

Abstract
Protein packing defects constitute structural singularities arising from backbone overexposure to water. Such features may be targeted in kinase-inhibitor design to achieve specificity, to control cross reactivity and to overcome drug resistant mutations.
Protein kinases constitute major targets in molecular cancer therapy. The structural conservation of kinases causes specificity problems in most drug inhibitors, often resulting in dangerous side effects. Here we survey recent approaches in drug design that exploit a molecular marker for specificity: the pattern of packing defects. These packing defects are solvent-exposed intramolecular hydrogen bonds that may be protected by drugs upon association. In this light, we review design strategies to achieve paralog discrimination, to control cross reactivity and to overcome drug resistance induced by target mutations. Introduction
Keywords: Kinases, Cancer, Small-Molecule Inhibitor, Selectivity, Cross Reactivity, Drug Specificity, Drug Resistance
Introduction
Specificity problems in kinase-inhibitor design
Protein kinases are quintessential signal transducers of the cell and have become important targets for cancer therapy [1–5]. Their evolutionary and structural relatedness [6–8], however, often results in unforeseen cross reactivities, yielding uncertain and at times health-threatening effects [9,10]. Although the relationship between specificity and anticancer activity is poorly understood and that specificity might not be essential for clinical activity, a lack of specificity generally underlies toxic side effects, due to off-target impact of the inhibitor [6,8]. A good example is the anticancer drug imatinib (Gleevec, STI 571) [3]. This drug, which has a relatively broad activity profile, has been a great therapeutic success, however, the lack of target specificity may lead to side effects such as cardiotoxicity [9].
Due to the structural conservation within the kinase superfamily, controlling specificity is a challenge. Figure 1 illustrates this issue showing the structural alignment of two kinases with 80% sequence identity: the insulin-like growth factor 1 receptor kinase (IGF1R) and the insulin receptor kinase (INSR). The structural similarity is reflected in the alignment (RMSD=1.2Å). Interestingly, whereas IGF1R kinase is a promising cancer target, as suggested by its overexpression in several tumors and by its role in cell growth and survival [11,12], the INSR kinase cannot be inhibited without introducing a health threat, since glucose uptake would be compromised. Thus, considerable effort is being devoted to control cross reactivity and the resulting side effects, motivated by the need to focus the inhibitory impact on clinically relevant targets.
Figure 1.
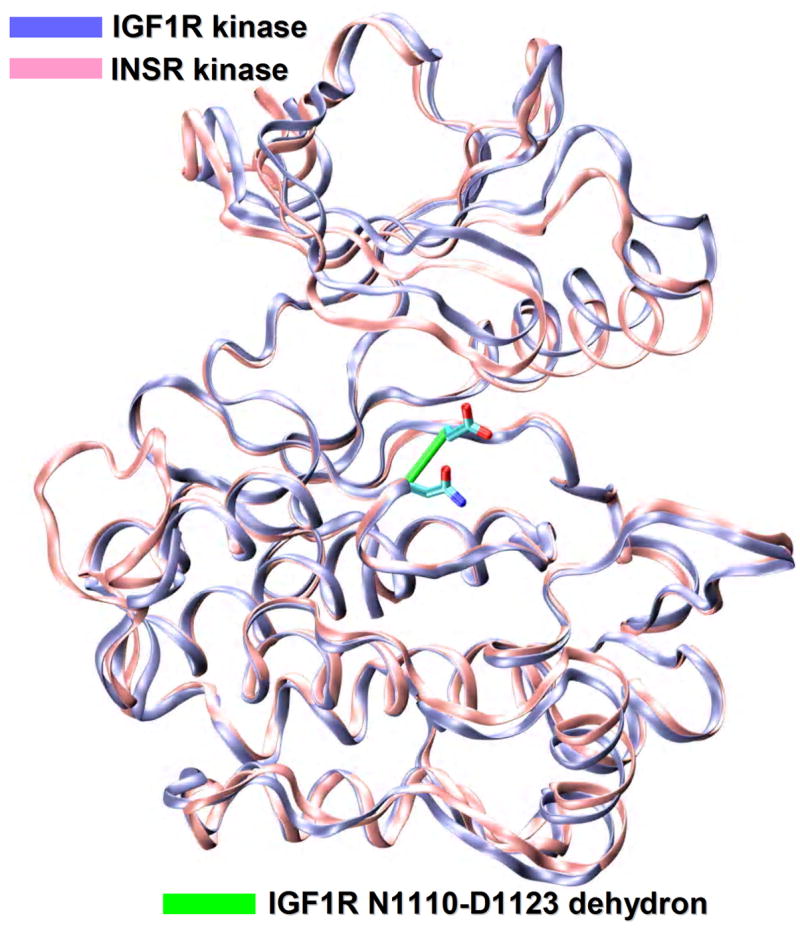
Specificity problems in kinase inhibition. Structural alignment (ribbon representation) of the IGF1R kinase (PDB.1K3A, ice blue), a proposed cancer target and the INSR kinase (PDB.1GAG, pink), a related target to be avoided. The degree of structural similarity between the two paralogs is staggeringly high (RMSD=1.2Å) as expected from the 80% sequence identity. The IGF1R nonconserved N1110-D1123 dehydron is also shown as a green virtual bond joining α-carbons (ρ=18 nonpolar groups for desolvation radius of 6.2Å).
