

Abstract
The objective of this study was to assess the potential interactions of the drug transporter P-glycoprotein with attention-deficit/hyperactivity disorder (ADHD) therapeutic agents atomoxetine — and the individual isomers of methylphenidate, amphetamine, and modafinil utilizing established in vitro assay. An initial ATPase assay indicated that both d- and l-methylphenidate have weak affinity for P-glycoprotein. The intracellular accumulation of P-glycoprotein substrates doxorubicin and rhodamine123 in the P-glycoprotein overexpressing cell line LLC-PK1/MDR1 was determined to evaluate potential inhibitory effects on P-glycoprotein. The results demonstrated that all compounds, except both modafinil isomers, significantly increased doxorubicin and rhodamine123 accumulation in LLC-PK1/MDR1 cells at higher concentrations. To investigate the P-glycoprotein substrate properties, the intracellular concentrations of the tested compounds in LLC-PK1/MDR1 and P-glycoprotein negative LLC-PK1 cells were measured in the presence and absence of the P-glycoprotein inhibitor PSC833. The results indicate that the accumulation of d-methylphenidate in LLC-PK1 cells was 32.0% higher than in LLC-PK1/MDR1 cells. Additionally, coadministration of PSC833 leads to 52.9% and 45.6% increases in d-modafinil and l-modafinil accumulation, respectively, in LLC-PK1/MDR1 cells. Further studies demonstrated that l-modafinil transport across LLC-PK1/MDR1 cell monolayers in the basolateral-to-apical (B–A) direction was significantly higher than in the apical-to-basolateral (A–B) direction. PSC833 treatment significantly decreased the transport of l-modafinil in B–A direction. In conclusion, our results suggest that all tested agents with the exception of modafinil isomers are relatively weak P-glycoprotein inhibitors. Furthermore, P-glycoprotein may play a minor role in the transport of d-methylphenidate, d-modafinil, and l-modafinil.
Keywords: P-glycoprotein, Methylphenidate, Amphetamine, Atomoxetine, Modafinil
1. Introduction
Attention-deficit/hyperactivity disorder (ADHD) is the most common neurobehavioral disorder affecting school-aged children. Recent data indicate that ADHD accounts for 30% to 50% of all mental health services referrals for children. Although once thought to be a disorder largely limited to childhood, it is now generally accepted that up to 50% or more of children diagnosed with ADHD may have symptoms persisting into adulthood (Weiss and Murray, 2003).
Recently established practice guidelines, algorithms, and consensus statements on the pharmacotherapy of ADHD have not identified an agent of first choice (Greenhill et al., 2002; Pliszka et al., 2000). However, methylphenidate (Leonard et al., 2004), together with the major amphetamine formulations such as d-amphetamine (James et al., 2001) and mixed isomers of amphetamine (Faraone et al., 2002) containing a 3:1 ratio of d-amphetamine to l-amphetamine, (Adderall®), and the selective norepinephrine reuptake inhibitor atomoxetine (Kratochvil et al., 2002) constitute the primary medications employed in the treatment of ADHD. Further, the nonsympathomimetic cognitive activator modafinil has emerged as a potential candidate for use in the treatment of ADHD and is frequently used “off label” for this indication. The use of all psychotropic medications to treat ADHD has recently shown a dramatic rise among every age group including preschoolers (Robison et al., 2002; Zito et al., 2000).
Large interindividual differences in pharmacokinetics and pharmacodynamics have been noted with the two major stimulants, methylphenidate and amphetamine (Greenhill et al., 2002; Wilens and Biederman, 1992). Predicting a therapeutic drug response in individual patients with ADHD is not presently possible as no reliable neurological, psychological or physical characteristics predict response to available therapeutic agents (Greenhill et al., 2002). Therapeutic drug monitoring has not proven clinically useful for any ADHD agent. Accordingly, there exists a recognized need for further exploration of sources of interindividual differences in pharmacokinetics of the various ADHD therapeutic agents.
Interindividual differences in drug transporter activity mediated by genetic polymorphisms and drug interactions are important determinants of individual drug response and disposition of drug and metabolites (Beringer and Slaughter, 2005; Bosch et al., 2006). P-glycoprotein is the most well-studied member of the adenosine triphosphate-binding cassette (ABC) transporter superfamily, and has been found in a large number of normal tissues including intestine, liver, placenta, kidney, and the blood–brain barrier. In the blood–brain barrier, P-glycoprotein prohibits substrate drug penetration into the central nervous system (CNS) (Mayer et al., 1997; van Asperen et al., 1997). Changes in P-glycoprotein expression or efflux activity induced by specific drug treatments or the result of genetic polymorphisms can have a significant influence on the disposition of therapeutic agents that are P-glycoprotein substrates (Hoffmeyer et al., 2000). Hence, the role of P-glycoprotein on drug disposition and pharmacodynamics has become an increasing focus of pharmacological research.
In a previous study, we investigated the role of P-glycoprotein in d-methylphenidate and d-amphetamine disposition by utilizing P-glycoprotein knockout mice models (Zhu et al., 2006a,b). After i.p. dosing of each compound, brain and plasma concentrations were determined at 10, 30, and 80 min postdosing timepoints. The results indicated no significant genotypic difference in brain concentrations and brain-to-plasma ratio of d-amphetamine. As for d-methylphenidate, the P-glycoprotein knockout mice had 33.0% higher brain concentrations (p<0.05) and 67.5% higher brain-to-plasma ratios (p<0.01) than wild-type control animals at the 10 min postdosing timepoint, indicating that d-methylphenidate is a relatively weak P-glycoprotein substrate. A recent in vitro study demonstrated that the amphetamine derivative 3,4-methylenedioxymethamphetamine (MDMA) is not the substrate of P-glycoprotein. Furthermore, in that study, amphetamine did not show any significant effect on the P-glycoprotein mediated transport of rhodamine123 in Caco-2 and LS180V cells at the concentrations of 10 and 100 μM suggesting that amphetamine is not a P-glycoprotein inhibitor at physiologically relevant concentrations (Bertelsen et al., 2006).
To fully understand the potential interactions of each of the primary agents employed in the treatment of ADHD with P-glycoprotein, a comprehensive in vitro screening of potential P-glycoprotein inhibitory effects of a variety of ADHD therapeutic agents and their less active isomers was undertaken including the agents d-methylphenidate, l-methylphenidate, d-amphetamine, l-amphetamine, atomoxetine, d-modafinil, and l-modafinil in the present study. Additionally, the P-glycoprotein substrate properties of these compounds were also evaluated through the use of P-glycoprotein transfected cell line and the selective P-glycoprotein inhibitor PSC833. This report, to our knowledge, is the first comprehensive assessment of the interaction of P-glycoprotein with each of these commonly used agents.
