

Abstract
Prostate cancer often relapses during androgen‐depletion therapy, even under conditions in which a drastic reduction of circulating androgens is observed. There is some evidence that androgens remain present in the tissues of hormone‐refractory prostate cancers (HRPC), and enzymes involved in the androgen and steroid metabolic pathway are likely to be active in HRPC cells. We previously carried out a genome‐wide gene expression profile analysis of clinical HRPC cells by means of cDNA microarrays in combination with microdissection of cancer cells and found dozens of transactivated genes. Among them, we here report the identification of a novel gene, SRD5A2L, encoding a putative 5α‐steroid reductase that produces the most potent androgen, 5α‐dihydrotestosterone (DHT), from testosterone. Liquid chromatography‐tandem mass spectrometry analysis following an in vitro 5α‐steroid reductase reaction validated its ability to produce DHT from testosterone, similar to type 1 5α‐steroid reductase. Because two types of 5α‐steroid reductase were previously reported, we termed this novel 5α‐steroid reductase ‘type 3 5α‐steroid reductase’ (SRD5A3). Reverse transcription–polymerase chain reaction and northern blot analyses confirmed its overexpression in HRPC cells, and indicated no or little expression in normal adult organs. Knockdown of SRD5A3 expression by small interfering RNA in prostate cancer cells resulted in a significant decrease in DHT production and a drastic reduction in cell viability. These findings indicate that a novel type 3 5α‐steroid reductase, SRD5A3, is associated with DHT production and maintenance of androgen–androgen receptor‐pathway activation in HRPC cells, and that this enzymatic activity should be a promising molecular target for prostate cancer therapy. (Cancer Sci 2008; 99: 81–86)
Prostate cancer (PC) is the most common malignancy in men and the second‐leading cause of cancer‐related death in the USA and Europe.( 1 ) Screening using serum prostate‐specific antigen has lead to a dramatic improvement in the early detection of PC and has resulted in an increase in the proportion of patients with a localized disease that could be curable by surgical or radiation therapies.( 1 , 2 ) However, 20–30% of PC patients suffer from relapsed disease, and androgen‐ablation therapy is usually used in this situation.( 3 , 4 ) The androgen–androgen receptor (AR) signaling pathway plays a central role in PC development and progression, and PC growth is usually androgen‐dependent. Hence, most patients with relapsed or advanced disease respond well to androgen‐ablation therapy, which suppresses testicular androgen production by medical or surgical castration. However, they eventually acquire a more aggressive and androgen‐independent phenotype that has been termed hormone‐refractory prostate cancer (HRPC). Recently, docetaxel has been established as a new standard therapy for HRPC patients,( 5 , 6 ) but its survival benefit is very limited. Hence, many groups are now attempting various approaches to identify novel molecular targets or signaling pathways that contribute to the growth of HRPC.( 7 )
The main circulating androgen is testosterone, which is produced in the testis under the control of luteinizing hormone. In several androgen target tissues such as the prostate, testosterone is converted to 5α‐dihydrotestosterone (DHT), which is the most potent natural androgen.( 8 ) Androgen‐ablation therapy can lead to a drastic reduction in the serum testosterone level (approximately 5%), but the intraprostatic concentration of DHT remains at ~40% in HRPC,( 9 , 10 , 11 ) indicating that HRPC cells preserve their level of DHT to maintain their AR signaling pathways and survive under the androgen‐depleted conditions. The conversion from testosterone to DHT is catalyzed by 5α‐steroid reductase enzymes, and two types of human 5α‐steroid reductase enzymes have been reported so far (types 1 and 2). Type 2 5α‐steroid reductase (SRD5A2) is found predominantly in androgen target organs such as the prostate, genital skin, and seminal vesicles, whereas type 1 5α‐steroid reductase (SRD5A1) is expressed predominantly in the skin, scalp, sebaceous gland, liver, and brain.( 8 , 12 ) In the normal prostate and benign prostate diseases, the type 2 isozyme is expressed dominantly. In contrast, the type 1 isozyme was shown to be upregulated in prostate cancers.( 13 ) Although there is some structural homology, the kinetic parameters are markedly different between the type 1 and 2 isozymes; the optimal pH is rather broad (pH 6–8.5) for type 1, whereas that for type 2 is narrow (pH 5.0–5.5) and acidic. Under optimal conditions, the type 2 isozyme has a higher Vmax/Km and higher activity as a 5α‐steroid reductase than the type 1 isozyme.( 8 )
In the present study, on the basis of a genome‐wide gene expression profile analysis of HRPC cells,( 14 ) we identified a novel 5α‐steroid reductase that was overexpressed specifically in HRPC cells, termed type 3 5α‐steroid reductase (SRD5A3). We demonstrated its activity to produce DHT from testosterone in vitro, and also its important roles in prostate cancer cell growth by small interfering RNA (siRNA) analysis. These findings implicate the novel 5α‐steroid reductase SRD5A3 to be associated with PC development and HRPC progression and to be a promising target for the development of molecular therapies for prostate cancers.