Approaches to achieve drug specificity
One approach to solving the specificity problem involves the rational design of selective kinase inhibitors [13] by using high-resolution crystal structures complexed with non-ATP ligands. Structural motifs unique to the purported targets have been identified that could be exploited to engineer specific inhibitors. For example, a small helical element (αT-helix) has been identified in the active site of the NEK2 kinase following the conserved DFG motif [14] or, a new activation-loop motif with a large helical insert [15] has been found in MPSK1 structure.
In addition to the structural conservation of kinases, their high degree of plasticity [16], especially in the activation loop, poses challenges to in-silico design due to induced fits, but also provides new features to engineer selective inhibitors [13]. Specific and clinically successful inhibitors that target inactive kinase conformations have been developed utilizing the pocket generated by the ‘DFG out’ conformation [17]. In this singular conformation, the position of the phenylalanine residue of the conserved DFG catalytic triad, located at the start of the activation loop, is flipped with respect to the active conformation, so that it points toward the ATP site, as in the case of imatinb binding to inactive Abl kinase [18]. All active kinases adopt similar conformations, while inactive kinases are more discernible. Therefore, to attain specificity, the inactive conformations are more attractive targets. While targeting the inactive conformation may appear to be a logical choice, there are also advantages in targeting the active conformation. The active conformation requires structure conservation, and hence it is less tolerant to drug-resistant mutations [18]. For example, the EGFR kinase inhibitor erlotinib (Tarceva) binds to the active conformation [18]. Furthermore, the size of the gatekeeper residue is a determinant of inhibitor selectivity: kinases with a threonine at this position are sensitive to a range of inhibitors, whereas those with a larger residue (methionine) are typically impervious [8].
Another way of tackling the specificity problem is the design of non-competitive kinase inhibitors [19]. These ligands are likely to be more specific, since they bind to residues outside the ATP pocket, which are less conserved [8]. Furthermore, they alter kinase conformation, preventing substrate binding. As an illustration, the crystal structural of a PD184352 analog in complex with MEK1 and ATP confirms that these compounds bind to a site adjacent to the ATP binding pocket. The low degree of sequence conservation in this region explains the high selectivity of these compounds [20]. Also, several classes of pyrazinones have been reported as being non-competitive inhibitors of Akt and show marked selectivity discriminating the isoforms Akt1 and Akt2 [21].
In spite of these significant advances, a rational control of kinase-inhibitor specificity remains a problem. In this review, we discuss how to attack this problem using a novel selectivity filter.
Novel molecular marker to achieve specificity
Drug design remains a semiempirical endeavor, essentially supplemented by structural considerations, and guided by the possibility of forming standard intermolecular hydrogen bonds, electrostatic or hydrophobic interactions. However, a ligand designed exclusively based on the possibility of promoting such interactions would likely be promiscuous due to the high degree of conservation of hydrogen-bond donors/acceptors and nonpolar residues on the kinase surface [7,22,23]. Thus, it is unlikely that the significant levels of cross reactivity detected in high-throughput screening experiments [6] will be tempered using rational design, unless a new approach is able to discern paralogs above and beyond what a structural characterization may reveal.