2. Materials and methods
2.1. Materials
The verapamil-stimulated vanadate-sensitive ATPase activity kit (Gentest Corporation, Woburn, MA) was used to determine the interaction of methylphenidate isomers with P-glycoprotein. The porcine kidney epithelial cell line LLC-PK1 and its human P-glycoprotein overexpressing mutant LLC-PK1/MDR1 cells were kindly provided by Dr. Kari Kivistö (Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Stuttgart, Germany). Fetal bovine serum, trypsin and Dulbecco’s Modified Eagle Medium (DMEM) containing 4500 mg/L glucose, 4 mM L-glutamine and sodium pyruvate were obtained from Hyclone Co. (Logan, UT). MEM Non-Essential Amino Acid Solution was from Stemcell Technologies Inc. (Vancouver, British Columbia, Canada). Dulbecco’s Phosphate-Buffered Saline (DPBS), penicillin and streptomycin were purchased from Mediatech Inc. (Herndon, VA). d-Amphetamine and l-amphetamine, as their respective sulfate salts, were provided by the National Institute of Drug Abuse (NIDA) Research Resource Drug Supply System; atomoxetine was a generous gift from Eli Lilly & Company (Indianapolis, IN); modafinil isomers, d- and l-modafinil were provided by Cephalon (West Chester, PA). PSC833 (valspodar, 6-[(2S,4R,6E)-4-Methyl-2-(methylamino)-3-oxo-6-octenoic Acid]cyclosporin D) was kindly provided as a gift by Novartis Pharmaceuticals (Switzerland). 4-(4,5-diphenyl-1H-imidazol-2-yl) benzoyl chloride (DIB-Cl) was purchased from Tokyo Kasei Kogyo Co. (Tokyo, Japan). d-methylphenidate, l-methylphenidate, 1-methyl-3-phenylpropylamine, quinine, vincristine, doxorubicin, and rhodamine123 were all obtained from Sigma Co. (St. Louis, MO). All other agents were of high analytical grade and commercially available.
2.2. ATPase assay
The ATPase assay was utilized to screen d- and l-methylphenidate for their affinity for P-glycoprotein by determining the increased inorganic phosphate (Pi) concentrations in the presence of 100 μM of ortho-vanadate as described previously (Boulton et al., 2002). Briefly, d- and l-methylphenidate and positive control verapamil were prepared freshly in Tris–MES buffer (pH 6.8) and added to the reaction mixture containing 40 μg P-glycoprotein membranes and 4 mM Mg-ATP at the final concentrations of 1, 10, 50, 100, 250, 500, 750 and 1000 μM. The reaction was initiated by adding ATP solution to the mixture. After incubation at 37 °C for 40 min, the reactions were terminated by the addition of 30 μl of ice-cold 10% sodium dodecylsulfate solution containing 0.1% Antifoam A. Finally, Pi was assayed by the ultraviolet (UV) absorption of the Pi-molybdate complex at 620 nm and the Pi concentrations were calculated from an eight-point standard curve established from 0–150 nM Pi standard solution. The Km and Vmax values were calculated by fitting Pi versus each compound concentration to the Michaelis–Menten equation.
2.3. Cell cultures
LLC-PK1 cells were cultured at 37 °C in DMEM supplemented with 10% fetal bovine serum, 1% MEM nonessential amino acids, 100 U/ml penicillin and 100 μg/ml streptomycin in an atmosphere of 5% CO2 and 95% relative humidity. LLC-PK1/MDR1 cells were cultured under the same conditions except 640 nM of vincristine was added to the culture medium to maintain P-glycoprotein expression (Schinkel et al., 1995). For intracellular uptake experiments, cells (1 ml) were seeded into 24-well plates at a density of 1 × 105 cells/ml. Culture medium was replaced every 2 days until cells reached confluence. For transport experiments, cells were seeded onto polyester membrane filters (0.4 μm pores, 1.12 cm2 growth area, Corning Inc. Corning, NY) of Transwell inserts at a density of 1 × 105 cells/cm2. Culture medium was likewise refreshed every 2 days. Transepithelial electric resistance (TEER) of cell monolayers was monitored using an EVOMeter® fitted with chopstick electrodes (World Precision Instruments, Sarasota, FL). Monolayers were suitable for transport studies 7 days postseeding when TEER reached 250 Ω cm2.
2.4. Intracellular uptake studies
To investigate the potential influence of these ADHD therapeutic drugs and their respective isomers (d-methylphenidate, l-methylphenidate, d-amphetamine, l-amphetamine, atomoxetine, d-modafinil, and l-modafinil) on P-glycoprotein activity, the intracellular accumulation of two classic P-glycoprotein fluorescent substrates doxorubicin and rhodamine123 was measured in LLC-PK1/MDR1 cells with LLC-PK1 included as a negative control. Briefly, after cells reached confluence, the culture medium was replaced by transport buffer (serum-free DMEM containing 25 mM HEPES, pH 7.4) for a 30 min preincubation at 37 °C. Next, vehicle control (0.5% dimethyl sulfoxide), positive control PSC833 (2 μM), quinine (200 μM), or relevant concentrations of tested compounds were added followed by addition of 10 μM doxorubicin or 5 μM rhodamine123 for an additional 60 min incubation. To investigate the P-glycoprotein substrate properties of ADHD therapeutic agents, 20 μM of each drug was added rather than the known P-glycoprotein substrates doxorubicin and rhodamine123 for a 60 min incubation period in the absence or presence of PSC833 (2 μM). After incubation, the solutions were discarded, and the cells were washed three times with ice-cold DPBS and solubilized with 1% Triton X-100. Cellular retention of tested compounds (doxorubicin, rhodamine123, and each ADHD therapeutic agent) was analyzed by HPLC assay. The concentration of tested compound in each sample was standardized with the protein content determined by a Pierce BCA Protein Kit (Rockford, Illinois). The inhibition rate (%) of tested ADHD agents were calculated using the following formula: (Ct −C0)/(Cp −C0) × 100%, where C0 is the intracellular concentrations of rhodamine123 or doxorubicin in the control group, Ct and Cp are rhodamine123 or doxorubicin concentrations after the treatment of the tested agent and positive P-glycoprotein inhibitor PSC833, respectively. The IC50 values were obtained using nonlinear dose–response fitting when the inhibitory efficiency of 2 μM PSC833 was defined as 100%.