Materials and Methods
Cell lines. COS7 cells and the prostate cancer cell lines LNCaP, 22Rv1, PC‐3, and DU‐145 were purchased from the American Type Culture Collection (Rockville, MD, USA). The LNCaP‐derived HRPC cell line C4‐2B was purchased from ViroMed Laboratories (Minnetonka, MN, USA). All of the cell lines except PC‐3 were grown in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA), and PC‐3 was grown in F‐12 (Invitrogen); these media were supplemented with 10% fetal bovine serum (Gemini Bio‐Products, West Sacramento, CA, USA) and 1% antibiotic and antimycotic solution (Sigma‐Aldrich, St Louis, MO, USA). The cells were maintained at 37°C in an atmosphere of humidified air with 5% CO2.
Semiquantitative reverse transcription–polymerase chain reaction. Purification of HRPC cells, hormone‐naïve prostate cancer (HNPC) cells, and normal prostatic epithelial (NP) cells by laser microbeam microdissection, RNA amplification, and cDNA generation were described previously.( 14 ) The primer sequences we used were forward 5′‐TTGGCTTGACTCAGGATTTA‐3′ and reverse 5′‐ATGCTATCACCTCCCCTGTG‐3′ for β‐actin (ACTB), forward 5′‐CCTGTTTGTTCTTTGTTGATTGAA‐3′ and reverse 5′‐CCAGATGAGATGATAAGGCAAAG‐3′ for SRD5A1, forward 5′‐ATCTGAACATACAGAGCCCACAT‐3′ and reverse 5′‐ATCCTCAGACCTTTCAAGTTTCC‐3′ for SRD5A2, forward 5′‐TTTAATCAGGCCCTGTCTGC‐3′ and reverse 5′‐GGGGTATAGAAATGGAATGGAGA‐3′ for SRD5A3 (GenBank accession no. NM_024592). Each polymerase chain reaction (PCR) regime involved a 98°C, 30‐s initial denaturation step followed by 22 cycles (for ACTB), 30 cycles (for SRD5A1), 25 cycles (for SRD5A2), or 26 cycles (for SRD5A3) at 98°C for 10 s, 55°C for 5 s, and 72°C for 30 s, on a Gene Amp PCR system 9600 (PE Applied Biosystems, Foster, CA, USA).
Northern blot analysis. We extracted total RNA from five PC cell lines using the RNeasy Kit (Qiagen) and carried out northern blot analysis. The mRNA was purified using the mRNA Purification Kit (GE Healthcare, Piscataway, NJ, USA), according to the manufacturer's protocols. A 1‐µg aliquot of each mRNA from the PC cell lines, as well as those isolated from normal human heart, lung, liver, kidney, brain, and prostate (BD Biosciences, Palo Alto, CA, USA), were separated on 1% denaturing agarose gels and transferred onto nylon membranes. The 804‐bp probe specific to SRD5A3 was prepared by PCR using the following primer set: forward 5′‐TACTCAATCTCTGTTCCTGGGAG‐3′ and reverse 5′‐GGGGTATAGAAATGGAATGGAGA‐3′. Hybridization with a random‐primed, α32P‐dCTP‐labeled probe was carried out according to the instructions for the Megaprime DNA labeling system (GE Healthcare). Prehybridization, hybridization, and washing were carried out according to the supplier's recommendations. The blots were autoradiographed with intensifying screens at –80°C for 7 days.
Small interfering RNA‐expressing constructs and transfection. To knockdown endogenous SRD5A3 expression in PC cells, we used the psiU6BX3.0 vector for expression of short hairpin RNA against a target gene as described previously.( 14 ) The target sequences of the synthetic oligonucleotides for SRD5A3 siRNA were as follows: si#524, 5′‐GACTCTTCGAGTGCCTCTA‐3′; si#790, 5′‐GCAGGAGTGGTCATTCACT‐3′; negative control scramble siRNA (si#SC), 5′‐GCGCGCTTTGTAGGATTCG‐3′; and si#EGFP, 5′‐GAAGCAGCACGACTTCTTC‐3′. The PC cell lines 22Rv1 and C4‐2B, which expressed SRD5A3, were plated onto 10‐cm dishes or six‐well plates, and transfected with plasmid designed to express siRNA to SRD5A3 (8 µg or 2 µg, respectively) using FuGENE6 (Roche, Basel, Switzerland) according to manufacturer's instructions. Cells were selected using 0.8 mg/mL geneticin (Sigma‐Aldrich) for 7 days, and then harvested to analyze the knockdown effect on SRD5A3 expression. Reverse transcription (RT)‐PCR to confirm SRD5A3 knockdown was carried out using the following primer set: forward 5′‐TACTCAATCTCTGTTCCTGGGAG‐3′ and reverse 5′‐GGGGTATAGAAATGGAATGGAGA‐3′. For the colony formation assay, transfectants expressing short hairpin RNA were grown for 20 days in media containing 0.8 mg/mL geneticin. After fixation with 100% methanol, transfected cells were stained with 0.1% crystal violet–H2O to assess colony formation. Using an 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay, cell viability was measured with Cell‐Counting Kit‐8 (Dojindo, Kumamoto, Japan) at 22 days after the transfection. Absorbance at 490 nm and at 630 nm as a reference was measured with a Microplate Reader 550 (Bio‐Rad, Hercules, CA, USA).