Recent progress along these lines is marked by the identification of a molecular marker for specificity: the packing defects in soluble proteins [24–27]. These defects consist of solvent-exposed backbone hydrogen bonds and are targetable features because of their inherent stickiness [25]. One most useful property from the perspective of drug design is their lack of conservation across proteins with common ancestry [7,28]. They are indicators of protein interactivity and constitute a determinant factor for macromolecular recognition [29–33]. They are termed dehydrons [24–27], since they promote their own dehydration as a mean to strengthen and stabilize the underlying electrostatic interaction. Thus, targeting these features by turning drugs into protectors or ‘wrappers’ (water-excluders) of packing defects [34,35] may control cross reactivity. The concept of drug-as-wrapper was initially introduced by Fernández et al. [34], when packing defects were exploited to design novel HIV-1-integrase inhibitors and rationalize the binding mode of existing HIV-1-protease inhibitors.
In this work we survey the molecular design strategies to engineer drugs that act as dehydron wrappers. Decisive in silico, in vitro and in vivo validation of the wrapping concept is surveyed [36,37]. Finally, we propose a molecular-design exercise as potential application of the dehydron-targeting principle: the differentiation of the close paralogs IGF1R and INSR kinases.
Packing defects in protein structure
Dehydrons constitute packing defects since they are identified by a dearth of nonpolar groups from the amino acid side chains in the spatial vicinity of a backbone hydrogen bond [24–27]. They are defined in terms of the effect on the dielectric environment due to the approach of a nonpolar group or wrapper [24–27]. Solvent exposed hydrogen bonds become strengthened and stabilized by the approach of a hydrophobic group, as in the case where a drug ligand binds to a protein. Thus, because of their propensity to attract nonpolar groups, or exclude water molecules, dehydrons constitute sticky spots that promote their own further dehydration [24–27].
Rigorously speaking, the term wrapping alludes to a clustering of hydrophobic groups framing an anhydrous microenvironment for an intramolecular backbone hydrogen bond within the structure of a soluble protein [24–27]. The extent of intramolecular hydrogen-bond desolvation, ρ, may be quantified by determining the number of nonpolar groups (carbonaceous, not covalently bonded to an electrophilic atom) contained within a desolvation domain. The desolvation domain is defined as two intersecting balls (Figure 2) of fixed radius centered at the α-carbons of the residues paired by the backbone hydrogen bond. The statistics of hydrogen-bond wrapping vary according to the desolvation radius adopted, but the tails of the distribution invariably single out the same dehydrons in a given structure over a 6–7Å radius range, a length scale that represents the thickness of three water layers. In folds for soluble proteins at least two thirds of the backbone hydrogen bonds are wrapped on average by ρ=26.6±7.5 nonpolar groups for desolvation radius of 6.2Å. Dehydrons are then defined as hydrogen bonds whose extent of wrapping lies in the tail of the distribution, i.e. with 19 or fewer hydrophobic groups in their desolvation domains, so their ρ-value is below the mean, minus one Gaussian dispersion.
Figure 2.