2.5. Transport studies
LLC-PK1/MDR1 cell monolayers cultured on Transwell inserts were utilized to assess the P-glycoprotein substrate properties of d-methylphenidate, d-modafinil, and l-modafinil, which emerged as candidates as P-glycoprotein substrates through the initial intracellular uptake studies described above. Briefly, the confluent cell monolayers were rinsed twice with DPBS and preincubated with transport buffer at 37 °C for 30 min. Next, 0.5% dimethyl sulfoxide (vehicle control) or 2 μM PSC833 was added to both sides of the monolayers, and each tested compound (5 μM) was loaded into the basolateral side for the basolateral to apical (B–A) transport study or apical side for the apical to basolateral (A–B) transport study. At designated times, 100 μl aliquots were removed from the receiver compartment, and immediately replaced by an equal volume of receiver compartment solution. The removed aliquots were analyzed by HPLC. Finally, apparent permeability coefficients, Papp (cm/s) were calculated according to the following equation:
where dQ/dt is the rate at which test compounds appear in the receiver compartment, A is the membrane area of Transwell insert (cm2), and C is the initial concentrations of tested compounds in the donor compartment.
2.6. HPLC analysis
Validated HPLC assays were utilized and developed for the quantification of rhodamine123, doxorubicin and all tested ADHD therapeutic agents. Analysis of rhodamine123, doxorubicin, d- and l-amphetamine, atomoxetine, and d- and l-modafinil was performed as previously described (Donovan et al., 2003; Zhu et al., 2006a,b; Zhu et al., 2007a,b). A novel HPLC-fluorescence detection method for d-methylphenidate and l-methylphenidate quantification was developed for these studies by using DIB-Cl as a fluorescence labeling reagent. Briefly, to a 100 μl sample, 50 μl of the internal standard 1-methyl-3-phenylpropylamine (2 μM) was added and vortexed, followed by the addition of 500 μl of 10 mM sodium carbonate buffer (pH 10.0) and 1.5 ml of butyl chloride: acetonitrile (4: 1). Samples were mixed for 5 min and then centrifuged for 10 min at 2000 ×g. The organic layer was transferred to fresh tubes and evaporated to dryness under nitrogen. The residues were derivatized by adding 10 μl of carbonate buffer (10 mM, pH 9.0) and 100 μl of 1 mM DIB-Cl. The mixture was vortexed briefly and the derivatization proceeded at room temperature for 60 min. The reaction was terminated by the addition of 10 μl of 28% ammonia solution. Lastly, 10 μl of the labeled sample was analyzed by HPLC with fluorescence detection. The HPLC system consisted of a Waters 2690 Separations module (Waters, Milford, MA), a Phenomenex Luna C18 (2) 250 × 4.6 mm, 5 μm reversed-phase column preceded by a 4 × 3 mm C18 guard column (Phenomenex, Torrance, CA) and a Waters 474 scanning fluorescence detector (Waters, Milford, MA). The separation was performed at room temperature using acetonitrile: water (73: 27, v: v) with the flow rate set at 1.0 ml/min. The excitation and emission wavelengths were set at 330 nm (λex), 460 nm (λem). This method was validated with the relative standard deviation of intra and interday precision less than 10% and an extraction recovery of approximately 65%.
2.7. Data analysis
The data are presented as mean±standard deviation (S.D.). The Papp values generated through the transport studies were analyzed by one-way ANOVA with a Tukey post hoc test. A two-tailed unpaired Student’s t test was used for the data analysis of all intracellular accumulation experiments. A P value of <0.05 was considered statistically significant.
3. Results
3.1. Effects of methylphenidate isomers on the P-glycoprotein ATPase activity
All of the tested compounds stimulated P-glycoprotein ATPase in a concentration-dependent manner (Fig. 1). d- and l-methylphenidate showed much weaker stimulative effects on P-glycoprotein ATPase relative to the positive control substrate verapamil, with resultant Vmax/Km values approximately an order of magnitude lower than that of verapamil (Table 1).
Fig. 1.
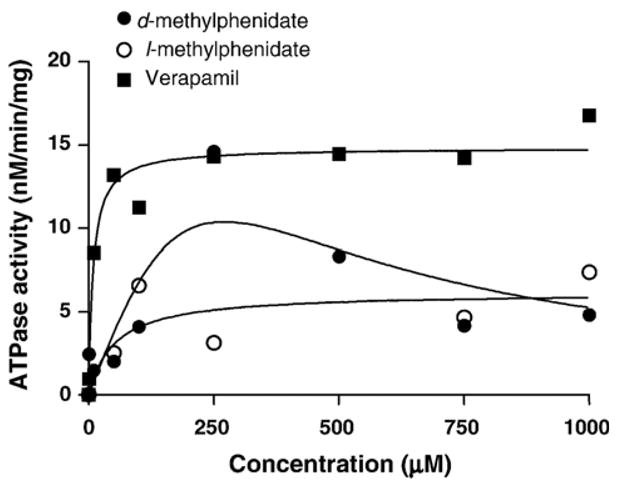
Concentration-dependency of ortho-vanadate sensitive substrate-induced P-glycoprotein ATPase activity as measured by inorganic phosphate release. Data points are expressed as the means of duplicate incubations. Lines represent the nonlinear regression lines of best fit for the Michaelis–Menten equation for d- and l-methylphenidate and positive control verapamil.
Table 1.
The Km, Vmax, and Clint (Vmax/Km) values of d- and l-methylphenidate on ortho-vanadate-sensitive ATPase activity in expressed P-glycoprotein membrane (data were mean of duplicate determinations)
| Km (μM) | Vmax (nmol/mg/min) | Clint(min−1 × 10−3) | |
|---|---|---|---|
| Verapamil | 8.7 | 14.8 | 1.7 |
| d-methylphenidate | 42.8 | 7.8 | 0.18 |
| l-methylphenidate | 52.0 | 6.2 | 0.12 |
3.2. Effect of ADHD therapeutic drugs on the intracellular accumulation of doxorubicin and rhodamine123
The potential inhibitory effects of the tested compounds on P-glycoprotein activity were evaluated by measuring the intracellular accumulation of known P-glycoprotein substrates doxorubicin and rhodamine123. As shown in Fig. 2A and B, all tested agents except d-modafinil and l-modafinil significantly increased the intracellular accumulation doxorubicin in LLC-PK1/MDR1 cells through inhibition of P-glycoprotein in a concentration-dependent manner. Atomoxetine and l-methylphenidate were found to be the most potent inhibitors relative to the other tested agents with IC50 values of 76.87±9.13 μM and 389.11±71.05 μM, respectively. Other compounds did not reach the level of 50% inhibition even at the highest tested concentration of 500 μM. The known P-glycoprotein inhibitors PSC833 (2 μM) and quinine (200 μM) had been used as positive controls and increased the intracellular concentration of doxorubicin in LLC-PK1/MDR1 cells to a level close to those in P-glycoprotein negative parental cells LLC-PK1 indicating that P-glycoprotein function could be abolished by these two inhibitors at the applied concentrations. As expected, both tested agents and positive controls did not show any significant effects on doxorubicin accumulation in LLC-PK1 cells which lack P-glycoprotein (Fig. 2C).