Inhibition of SRD5A3 activity. To inhibit the SRD5A3 expression in PC cells more efficiently, we synthesized an RNA duplex corresponding to the target sequence described above as si#790 (5′‐GCAGGAGUGGUCAUUCACU‐3′), as well as control RNA duplex for si#EGFP (5′‐GAAGCAGCACGACUUCUUC‐3′). Cells seeded onto 10‐cm dishes were incubated with si#790 or control si#EGFP RNA duplex in lipofectamine2000 (Invitrogen) at a final concentration of 100 nM. At 4 h after transfection, the siRNA–lipofectamine2000 mixtures were replaced with fresh medium. After an additional 48‐h incubation, the knockdown effect on SRD5A1, SRD5A2, and SRD5A3 was evaluated by RT‐PCR using the primers described above, and the cells and their conditioned media were collected to measure their DHT and testosterone levels by liquid chromatography‐tandem mass spectrometry (LC‐MS/MS).
Construction of expression vectors. To construct the SRD5A1 and SRD5A3 expression vectors, the entire coding sequences of the SRD5A1 (GenBank accession no. NM_001047) and SRD5A3 (GenBank accession no. NM_024592) cDNA were amplified by PCR using Prime STAR DNA polymerase (Takara, Kyoto, Japan). The primer sets were as follows: forward 5′‐ATGGCAACGGCGACGGG‐3′ and reverse 5′‐TTAAAACAAAAATGGAATTATAATTTTCTGAAC‐3′ for SRD5A1; and forward 5′‐ATGGCTCCCTGGGCGGAG‐3′ and reverse 5′‐TTAAAACAAAAATGGTAGGAAAGCTTTC‐3′ for SRD5A3. These PCR products were cloned into the multiple cloning site of the pCAGGS vector. We also constructed two expression vectors of mutant SRD5A3; one was a deletion mutant of the N‐terminal‐58 residues (ΔN), the other was H296A‐mutated. The SRD5A3‐ΔN vector was amplified by PCR using the primer sets: forward 5′‐ATGGAGCCGTCGCGCCC‐3′ and reverse 5′‐TTAAAACAAAAATGGTAGGAAAGCTTTC‐3′. The H296A‐mutated SRD5A3 was subjected to site‐directed mutagenesis (Stratagene, La Jolla, CA, USA). Both of the products were cloned into the multiple cloning site of the same vector. All of the constructs were confirmed by DNA sequencing (ABI3700; PE Applied Biosystems).
In vitro 5α‐steroid reductase reaction. The enzyme source was obtained from COS7 cells transfected with each of five hemagglutinin (HA)‐tagged expression vectors (SRD5A1, wild‐type SRD5A3, SRD5A3‐ΔN, SRD5A3‐H296A, and mock). COS7 cells (1 × 107) were transfected with each vector using FuGENE6 according to the manufacturer's instructions, and after 48 h incubation, the transfected COS7 cells were harvested and homogenized in 500 µL of 40 mM sodium phosphate buffer (pH 6.9) using a digital homogenizer. All assays were carried out using a freshly prepared enzyme source. The substrate 1000 ng testosterone (Teikoku Hormone MFG, Kawasaki, Japan) and 500 µg NADPH (Sigma‐Aldrich) were added to each of the cell lysates and incubated for 30 min at 37°C. The reaction was quenched by icing. The expression of exogenous 5α‐steroid reductases and their mutant forms were evaluated by western blot analysis using anti‐HA tag antibody (Sigma‐Aldrich).