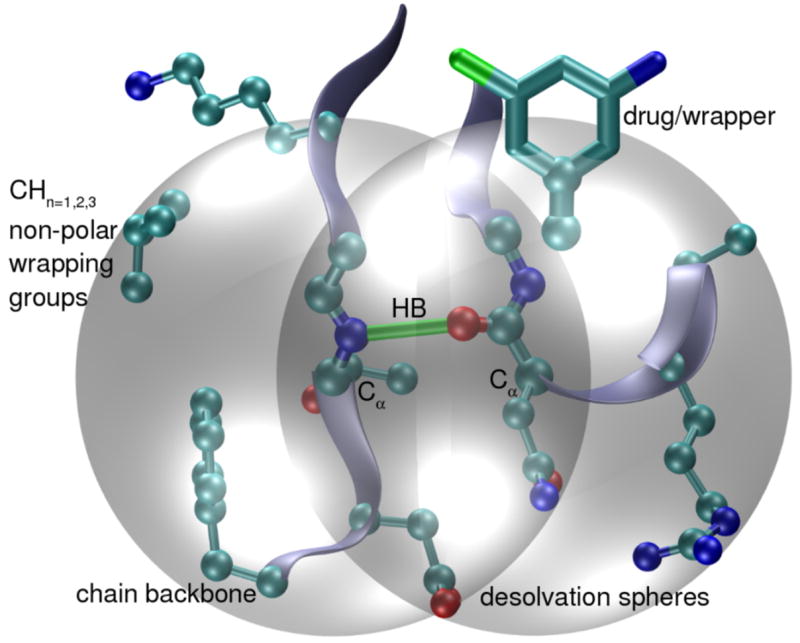
Dehydron concept. Intramolecular backbone hydrogen bonds in target proteins (green bond) prevail only if protected from water attack. Their extent of intramolecular wrapping, ρ, is defined by counting the nonpolar groups (carbonaceous atoms, cyan) contained within the desolvation domain (grey spheres). Poorly wrapped hydrogen bonds (dehydrons) are defined with 19 or fewer hydrophobic groups in their desolvation domains (desolvation radius of 6.2Å). These packing defects consist of solvent-exposed backbone hydrogen bonds and are targetable features because of their inherent stickiness, since they promote the removal of surrounding water molecules as a mean to strengthen and stabilize the underlying electrostatic interaction.
Dehydrons are invariant across complexes of the same protein with different ligands and invariant across different crystallization forms, except in cases where the different ligands produce different induced fits. Thus, due to the high conformational plasticity of kinases [16], targeting dehydrons in induced fits or flexible regions (activation loop, P-loop, catalytic loop) requires performing molecular dynamic (MD) simulations to identify the binding mode of the ligand. On the other hand, the effect of structural fluctuations on the persistence of dehydrons has been addressed through long MD simulations [36,37].
A shortcoming in the engineering of selective drugs geared at wrapping nonconserved packing defects arises from the need for structural information on the targets. In addressing this problem, we note that comparing the wrapping pattern of targets of common ancestry is actually feasible, even in the absence of structure. This is so, since a surrogate for dehydrons, reliably predicted from sequence, may be used to infer the wrapping patterns which can be subsequently aligned using homology threading [7]. Thus, a sequence-based disorder score, measuring the propensity for inherent disorder of the peptide chain [28] correlates with the wrapping of individual residues engaged in backbone hydrogen bonds, with dehydrons belonging to the twilight region between order and disorder [38]. The disorder score has been used to predict dehydrons for all tyrosine kinases, even those with unreported structure and the predictions on cross reactivity resulting thereof have been validated against high-throughput screening data [7]. This dehydron predictor based on the disorder score is operational only for homologs likely to possess high structure similarity with PDB reported proteins, as in the case of the kinase superfamily.
Targeting non-conserved kinase packing defects
Specificity and promiscuity in the druggable kinome
In a recent bioinformatics contribution [7], it has been demonstrated that there is a relationship between the pharmacological differences between kinases against a background of available drugs and the differences between their respective patterns of packing defects. To compare the dehydron patterns from different kinases, a common region was introduced for the aligned target structures. In this way, all kinases were compared within a region defined by alignment of all residues framing the desolvation domains of dehydrons that, in turn, are environmentally affected by ligands in PDB-reported complexes. Within this framework of comparison, an environmental or wrapping distance between kinases i and j was obtained by contrasting their aligned hydrogen-bond environments that compares local dehydration propensities associated with dehydrons patterns in kinase pairs. The enviromental distance matrix was obtained across the 119 kinases for which drug-affinity fingerprinting was experimentally and independently obtained [6]. This distance matrix has been contrasted with a pharmacological distance matrix assessing similarities in the affinity profiling of kinases. 19 inhibitors were selected from a pool of 20 that have been assayed for cross reactivity against the set of 119 kinases [6]. The highly promiscuous ligand staurosporine has been excluded since it is not pharmacologically relevant [23]. The pharmacological matrix was obtained by computing the Euclidean distance between ligand-affinity vectors in R19 with entries given in –ln scale (or dimensionless ΔG/RT units; ΔG=Gibbs free energy of binding, R=gas constant, T=temperature).