Fig. 2.



Effect of ADHD therapeutic agents on the intracellular accumulation of doxorubicin and rhodamine123 in LLC-PK1/MDR1 and LLC-PK1 cells. Inhibitory effects on P-glycoprotein function in LLC-PK1/MDR1 cells were assessed by measuring the intracellular concentrations of doxorubicin A, B and rhodamine123 D, E after incubation for 1 h in the presence or absence of specific concentrations of each tested drug. PSC833 (2 μM) and quinine (200 μM) were included as positive controls. The inhibitory efficiency of PSC833 was defined as 100%. LLC-PK1 cells were also incorporated as the negative controls C, F. Each data value is the mean with a bar representing S.D., n=3. *P<0.05, **P<0.01 versus control.
In a similar experiment, when doxorubicin was replaced by a second frequently utilized P-glycoprotein fluorescence substrate, rhodamine123, all tested compounds except d, and l-modafinil produced significant inhibitory effects on the P-glycoprotein efflux function in a concentration-dependent manner (Fig. 2D and E). However, their inhibitory potencies were much less than that observed in the doxorubicin uptake assay since none of these tested drugs reached a 50% inhibition even at the highest concentration tested (i.e. 500 μM). In contrast to the doxorubicin uptake studies, quinine (200 μM) did not increase intracellular rhodamine123 concentration in LLC-PK1/MDR1 to a level comparable to that of PSC833 (2 μM). Unexpectedly, atomoxetine at concentrations above 100 μM, d-amphetamine (500 μM), and quinine (200 μM) significantly decreased the intracellular accumulation of rhodamine123 in LLC-PK1 cells (Fig. 2F).
3.3. Evaluation of ADHD therapeutic agents as substrates of P-glycoprotein
P-glycoprotein substrate properties of ADHD therapeutic drugs were assessed by measuring the intracellular drug accumulations in LLC-PK1/MDR1 and LLC-PK1 cells in the presence or absence of the P-glycoprotein selective inhibitor PSC833 (2 μM). The results shown in Fig. 3 indicated that the intracellular concentration of d-methylphenidate in LLC-PK1 cells was 32.0% higher than that in LLC-PK1/MDR1 cells. Additionally, treatment with 2 μM PSC833 resulted in 52.9% and 45.6% increases of d-modafinil and l-modafinil accumulation, respectively, in LLC-PK1/MDR1 cells. Beyond those agents mentioned above, all other tested ADHD drugs displayed a relatively similar pattern of intracellular accumulation in LLC-PK1, LLC-PK1/MDR1, and PSC833 treated cells. These results suggest that P-glycoprotein may play a role in the transport of d-methylphenidate, d-modafinil, and l-modafinil across the cellular membrane.
Fig. 3.

P-glycoprotein substrate properties of ADHD therapeutic agents were assessed by measuring the intracellular accumulation of tested drugs in LLC-PK1 and LLC-PK1/MDR1 cells. Intracellular concentrations of tested agents were measured after incubation for 1 h in the presence or absence of the P-glycoprotein selective inhibitor PSC833 (2 μM) in LLC-PK1 (solid bars) and LLC-PK1/MDR1 (open bars). Data are presented as mean± S.D. (n=3). *P<0.05, **P<0.01 versus control.
The classic P-glycoprotein substrates rhodamine123 and doxorubicin exhibited 495.7% and 426.3% higher intracellular accumulation in LLC-PK1 cells than in LLC-PK1/MDR1 cells. Additionally, coincubation with PSC833 (2 μM) increased the intracellular accumulation of rhodamine123 and doxorubicin in LLC-PK1/MDR1 cells to 568.0% and 526.1% of control, respectively. Overall, when compared to rhodamine123 or doxorubicin, d-methylphenidate, d-modafinil, and l-modafinil could only be considered as relatively weak P-glycoprotein substrates, and the other tested compounds d-amphetamine, l-amphetamine, l-methylphenidate, and atomoxetine appear unlikely to be transported by P-glycoprotein.
3.4. Transport across LLC-PK1/MDR1 monolayers
To further investigate the P-glycoprotein substrate properties of d-methylphenidate, d-modafinil, and l-modafinil, LLC-PK1/MDR1 monolayers cultured on Transwell insert membranes were utilized to measure the transport of these three compounds in both B–A and A–B directions. Typical P-glycoprotein substrates exhibit significant directional transport across LLC-PK1/MDR1 monolayers, i.e. Papp B–A>Papp A–B, which can be attenuated by coincubation of known P-glycoprotein inhibitors. As shown in Table 1, significantly higher transport rates of l-modafinil were found in B–A direction with the ratio of Papp B–A-to-Papp A–B of 1.17. Coincubation of PSC833 (2 μM) significantly decreased the transport of l-modafinil in B–A direction, but did not alter the transport in the A–B direction. These transport data are consistent with that of intracellular accumulation studies described above which taken together, suggest that l-modafinil, at least to some degree, is a P-glycoprotein substrate. In addition, d-modafinil and d-methylphenidate showed similar transport parameters in B–A and A–B directions. Further, PSC833 did not alter d-modafinil and d-methylphenidate transport in either direction. As a comparator, the classic P-glycoprotein substrate rhodamine123 yielded a Papp B–A-to-Papp A–B ratio of 11.11 indicating that P-glycoprotein plays a major role in rhodamine123 transport across LLC-PK1/MDR1 monolayers. PSC833 significantly decreased rhodamine123 transport in B–A direction, but increased transport in the reverse direction resulting in a decreased Papp B–A-to-Papp A–B ratio of 3.68 (Table 2).
Table 2.