Quantitative analysis of testosterone and DHT by LC‐MS/MS. The internal standards 1 ng T‐d3 [T‐19‐C2H3], and 1 ng DHT‐d3 [17,16,16–2H3]‐DHT were added to the individual homogenized cells or the conditioned media, which was extracted with ether. The organic layer was evaporated and the extracts were dissolved in 1 mL of 20% acetonitrile–H2O in an ultrasonic bath and applied to a 3‐mL Bond Elut C18 cartridge column (Varian, Harbor City, CA, USA). These columns were then washed successively with 1 mL water and 3 mL of 30% acetonitrile–H2O, and the steroidal fraction was eluted with 2.5 mL of 70% acetonitrile–H2O and dried using a centrifugal evaporator. To increase the sensitivity of MS analysis, the dried steroidal fraction was reacted with 100 µL reagent mixture (20 mg 2‐methyl‐6‐nitrobenzoic anhydride, 10 mg 4‐dimethyl‐aminopyridine, and 20 mg picolinic acid in 1 mL tetrahydrofuran) and 15 µL triethylamine for 60 min at room temperature. The reaction mixture diluted with 1% acetic acid was applied to a 3‐mL Bond Elut C18 cartridge column. The columns were washed with distilled water and 30% acetonitrile–H2O, and the steroidal fraction was eluted with 3 mL of 70% acetonitrile–H2O. The collected fraction was evaporated and dissolved in 100 µL of 40% acetonitrile–H2O. Ten microliters was applied to the LC‐MS/MS instrument: ABI‐4000 (Applied Biosystems, Foster City, CA) equipped with an ESI ion source and a Shimadzu high‐performance liquid chromatography (HPLC) system (Shimazu, Kyoto, Japan). The HPLC column was a Cadenza CD‐C18 (150 mm × 2 mm, inner diameter [i.d.], 3 µm; Imtakt, Kyoto, Japan). The mobile phase consisting of acetonitrile–methanol (50:50 v/v, solvent A) and 0.1% formic acid (solvent B) was used with a gradient elution of A : B = 60 : 40–100 : 0) at a flow rate of 0.4 mL/min. The electrospray (ESI)/MS conditions were as follows: spray voltage, 3300 V; Collison gas, 1.5 psi (gas pressure) nitrogen; curtain gas nitrogen, 11 psi (gas pressure); ion source temperature, 600°C; and ion polarity, positive. For DHT determination, m/z 382.3 was activated as a precursor ion, and the product m/z 255 ions were monitored. For testosterone determination, m/z 380.3 was activated as a precursor ion and decomposed. Among the product ions, m/z 253 was monitored. The produced ion mass monitored for the internal standards T‐d3 and DHT‐d3 were 271 and 273 m/z, respectively. The assay was validated to ensure that the result was within the 20% range of accuracy and precision. The lower limit values were 1 pg for DHT and testosterone.
Results
SRD5A3 overexpression in HRPC cells. Through detailed expression profile analysis of HRPC cells, we previously identified dozens of transactivated genes in HRPC.( 14 ) Among them, we focused on a putative 5α‐steroid reductase (SRD5A2L; GenBank accession no. NM_024592), which we termed type 3 5α‐steroid reductase (SRD5A3), because it is possibly linked to the hormone‐refractory nature of PC cells. Fig. 1a shows the alignment of the amino acid sequence of human SRD5A3 (type 3) with those of human SRD5A1 (type 1) and human SRD5A2 (type 2). Their C‐terminal regions were conserved and are likely to be responsible for the catalytic activity of the 5α‐steroid reductase.( 15 , 16 ) We analyzed the RNA expression pattern of the three isozymes of 5α‐steroid reductase using RNA obtained from microdissected HRPC cells, HNPC cells, and NP cells, by semiquantitative RT‐PCR. The expression level of SRD5A3 was significantly elevated in HRPC cells, compared with HNPC and NP cells (Fig. 1b) and was similar to that of type 1 5α‐steroid reductase (SRD5A1). Further investigation of the SRD5A3 expression patterns in PC cell lines and normal tissues by northern blot analysis confirmed the elevated expression of SRD5A3 in all of the PC cell lines we analyzed (Fig. 1c), compared with the normal prostate. Multiple tissue northern blot analysis revealed no or very low expression of SRD5A3 in most normal adult organs, and low‐level expression in the normal pancreas (Fig. 1d).
Figure 1.
