998.80.5.2797. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. Journal of Neuroscience. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-X, Roper SN. PKA and PKC enhance excitatory synaptic transmission in human dentate gyrus. J Neurophysiol. 2003;89:2482–2488. doi: 10.1152/jn.01031.2002. [DOI] [PubMed] [Google Scholar]
- Corradetti R, Pugliese AM, Ropert N. The protein kinase C inhibitor 1-(5-isoquinolinesulphonyl)-2-methylpiperazine (H-7) disinhibits CA1 pyramidal cells in rat hippocampal slices. Br J Pharmacol. 1989;98:1376–1382. doi: 10.1111/j.1476-5381.1989.tb12687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO, Fields HL. Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J Neurophysiol. 2003;89:2389–2395. doi: 10.1152/jn.01115.2002. [DOI] [PubMed] [Google Scholar]
- Kumakura K, Sasaki K, Sakurai T, Ohara-Imaizumi M, Misonou H, Nakamura S, Matsuda Y, Nonomura Y. Essential role of myosin light chain kinase in the mechanism for MgATP-dependent priming of exocytosis in adrenal chromaffin cells. J Neurosci. 1994;14:7695–7703. doi: 10.1523/JNEUROSCI.14-12-07695.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy JC, Vallano ML. Differential effects of isoquinolinesulfonamide protein kinase inhibitors on CA1 responses in hippocampal slices. Neuroscience. 1991;44:361–370. doi: 10.1016/0306-4522(91)90061-r. [DOI] [PubMed] [Google Scholar]
- Leenders AG, Sheng ZH. Modulation of neurotransmitter release by the second messenger-activated protein kinases: implications for presynaptic plasticity. Pharmacol Ther. 2005;105:69–84. doi: 10.1016/j.pharmthera.2004.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Madison DV, Tsien RW. Persistent protein kinase activity underlying long-term potentiation. Nature. 1988;335:820–824. doi: 10.1038/335820a0. [DOI] [PubMed] [Google Scholar]
- Mochida S, Kobayashi H, Matsuda Y, Yuda Y, Muramoto K, Nonomura Y. Myosin II is involved in transmitter release at synapses formed between rat sympathetic neurons in culture. Neuron. 1994;13:1131–1142. doi: 10.1016/0896-6273(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Muller D, Buchs PA, Dunant Y, Lynch G. Protein kinase C activity is not responsible for the expression of long-term potentiation in hippocampus. Proc Natl Acad Sci U S A. 1990;87:4073–4077. doi: 10.1073/pnas.87.11.4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola SM, Malenka RC. Dopamine depresses excitatory and inhibitory synaptic transmission by distinct mechanisms in the nucleus accumbens. J Neurosci. 1997;17:5697–5710. doi: 10.1523/JNEUROSCI.17-15-05697.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TA. Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J Neurosci. 1999;19:1317–1323. doi: 10.1523/JNEUROSCI.19-04-01317.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self DW, Genova LM, Hope BT, Barnhart WJ, Spencer JJ, Nestler EJ. Involvement of cAMP-dependent protein kinase in the nucleus accumbens in cocaine self-administration and relapse of cocaine-seeking behavior. J Neurosci. 1998;18:1848–1859. doi: 10.1523/JNEUROSCI.18-05-01848.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Tokuoka H, Goda Y. Myosin light chain kinase is not a regulator of synaptic vesicle trafficking during repetitive exocytosis in cultured hippocampal neurons. J Neurosci. 2006;26:11606–11614. doi: 10.1523/JNEUROSCI.3400-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxham MN, Malenka RC, Kelly PT, Mauk MD. Calcium/calmodulin-dependent protein kinase II regulates hippocampal synaptic transmission. Brain Res. 1993;609:1–8. doi: 10.1016/0006-8993(93)90847-g. [DOI] [PubMed] [Google Scholar]