The apparent permeability coefficients (Papp) values of ADHD therapeutic drugs (5 μM) across LLC-PK1/MDR1 monolayers
| Substrate | Inhibitor |
Papp(cm s−1 × 10−6) |
Ratio (Papp B–A/Papp A–B) |
|
|---|---|---|---|---|
| B–A | A–B | |||
| d-methylphenidate | Vehicle | 14.29±2.12 | 14.28±4.20 | 1.00 |
| 2 μM PSC833 | 11.00±0.79 | 12.68±1.57 | 0.87 | |
| d-modafinil | Vehicle | 12.49±0.61 | 10.11±1.26 | 1.24 |
| 2 μM PSC833 | 10.39±0.38 | 12.19±1.23 | 0.85 | |
| l-modafinil | Vehicle | 11.97±0.42 | 10.26±0.15c | 1.17 |
| 2 μM PSC833 | 9.95±0.14b | 10.99±0.54 | 0.91 | |
| Rhodamine123 | Vehicle | 6.42±0.53 | 0.58±0.05c | 11.11 |
| 2 μM PSC833 | 3.63±0.67b | 0.99±0.08b, c | 3.68 | |
Results are reported as mean±S.D., n=3.
P<0.01 versus vehicle control,
P<0.01 versus B–A direction.
4. Discussion
In recent years, it has been demonstrated that a number of structurally diverse psychotropic medications including risperidone, its active metabolite 9-hydroxyrisperidone, methadone, olanzapine, and others are significant P-glycoprotein substrates (Uhr et al., 2003; Wang et al., 2004a,b). The entry of these drugs into CNS is significantly limited by P-glycoprotein. Furthermore, some of psychotropic agents were also found to be potent P-glycoprotein inhibitors and may influence the disposition of coadministered drugs which are likewise substrates of P-glycoprotein (Wang et al., 2006; Zhu et al., 2007b).
ADHD therapeutic agents are the most commonly used psychotropic drugs prescribed. Clinical experience has demonstrated that at least 30% of patients are either nonresponders or are intolerant to a major psychostimulant medication (methylphenidate or amphetamine) (Greenhill et al., 1999; Patrick and Markowitz, 1998). Additionally, up to 70% of children and adolescents with ADHD also have a comorbid disorder such as conduct disorder, or anxiety (Spencer et al., 1999). These situations often lead to the use of various drugs in combination. A growing trend towards combination pharmacotherapy in all of child, adolescent and adult psychiatric practice has been well documented over the past decade (Connor et al., 1997; Safer et al., 2003; Wilens et al., 1995). Thus, potential P-glycoprotein mediated drug interactions occurring with combined pharmacotherapy on an acute or chronic basis could have therapeutic implications for patients receiving medications for ADHD (Markowitz and Patrick, 2001).
In the present investigations, our initial study utilizing an ATPase screening assay demonstrated that both d- and l-methylphenidate have weak affinity for P-glycoprotein. Further experiments were conducted to investigate the interaction of P-glycoprotein with each of the primary ADHD therapeutic agents and their respective isomers utilizing the P-glycoprotein overexpressing cell line LLC-PK1/MDR1. Our data indicate that all tested agents with the exception of d- and l-modafinil increased the intracellular accumulation of doxorubicin in LLC-PK1/MDR1 cells at relatively high concentrations in a concentration-dependent manner. In comparison, no tested compound significantly influenced the doxorubicin accumulation in P-glycoprotein negative LLC-PK1 cells indicating that the increased doxorubicin accumulation in LLC-PK1/MDR1 cells was due to the inhibition of P-glycoprotein. Studies were extended to a second recognized P-glycoprotein fluorescent substrate, rhodamine123, and demonstrated that all the tested compounds, again with the exception of the two modafinil isomers, increased intracellular rhodamine123 accumulation in LLC-PK1/MDR1 cells in a concentration-dependent manner. Among these, d-amphetamine and l-amphetamine failed to influence the uptake of rhodamine123 in LLC-PH1/MDR cells at or under the concentration of 100 μM, which is consistent with the recent published study that indicates that amphetamine is not a P-glycoprotein inhibitor at physiologically relevant conditions (Bertelsen et al., 2006). Unexpectedly, atomoxetine at concentrations above 100 μM, d-amphetamine (500 μM), and quinine (200 μM) significantly decreased the intracellular accumulation of rhodamine123 in LLC-PK1 cells. Due to its intense fluorescence, rhodamine123 has been widely used as a substrate for evaluation of P-glycoprotein function. However, it should be noted that, in addition to P-glycoprotein, other transporters (e.g. mutant BCRP and OCTs) have also been reported to transport rhodamine123 across the cellular membrane (Alqawi et al., 2004; Masereeuw et al., 1997; van der Sandt et al., 2000). van der Sandt and coworkers (2000) reported that rhodamine123, but not doxorubicin, could be transported by OCTs expressed in LLC-PK1 cells. Furthermore, quinine is a dual P-glycoprotein and OCT inhibitor, and is widely incorporated as a positive control for OCT inhibition studies. In view of these overlapping activities, we suspect that the decreased intracellular accumulation of rhodamine123 observed in LLC-PK1 cells following the treatments with atomoxetine, d-amphetamine, and quinine could be potentially explained by the inhibition of OCT mediated influx transport. To further investigate the potential interactions of these compounds with OCTs, we are undertaking studies of the role of OCTs in the translocation of d-amphetamine across the cellular membrane. The preliminary results indicated that the typical OCTs inhibitors quinine, verapamil, desipramine, and prazosin significantly decreases the intracellular concentration of d-amphetamine, ostensibly through inhibition of one or more OCTs present in LLC-PK1, MDCK, and Caco-2 cell lines (unpublished observation). These data are consistent with that of our presently described rhodamine123 accumulation studies suggesting that atomoxetine and d-amphetamine may exhibit binding affinity to OCTs. It should be noted that the mitochondria is the major organelle wherein rhodamine123 is enriched in cells (Johnson et al., 1980). Thus, the possibility cannot be excluded that the observed decrease in rhodamine123 accumulation in LLC-PK1 cells could be caused by decreased mitochondrial activity induced by the tested ADHD agents.