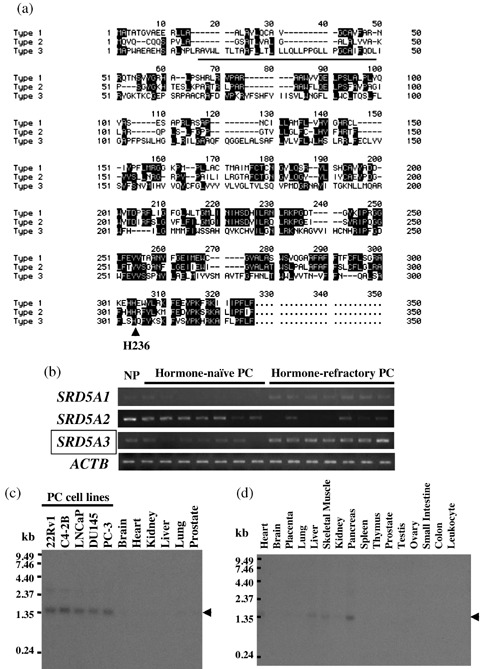
(a) Alignment of the amino acid sequence of human SRD5A3 (type 3; GenBank accession no. NP_078868) with those of human SRD5A1 (type 1; GenBank accession no. GenBank accession no. NP_001038), and human SRD5A2 (type 2, NP_000339). The underlined sequence of SRD5A3 is a hydrophobic region that was deleted in SRD5A3‐ΔN (1–58) and the arrow indicates H296, which is one of the most conserved residues in all three 5α‐steroid reductases. (b) Semiquantitative reverse transcription–polymerase chain reaction validated that SRD5A3 expression was upregulated in the microdissected hormone‐refractory prostate cancer (HRPC) cells, compared with hormone‐naïve prostate cancer (HNPC) cells and normal prostatic epithelial (NP) cells, which were also microdissected. SRD5A1 expression was also upregulated in HRPC cells, whereas SRD5A2 expression was suppressed in HNPC and HRPC cells. Expression of ACTB served as the quantitative control. (c) Northern blot analysis showed a high level of SRD5A3 expression in five prostate cell lines (lanes 1–5), whereas its expression was hardly detectable in vital adult organs, including heart, lung, liver, kidney, brain, and prostate (lanes 6–11). (d) Multiple tissue northern blot analysis showed faint expression only in the normal pancreas and no or very low expression of SRD5A3 in adult normal organs.
5α‐steroid reductase function of SRD5A3. To investigate whether this putative 5α‐steroid reductase could have an actual enzymatic activity as a 5α‐steroid reductase, we constructed the expression vector of SRD5A3 and SRD5A1 as a positive control, and prepared the lysates of the transfected cells with each of the expression vectors as enzyme sources of in vitro 5α‐steroid reductase reaction. Exogenous SRD5A3 and SRD5A1 protein expression was confirmed by western blot analysis using anti‐HA tag antibody (Fig. 2a). After in vitro 5α‐steroid reductase reaction using testosterone as a substrate, highly sensitive LC‐MS/MS analysis( 17 ) detected the production of DHT in the cell lysates transfected with SAR5A3 as well as SRD5A1 (Fig. 2b, panels 1–3). To further investigate the specificity of this enzymatic activity and the functional domain of SRD5A3, we constructed two mutated SRD5A3 expression vectors and prepared the cell lysates of the transfected cells as enzyme sources of the in vitro 5α‐steroid reductase reaction. Previous studies suggested that the N‐terminal hydrophobic region of the type 1 and type 2 5α‐steroid reductases could interact with the aliphatic and aromatic side chains of substrates, and that some of the C‐terminal resides, such as H232, could play critical roles in the catalytic activity and Km of testosterone.( 15 , 16 ) Type 3 5α‐steroid reductase conserved its hydrophobic region at its N‐terminus (Fig. 1a, underlined) and the histidine residue at codon 296 (Fig. 1a, arrow). As shown in Fig. 2b (panels 4 and 5), the mutant SRD5A3 proteins lacking in the N‐terminal hydrophobic region or with the conserved H296 residue replaced with alanine lost their 5α‐steroid reductase enzymatic activity, indicating the critical roles of the domain or the residue in the 5α‐steroid reductase activity of SRD5A3 as well as the other two isozymes.
Figure 2.
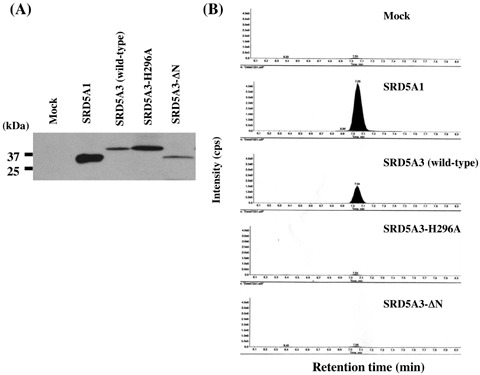
In vitro 5α‐reductase activity. (a) The enzyme source was obtained from COS7 cells transfected with each of five hemagglutinin (HA)‐tagged expression vectors (SRD5A1, wild‐type SRD5A3, SRD5A3‐ΔN, SRD5A3‐H296A, and Mock). Western blot analysis using HA‐antibody validated their expression. (b) The liquid chromatography‐tandem mass spectrometry chromatogram demonstrated 5α‐dihydrotestosterone (DHT) production in SRD5A3‐transfected cells (panel 3) as well as SRD5A1‐transfected cells (panel 2). Panel 1 shows DHT production in Mock‐transfected COS7 cells as the background. The mutant SRD5A3 (SRD5A3‐ΔN and SRD5A3‐H296A) did not produce DHT (panels 4 and 5).