It is well recognized that beyond the major role of P-glycoprotein in tumor multidrug resistance, this transporter is also an important determinate of drug absorption after oral dosing as well as drug distribution into the brain depending on substrate specificity. A number of agents have been developed in an effort to modulate transporter activity including the potent and selective P-glycoprotein inhibitors valspodar (PSC833), elacridar (GF120918), zosuquidar (LY335979), and tariquidar (XR9576). These agents are intended to reverse multidrug resistance and/or improve oral bioavailability and CNS penetration (Breedveld et al., 2006; Thomas and Coley, 2003). In addition to these specific compounds targeting P-glycoprotein, a number of therapeutic agents such as the calcium channel blocker verapamil and the immunosuppressant cyclosporine A have been found to significantly inhibit P-glycoprotein activity in vivo within normal dosing ranges (Nakagami et al., 2005; Schmitt et al., 2006). The ultimate clinical relevance of P-glycoprotein inhibition is related to the inhibitor’s potency and its concentration in pertinent P-glycoprotein expressing tissues. After a single 10 mg oral dose of racemic methylphenidate, methylphenidate is rapidly absorbed and typically attains a maximum plasma concentration (Cmax) of approximately 4.0 ng/ml (0.017 μM) between 2 and 5 h after dosing. Due to enantioselective metabolism, only a minor amount of l-methylphenidate is detected in blood with d-methylphenidate being the major circulating species of methylphenidate (Markowitz et al., 2003). The Cmax values of d-amphetamine after 0.25 or 0.5 mg/kg doses are approximately 40 and 70 ng/ml (0.30 and 0.52 μM), respectively (Angrist et al., 1987). It has been demonstrated in the present study that both methylphenidate and amphetamine isomers display P-glycoprotein inhibitory effects only at or above the concentrations of 50 to 100 μM. Therefore, it is not likely that methylphenidate and amphetamine could influence the pharmacokinetics of coadministered P-glycoprotein substrates by inhibiting P-glycoprotein activity in vivo even if these two agents could reach relatively high concentrations in some tissues with local accumulation. Atomoxetine is well absorbed in gastrointestinal tract and predominantly metabolized by cytochrome P4502D6 (CYP2D6). After oral administration of 20 mg twice daily in adults, the Cmax have been observed in the range of 160–180 ng/ml (0.55–0.62 μM) in CYP2D6 extensive metabolizers and up to 925 ng/ml (3.17 μM) in genetically deficient CYP2D6 metabolizers (Sauer et al., 2003). In our in vitro studies, atomoxetine proved to be the most potent P-glycoprotein inhibitor of all the tested agents potentially utilized to treat ADHD. The minimum concentration at which atomoxetine significantly increased intracellular doxorubicin accumulation in LLC-PK1/MDR1 cells is 10 μM. Therefore, the potential interaction of atomoxetine at high concentrations with coadministered P-glycoprotein substrate drugs cannot be excluded.
The P-glycoprotein substrate properties of ADHD therapeutic agents were evaluated by determining the effect of P-glycoprotein on drug intracellular accumulation and transport across cell monolayers. The results showed that the intracellular accumulation of d-methylphenidate in LLC-PK1 cells was 32.0% higher than in LLC-PK1/MDR1 cells. Since P-glycoprotein expression is generally considered as the only functional difference between LLC-PK1 and LLC-PK1/MDR1 cells, the data suggest that P-glycoprotein plays a role in the transport of d-methylphenidate across the cellular membrane. Further transport studies did not reveal any statistical differences in d-methylphenidate transport between in B–A and in A–B directions across LLC-PK1/MDR1 cell monolayers. Also, the potent P-glycoprotein inhibitor PSC833 did not change d-methylphenidate transport rates in both B–A and A–B directions. In totality, these data suggest that P-glycoprotein only plays a minor role (if any) in the disposition of d-methylphenidate, a finding consistent with our previous investigation utilizing P-glycoprotein knockout mice (Zhu et al., 2006a,b). With regard to modafinil, treatment with 2 μM of PSC833 resulted in 52.9% and 45.6% increases of d-modafinil and l-modafinil accumulation in LLC-PK1/MDR1 cells but not in P-glycoprotein negative LLC-PK1 cells. Furthermore, a significantly higher transport rate of l-modafinil, but not d-modafinil was found in the B–A direction with the ratio of Papp B–A-to-Papp A–B of 1.17. Coincubation of PSC833 significantly decreased the transport of l-modafinil in the B–A direction. These results suggest that, similar to the findings with d-methylphenidate, modafinil isomers are also likely to be minimally transported by P-glycoprotein. The known P-glycoprotein substrates rhodamine123 and doxorubicin were incorporated as positive controls in the present study. After the treatment with PSC833 (2 μM), the intracellular accumulation of rhodamine123 and doxorubicin in LLC-PK1/MDR1 cells were increased to the levels similar to that in the P-glycoprotein negative LLC-PK1 cells indicating that the P-glycoprotein efflux function could be completely inhibited by 2 μM PSC833. Interestingly, in the LLC-PK1/MDR1 cell monolayers transport studies, PSC833 also dramatically decreased the Papp B–A-to-Papp A–B ratio of rhodamine123 across LLC-PK1/MDR1 cell monolayers from 11.11 to 3.68, but failed to abolish the directional transport of rhodamine123 i.e. did not decrease the Papp B–A-to-Papp A–B ratio to around 1.00. Our current data are not sufficient to provide a firm explanation of this phenomenon, but we speculate that it might be caused by the participation of other transporters in rhodamine123 transport since rhodamine123 has been reported to be also the substrate of other transporters such as OCTs (Masereeuw et al., 1997; van der Sandt et al., 2000).
The present study represents a comprehensive in vitro assessment of the potential interactions of the primary agents employed in the treatment of ADHD with P-glycoprotein. Our data suggest that all of the assessed agents, except d- and l-modafinil, are relatively weak P-glycoprotein inhibitors. Given their inhibitory potency and likely in vivo concentrations following typical therapeutic dosing, all tested compounds, with the possible exception of atomoxetine, are unlikely to interfere with the pharmacokinetics of other P-glycoprotein substrates via P-glycoprotein inhibition during clinical use. Finally, d-methylphenidate, d-modafinil, and l-modafinil were found to be relatively weak P-glycoprotein substrates. Although the results of the present study were generally negative with respect to interactions of ADHD therapeutic agents and P-glycoprotein, two important issues were addressed. First, the in vitro methods for assessing P-glycoprotein substrate specificity were in good agreement with our previous in vivo data generated in the transgenic mouse in which we assessed the two most commonly prescribed agents, d-methylphenidate and d-amphetamine (Zhu et al., 2006a,b). This further validates the predictive value of cell culture models in evaluating potential P-glycoprotein substrate properties of other AHDH therapeutic agents. Secondly, the overall broad survey of the potential interactions of ADHD agents with P-glycoprotein should provide clinicians some degree of comfort when prescribing these agents to patients as monotherapy or in combination with other agents since the results suggest P-glycoprotein mediated effects on drug disposition are unlikely.
Acknowledgments
This work was supported in part by NICHD Grant R21HD 047810-01.