Knockdown of SRD5A3 by siRNA in HRPC cell lines. To investigate a potential growth‐promoting role of SRD5A3 overexpression in PC cells, we constructed several siRNA expression vectors to examine their knockdown effects on the SRD5A3‐overexpressing PC cells 22Rv1. Semiquantitative RT‐PCR experiments after transfection of each of these siRNA‐expressing constructs into 22Rv1 cells revealed a significant knockdown effect on the cells transfected with either of the si#524 or si#790 constructs, but no effect on those transfected with either of the other constructs (Fig. 3a). After culture in medium containing geneticin, we carried out MTT (Fig. 3b) and colony‐formation (Fig. 3c) assays, and observed that introduction of si#524 or si#790 into 22Rv1 cells drastically attenuated their cell growth and viability, whereas that of either of the other siRNA, which did not affect SRD5A3 expression, did not affect cell growth, Similar results were obtained when we used another HRPC cell line, C4‐2B (data not shown), indicating that SRD5A3 is likely to play important roles in PC cell viability.
Figure 3.
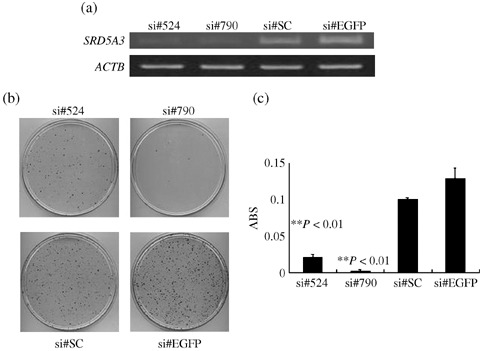
Effect of SRD5A3 knockdown by small interfering RNA (siRNA) on the growth of prostate caner cells. (a) The siRNA expression vectors specific to the SRD5A3 transcript (si#524 and si#790) or scrambled siRNA (si#SC) and enhanced green fluorescent protein (EGFP) siRNA expression vector (si#EGFP) as negative controls were transfected into 22Rv1 cells. The knockdown effect on the SRD5A3 transcript was validated by reverse transcription–polymerase chain reaction, with ACTB expression as a quantitative control. Transfection with si#524 and si#790 showed a significant knockdown effect, whereas si#SC or si#EGFP showed no effect on the level of SRD5A3 transcript. (b) Transfection with si#524 or si#790 resulted in a drastic reduction in the number of viable cells evaluated by colony‐formation assay, compared with the cells transfected with either of the other siRNA expression vectors that showed no knockdown effect on SRD5A3 expression. (c) Transfection with si#524 or si#790 resulted in a drastic reduction in the number of viable cells measured by MTT assay (P < 0.01, Student's t‐test). y‐axis, absorbance (ABS) at 490 nm (MTT assay), and at 630 nm as a reference, measured with a microplate reader.
Subsequently, to evaluate the endogenous activity of 5α‐steroid reductase, we used sensitive LC‐MS/MS analysis to measure the amounts of DHT and testosterone in the culture media and within C4‐2B cells, which were treated with duplex RNA corresponding to si#790 and the control siRNA. The treatment with siRNA for SRD5A3 significantly reduced the SRD5A3 expression level, whereas it did not affect the expression level of SRD5A1 (Fig. 4a). SRD5A2 was not expressed in C4‐2B cells. The ratio of the total amounts of DHT to testosterone was significantly decreased in the cells in which SRD5A3 was knocked down (Fig. 4b; P < 0.05), suggesting that SRD5A3 (type 3) could have a critical role in DHT production in HRPC cells.
Figure 4.
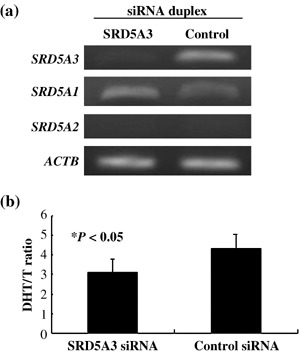
Suppression of SRD5A3 expression attenuated 5α‐dihydrotestosterone (DHT) production in C4‐2B cells. (a) Reverse transcription–polymerase chain reaction validated the knockdown effect of SRD5A3 expression and almost unchanged expression of SRD5A1 in C4‐2B cells by SRD5A3 small interfering RNA (siRNA). SRD5A2 was not expressed in C4‐2B cells. ACTB expression served as a quantitative control. (b) The ratio of the total amounts of DHT and testosterone in prostate caner cells and their media. Transfection of the RNA duplex to SRD5A3 (si#790) significantly suppressed the conversion from testosterone to DHT in C4‐2B cells, compared to the control RNA duplex si#EGFP (P < 0.05, Student's t‐test). The y‐axis reveals the ratio of the total DHT to the total testosterone (T).