Footnotes
Publisher's Disclaimer: This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author’s institution, sharing with colleagues and providing to institution administration.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases the authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit:
References
- Alqawi O, Bates S, Georges E. Arginine482 to threonine mutation in the breast cancer resistance protein ABCG2 inhibits rhodamine 123 transport while increasing binding. Biochem J. 2004;382:711–716. doi: 10.1042/BJ20040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angrist B, Corwin J, Bartlik B, Cooper T. Early pharmacokinetics and clinical effects of oral D-amphetamine in normal subjects. Biol Psychiatry. 1987;22:1357–1368. doi: 10.1016/0006-3223(87)90070-9. [DOI] [PubMed] [Google Scholar]
- Beringer PM, Slaughter RL. Transporters and their impact on drug disposition. Ann Pharmacother. 2005;39:1097–1108. doi: 10.1345/aph.1E614. [DOI] [PubMed] [Google Scholar]
- Bertelsen KM, Greenblatt DJ, von Moltke LL. Apparent active transport of MDMA is not mediated by P-glycoprotein: a comparison with MDCK and Caco-2 monolayers. Biopharm Drug Dispos. 2006;27:219–227. doi: 10.1002/bdd.501. [DOI] [PubMed] [Google Scholar]
- Bosch TM, Meijerman I, Beijnen JH, Schellens JH. Genetic polymorphisms of drug-metabolising enzymes and drug transporters in the chemotherapeutic treatment of cancer. Clin Pharmacokinet. 2006;45:253–285. doi: 10.2165/00003088-200645030-00003. [DOI] [PubMed] [Google Scholar]
- Boulton DW, DeVane CL, Liston HL, Markowitz JS. In vitro P-glycoprotein affinity for atypical and conventional antipsychotics. Life Sci. 2002;71:163–169. doi: 10.1016/s0024-3205(02)01680-6. [DOI] [PubMed] [Google Scholar]
- Breedveld P, Beijnen JH, Schellens JH. Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugs. Trends Pharmacol Sci. 2006;27:17–24. doi: 10.1016/j.tips.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Connor DF, Ozbayrak KR, Kusiak KA, Caponi AB, Melloni RH., Jr Combined pharmacotherapy in children and adolescents in a residential treatment center. J Am Acad Child Adolesc Psych. 1997;36:248–254. doi: 10.1097/00004583-199702000-00016. [DOI] [PubMed] [Google Scholar]
- Donovan JL, Malcolm RJ, Markowitz JS, DeVane CL. Chiral analysis of d- and l-modafinil in human serum: application to human pharmacokinetic studies. Ther Drug Monit. 2003;25:197–202. doi: 10.1097/00007691-200304000-00009. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Biederman J, Roe C. Comparative efficacy of Adderall and methylphenidate in attention-deficit/hyperactivity disorder: a meta-analysis. J Clin Psychopharmacol. 2002;22:468–473. doi: 10.1097/00004714-200210000-00005. [DOI] [PubMed] [Google Scholar]
- Greenhill LL, Halperin JM, Abikoff H. Stimulant medications. J Am Acad Child Adolesc Psych. 1999;38:503–512. doi: 10.1097/00004583-199905000-00011. [DOI] [PubMed] [Google Scholar]
- Greenhill LL, Pliszka S, Dulcan MK. Practice parameters for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psych. 2002;41(Suppl):26S–49S. doi: 10.1097/00004583-200202001-00003. [DOI] [PubMed] [Google Scholar]
- Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmoller J, Johne A, Cascorbi I, Gerloff T, Roots I, Eichelbaum M, Brinkmann U. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A. 2000;97:3473–3478. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James RS, Sharp WS, Bastain TM, Lee PP, Walter JM, Czarnolewski M, Castellanos FX. Double-blind, placebo-controlled study of single-dose amphetamine formulations in ADHD. J Am Acad Child Adolesc Psych. 2001;40:1268–1276. doi: 10.1097/00004583-200111000-00006. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Walsh ML, Chen LB. Localization of mitochondria in living cells with rhodamine 123. Proc Natl Acad Sci U S A. 1980;77:990–994. doi: 10.1073/pnas.77.2.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochvil CJ, Heiligenstein JH, Dittmann R, Spencer TJ, Biederman J, Wernicke J, Newcorn JH, Casat C, Milton D, Michelson D. Atomoxetine and methylphenidate treatment in children with ADHD: a prospective, randomized, open-label trial. J Am Acad Child Adolesc Psych. 2002;41:776–784. doi: 10.1097/00004583-200207000-00008. [DOI] [PubMed] [Google Scholar]
- Leonard BE, McCartan D, White J, King DJ. Methylphenidate: a review of its neuropharmacological, neuropsychological and adverse clinical effects. Hum Psychopharmacol. 2004;19:151–180. doi: 10.1002/hup.579. [DOI] [PubMed] [Google Scholar]
- Markowitz JS, Patrick KS. Pharmacokinetic and pharmacodynamic drug interactions in the treatment of attention-deficit hyperactivity disorder. Clin Pharmacokinet. 2001;40:753–772. doi: 10.2165/00003088-200140100-00004. [DOI] [PubMed] [Google Scholar]
- Markowitz JS, Straughn AB, Patrick KS. Advances in the pharmacotherapy of attention-deficit-hyperactivity disorder: focus on methylphenidate formulations. Pharmacotherapy. 2003;23:1281–1299. doi: 10.1592/phco.23.12.1281.32697. [DOI] [PubMed] [Google Scholar]
- Masereeuw R, Moons MM, Russel FG. Rhodamine 123 accumulates extensively in the isolated perfused rat kidney and is secreted by the organic cation system. Eur J Pharmacol. 1997;321:315–323. doi: 10.1016/s0014-2999(96)00957-0. [DOI] [PubMed] [Google Scholar]
- Mayer U, Wagenaar E, Dorobek B, Beijnen JH, Borst P, Schinkel AH. Full blockade of intestinal P-glycoprotein and extensive inhibition of blood–brain barrier P-glycoprotein by oral treatment of mice with PSC833. J Clin Invest. 1997;100:2430–2436. doi: 10.1172/JCI119784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagami T, Yasui-Furukori N, Saito M, Tateishi T, Kaneo S. Effect of verapamil on pharmacokinetics and pharmacodynamics of risperidone: in vivo evidence of involvement of P-glycoprotein in risperidone disposition. Clin Pharmacol Ther. 2005;78:43–51. doi: 10.1016/j.clpt.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Patrick KS, Markowitz JS. Pharmacology of methylphenidate, amphetamine enatiomers and pemoline in attention-deficit hyperactivity disorder. Hum Psychopharmacol. 1998;12:527–546. [Google Scholar]