Discussion
Although androgen‐ablation therapy can drastically reduce the serum testosterone level to less than 5%, the intraprostatic concentration of DHT in HRPC remains at ~40%,( 9 , 10 , 11 ) indicating that HRPC cells could produce the most potent androgen, DHT, from testosterone or other steroids, which are likely to be derived from the adrenal grand or other sources. 5α‐steroid reductase enzymes catalyze the conversion from testosterone to DHT, and in the normal prostate and benign prostate diseases, the type 2 isozyme (SRD5A2) is predominant( 12 ) and is strongly associated with benign prostatic hyperplasia (BPH)( 18 ) and prostate cancer risk.( 19 ) However, our study and other reports( 13 , 20 , 21 ) have demonstrated that expression of the type 2 isozyme is decreased drastically in PC cells and suppressed more significantly in HRPC cells, whereas the type 1 isozyme (SRD5A1) seems to be expressed dominantly in PC cells and HRPC cells. In addition to this switching of 5α‐steroid reductase isozymes, we here demonstrated that a novel 5α‐steroid reductase type 3 was overexpressed specifically in HRPC cells and played important roles in HRPC growth and progression.
The selective inhibitor to type 2 5α‐steroid reductase, finasteride, is shown to be effective to BPH by inducing apoptosis,( 18 , 22 ) and the Prostate Cancer Prevention Trial demonstrated that finasteride treatment decreased the rate of PC incidence by 24.8%.( 19 ) However, because SRD5A2 activity was decreased in PC cells, it is very unlikely to expect the effect of finasteride for treatment of PC. In fact, there is no conclusive report to indicate a benefit of finasteride for treatment of established PC.( 21 ) Furthermore, a dual inhibitor to both type 1 and type 2 5α‐steroid reductases, dutasteride, was recently developed and expected to be more effective than finasteride in DHT depletion.( 23 ) Two reports have indicated that treatment with dutasteride caused a 97% decrease in intraprostatic DHT and induced a decrease in the tumor volume of early or operable PC.( 23 , 24 ) However, it is still uncertain whether dutasteride can induce regression of advanced PC or HRPC. Although we do not know whether these 5α‐steroid reductase inhibitors can inhibit the activity of the type 3 isozyme, an inhibitor of the type 3 isozyme in HRPC cells should be expected to cause DHT depletion and enhance the therapeutic effect of HRPC.
In addition to the 5α‐reduction of testosterone, other types of steroids are subject to 5α‐reduction, and type 1 or type 2 5α‐steroid reductase was reported to be involved in 5α‐reduction of progesterone and androstenedione as well as testosterone.( 8 , 25 ) Other types of steroids have been implicated to play important roles in the viability of HRPC cancer under serum androgen depletion;( 17 , 26 ) HRPC cells often maintain their growth signaling through overexpression of AR, mutated AR, and other steroid receptors. Although other substrates of this novel 5α‐steroid reductase reported here are unclear, some 5α‐reduced steroids, such as neurosteroids,( 27 ) might play critical roles in HRPC growth and progression. Identification and characterization of such a novel steroid associated with HRPC and type 3 5α‐steroid reductase would shed light on a better and novel understanding of the molecular mechanisms underlying HRPC progression and contribute to the development of novel therapies for HRPC.
Acknowledgments
We thank Ms Satomi Uchida, Ms Mami U, and Ms Lian‐hua Piao for their technical assistance, and Dr Kazuo Nishimura and Dr Norio Nonomura at Osaka University for their helpful discussions. This work was supported in part by Research Grants #18590323 (H. Nakagawa) and #00L01402 (Y. Nakamura) from the Japan Society for the Promotion of Science.
References
- 1. Gronberg H. Prostate cancer epidemiology. Lancet 2003; 361: 859–64. [DOI] [PubMed] [Google Scholar]
- 2. Hsing AW, Devesa SS. Trends and patterns of prostate cancer: what do they suggest? Epidemiol Rev 2001; 23: 3–13. [DOI] [PubMed] [Google Scholar]
- 3. Feldman BJ, Feldman D. The development of androgen‐independent prostate cancer. Nat Rev Cancer 2001; 1: 34–45. [DOI] [PubMed] [Google Scholar]
- 4. Han M, Partin AW, Piantadosi S et al . Era specific biochemical recurrence‐free survival following radical prostatectomy for clinically localized prostate cancer. J Urol 2001; 166: 416–19. [PubMed] [Google Scholar]
- 5. Petrylak DP, Tangen CM, Hussain MH et al . Docetaxel and estramustine compared with mitoxantrone and predonisone for advanced refractory prostate cancer. N Engl J Med 2004; 351: 1513–20. [DOI] [PubMed] [Google Scholar]
- 6. Tannock IF, De Wit R, Berry WR et al . Docetaxel plus predonisone or mitoxantrone plus predonisone for advanced prostate cancer. N Engl J Med 2004; 351: 1502–12. [DOI] [PubMed] [Google Scholar]
- 7. Scher HI, Sawyers CL. Biology of progressive, castration‐resistant prostate cancer: directed therapies targeting the androgen‐receptor signaling axis. J Clin Oncol 2006; 23: 8253–61. [DOI] [PubMed] [Google Scholar]
- 8. Chang C. Androgen and Androgen Receptor, 1st edn. Boston, MA: Kluwer Academic Publishers, 2002. [Google Scholar]
- 9. Mohler JL, Gregory CW, Ford OH 3rd et al . The androgen axis in recurrent prostate cancer. Clin Cancer Res 2004; 10: 440–8. [DOI] [PubMed] [Google Scholar]
- 10. Nishiyama T, Hashimoto Y, Takahashi K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clin Cancer Res 2004; 10: 7121–6. [DOI] [PubMed] [Google Scholar]
- 11. Titus MA, Schell MJ, Lih FB et al . Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res 2005; 11: 4653–7. [DOI] [PubMed] [Google Scholar]
- 12. Thigpen AE, Silver RI, Guileyardo JM et al . Tissue distribution and ontogeny of steroid 5α reductase isozyme expression. J Clin Invest 1993; 92: 903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Titus MA, Gregory CW, Ford OH 3rd et al . Steroid 5α‐reductase isozymes I and II in recurrent prostate cancer. Clin Cancer Res 2005; 11: 4365–71. [DOI] [PubMed] [Google Scholar]
- 14. Tamura K, Furihata M, Tsunoda T et al . Molecular features of hormone‐refractory prostate cancer cells by genome‐wide gene‐expression profiles. Cancer Res 2007; 67: 5117–25. [DOI] [PubMed] [Google Scholar]
- 15. Wigley WC, Prihoda JS, Mowszowicz I et al . Natural mutagenesis study of the human steroid 5α‐reductase 2 isozyme. Biochemistry 1994; 33: 1265–70. [DOI] [PubMed] [Google Scholar]
- 16. Wang M, Bhattacharyya AK, Taylor MF et al . Site‐directed mutagenesis studies of NADPH‐binding domain of rat steroid 5α‐reductase (isozyme‐1); analysis of aromatic and hydroxylated amino acid residues. Steroids 1999; 64: 356–62. [DOI] [PubMed] [Google Scholar]
- 17. Mizokami A, Koh E, Fujita H et al . The adrenal androgen androstenediol is present in prostate cancer tissue after androgen deprivation therapy and activates mutated androgen receptor. Cancer Res 2004; 64: 765–71. [DOI] [PubMed] [Google Scholar]
- 18. Bartsch G, Rittmaster RS, Klocker H. Dihydrotestosterone and the concept of 5α‐reductase inhibition in human benign prostatic hyperplasia. Eur Urol 2000; 37: 367–80. [DOI] [PubMed] [Google Scholar]
- 19. Thompson IM, Goodman PJ, Tangen CM et al . The influence of finasteride on the development of prostate cancer. N Engl J Med 2003; 349: 215–24. [DOI] [PubMed] [Google Scholar]
- 20. Iehle C, Radvanyi F, Gil Diez de Medina S et al . Differences in steroid 5α‐reductase iso‐enzymes expression between normal and pathological human prostate tissue. J Steroid Biochem Mol Biol 1999; 68: 189–95. [DOI] [PubMed] [Google Scholar]
- 21. Luo J, Dunn TA, Ewing CM et al . Decreased gene expression of steroid 5α‐reductase 2 in human prostate cancers: implications for finasteride therapy of prostate carcinoma. Prostate 2003; 57: 134–9. [DOI] [PubMed] [Google Scholar]
- 22. McConnell JD, Bruskewitz R, Walsh P et al . The effect of finasteride on the risk of acute urinary retention and the need for surgical treatment among men with benign prostatic hyperplasia. N Engl J Med 1998; 338: 557–63. [DOI] [PubMed] [Google Scholar]
- 23. Andriole GL, Humphrey P, Ray P et al . Effect of the dual 5α‐reductase inhibitor dutasteride on markers of the tumor regression in prostate cancer. J Urol 2004; 172: 915–19. [DOI] [PubMed] [Google Scholar]
- 24. Gleave M, Qian J, Andreou C et al . The effects of the dual 5α‐reductase inhibitor dutasteride on localized prostate cancer. Prostate 2006; 66: 1674–85. [DOI] [PubMed] [Google Scholar]
- 25. Normington K, Russell DW. Tissue distribution and kinetic characteristics of rat steroid 5α‐reductase isozymes. J Biol Chem 1992; 267: 19 548–54. [PubMed] [Google Scholar]
- 26. Zhao XY, Malloy PJ, Krishnan AV et al . Glucocorticoids can promote androgen‐independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med 2000; 6: 703–6. [DOI] [PubMed] [Google Scholar]
- 27. Stoffel‐Wagner B. Neurosteroid biosynthesis in the human brain and its clinical implications. Ann N Y Acad Sci 2003; 1007: 64–78. [DOI] [PubMed] [Google Scholar]