- Pliszka SR, Greenhill LL, Crismon ML, Sedillo A, Carlson C, Conners CK, McCracken JT, Swanson JM, Hughes CW, Llana ME, Lopez M, Toprac MG. The Texas Children’s Medication Algorithm Project: Report of the Texas Consensus Conference Panel on Medication Treatment of Childhood Attention-Deficit/Hyperactivity Disorder. Part I Attention-Deficit/Hyperactivity Disorder. J Am Acad Child Adolesc Psych. 2000;39:908–919. doi: 10.1097/00004583-200007000-00021. [DOI] [PubMed] [Google Scholar]
- Robison LM, Skaer TL, Sclar DA, Galin RS. Is attention deficit hyperactivity disorder increasing among girls in the US? Trends in diagnosis and the prescribing of stimulants CNS. Drugs. 2002;16:129–137. doi: 10.2165/00023210-200216020-00005. [DOI] [PubMed] [Google Scholar]
- Safer DJ, Zito JM, DosReis S. Concomitant psychotropic medication for youths. Am J Psych. 2003;160:438–449. doi: 10.1176/appi.ajp.160.3.438. [DOI] [PubMed] [Google Scholar]
- Sauer JM, Ponsler GD, Mattiuz EL, Long AJ, Witcher JW, Thomasson HR, Desante KA. Disposition and metabolic fate of atomoxetine hydrochloride: the role of CYP2D6 in human disposition and metabolism. Drug Metab Dispos. 2003;31:98–107. doi: 10.1124/dmd.31.1.98. [DOI] [PubMed] [Google Scholar]
- Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest. 1995;96:1698–1705. doi: 10.1172/JCI118214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt U, Abou El-Ela A, Guo LJ, Glavinas H, Krajcsi P, Baron JM, Tillmann C, Hiemke C, Langguth P, Hartter S. Cyclosporine A (CsA) affects the pharmacodynamics and pharmacokinetics of the atypical antipsychotic amisulpride probably via inhibition of P-glycoprotein (P-gp) J Neural Transm. 2006;113:787–801. doi: 10.1007/s00702-005-0367-4. [DOI] [PubMed] [Google Scholar]
- Spencer T, Biederman J, Wilens T. Attention-deficit/hyperactivity disorder and comorbidity. Pediatr Clin North Am. 1999;46:915–927. vii. doi: 10.1016/s0031-3955(05)70163-2. [DOI] [PubMed] [Google Scholar]
- Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10:159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- Uhr M, Grauer MT, Holsboer F. Differential enhancement of antidepressant penetration into the brain in mice with abcb1ab (mdr1ab) P-glycoprotein gene disruption. Biol Psychiatry. 2003;54:840–846. doi: 10.1016/s0006-3223(03)00074-x. [DOI] [PubMed] [Google Scholar]
- van Asperen J, Mayer U, van Tellingen O, Beijnen JH. The functional role of P-glycoprotein in the blood–brain barrier. J Pharm Sci. 1997;86:881–884. doi: 10.1021/js9701364. [DOI] [PubMed] [Google Scholar]
- van der Sandt IC, Blom-Roosemalen MC, de Boer AG, Breimer DD. Specificity of doxorubicin versus rhodamine-123 in assessing P-glycoprotein functionality in the LLC-PK1, LLC-PK1:MDR1 and Caco-2 cell lines. Eur J Pharm Sci. 2000;11:207–214. doi: 10.1016/s0928-0987(00)00097-x. [DOI] [PubMed] [Google Scholar]
- Wang JS, Ruan Y, Taylor RM, Donovan JL, Markowitz JS, DeVane CL. Brain penetration of methadone (R)- and (S)-enantiomers is greatly increased by P-glycoprotein deficiency in the blood–brain barrier of Abcb1a gene knockout mice. Psychopharmacology (Berl) 2004a;173:132–138. doi: 10.1007/s00213-003-1718-1. [DOI] [PubMed] [Google Scholar]
- Wang JS, Taylor R, Ruan Y, Donovan JL, Markowitz JS, Lindsay De Vane C. Olanzapine penetration into brain is greater in transgenic Abcb1a P-glycoprotein-deficient mice than FVB1 (wild-type) animals. Neuropsychopharmacology. 2004b;29:551–557. doi: 10.1038/sj.npp.1300372. [DOI] [PubMed] [Google Scholar]
- Wang JS, Zhu HJ, Markowitz JS, Donovan JL, Devane CL. Evaluation of antipsychotic drugs as inhibitors of multidrug resistance transporter P-glycoprotein. Psychopharmacology (Berl) 2006;187:415–423. doi: 10.1007/s00213-006-0437-9. [DOI] [PubMed] [Google Scholar]
- Weiss M, Murray C. Assessment and management of attention-deficit hyperactivity disorder in adults. CMAJ. 2003;168:715–722. [PMC free article] [PubMed] [Google Scholar]
- Wilens TE, Biederman J. The stimulants. Psychiatr Clin North Am. 1992;15:191–222. [PubMed] [Google Scholar]
- Wilens TE, Spencer T, Biederman J, Wozniak J, Connor D. Combined pharmacotherapy: an emerging trend in pediatric psychopharmacology. J Am Acad Child Adolesc Psych. 1995;34:110–112. doi: 10.1097/00004583-199501000-00021. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Wang JS, DeVane CL, Williard RL, Donovan JL, Middaugh LD, Gibson BB, Patrick KS, Markowitz JS. The role of the polymorphic efflux transporter P-glycoprotein on the brain accumulation of d-methylphenidate and d-amphetamine. Drug Metab Dispos. 2006a;34:1116–1121. doi: 10.1124/dmd.106.009605. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Wang JS, Donovan JL, DeVane CL, Gibson BB, Markowitz JS. Sensitive quantification of atomoxetine in human plasma by HPLC with fluorescence detection using 4-(4,5-diphenyl-1H-imidazole-2-yl) benzoyl chloride derivatization. J Chromatogr, B Analyt Technol Biomed Life Sci. 2007a;846:351–354. doi: 10.1016/j.jchromb.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Wang JS, Markowitz JS, Donovan JL, Gibson BB, DeVane CL. Risperidone and paliperidone inhibit p-glycoprotein activity in vitro. Neuropsychopharmacology. 2007b;32:757–764. doi: 10.1038/sj.npp.1301181. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Wang JS, Markowitz JS, Donovan JL, Gibson BB, Gefroh HA, Devane CL. Characterization of P-glycoprotein inhibition by major cannabinoids from marijuana. J Pharmacol Exp Ther. 2006b;317:850–857. doi: 10.1124/jpet.105.098541. [DOI] [PubMed] [Google Scholar]
- Zito JM, Safer DJ, dosReis S, Gardner JF, Boles M, Lynch F. Trends in the prescribing of psychotropic medications to preschoolers. JAMA. 2000;283:1025–1030. doi: 10.1001/jama.283.8.1025. [DOI] [PubMed] [Google Scholar]