A strong correlation (R2=0.917) was established between the environmental and pharmacological distance for each pair of kinases fingerprinted for affinity against a background of 19 drugs [7]. This correlation reveals that the pattern of packing defects is an operational selectivity filter for drug design, even though individual drugs were not purposely designed to wrap packing defects: pharmacological differences are essentially dictated by packing differences among targets. Thus, we can take advantage of this hitherto overlooked design feature to simplify the drug development effort and rationally enhance selectivity towards clinically significant targets.
Turning a promiscuous drug into a paralog discriminator
The feasibility of achieving specificity by designing a wrapping drug has been tested experimentally [23,35–37]. A preliminary effort was geared at redesigning commercially available inhibitors to discriminate paralog kinases. This endeavor started with the introduction of wrapping modifications in imatinib to enhance its specificity towards its primary target, the Abelson kinase (Abl) [35]. The nonconserved dehydrons Q300-E316, G249-Q252 in Abl were selectively targeted by a suitable wrapping modification of imatinib, sculpting into the ligand the dehydration hot spots of the kinase.
In a more stringent test [23], staurosporine, the most promiscuous kinase ligand available [6] was re-engineered to elicit a selective inhibitory impact. Four PDB-reported staurosporine-binding kinases were chosen: Src kinase (PDB.1BYG), CDK2 (PDB.1AQ1), Chk1 (PDB.1NVR) and PDK1 (PDB.1OKY). A wrapping analysis revealed that only the Src kinase possesses a targetable nonconserved dehydron (Q250-E267) that may be wrapped by specific methylation of staurosporine at the imide N6-position of the indole ring (Figure 3a). Upon structural alignment, the Src Q250-E267 dehydron maps into well-wrapped backbone hydrogen bonds: K65-E81 in CDK2, K69-E85 in Chk1 and K144-S160 in PDK1 (Figure 3a) [23]. Thus, selectivity for Src kinase may be achieved by redesigning staurosporine to turn it into a wrapper of the Q250-E267 dehydron. The inhibition of Src by the staurosporine derivative improved when compared with staurosporine levels and its impact became selective for Src to the extent expected from the limited set of targets analyzed [23]. These results demonstrate that the packing differences across paralogs may guide molecular design to significantly enhance specificity.
Figure 3.
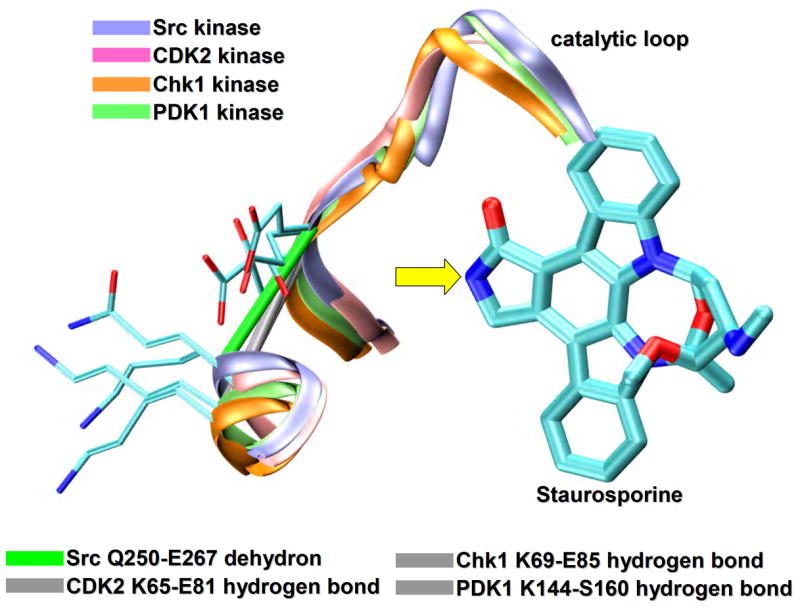
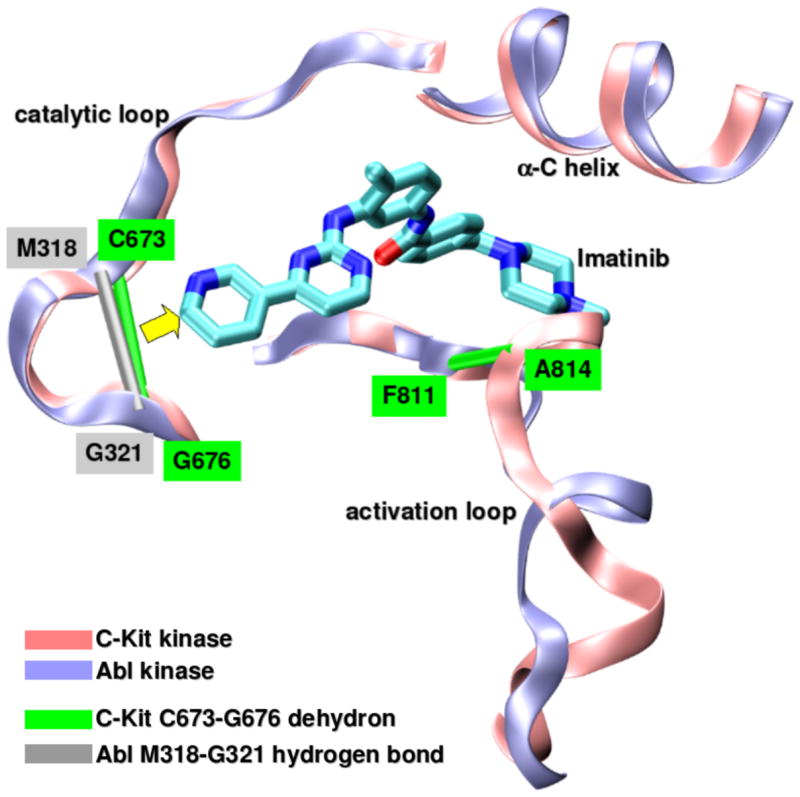
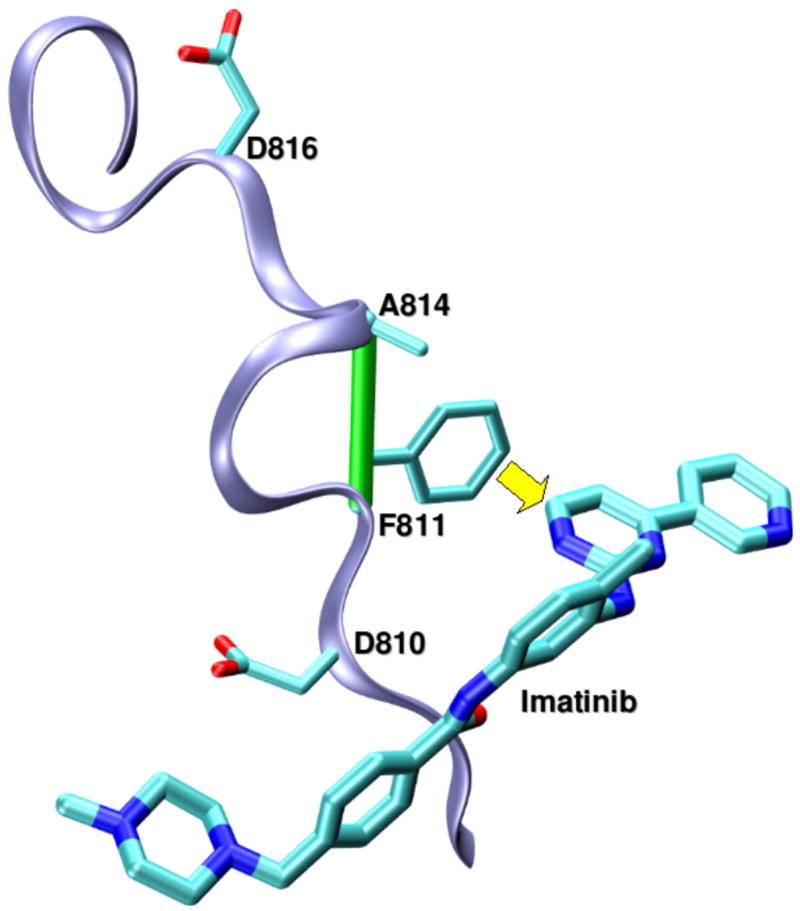
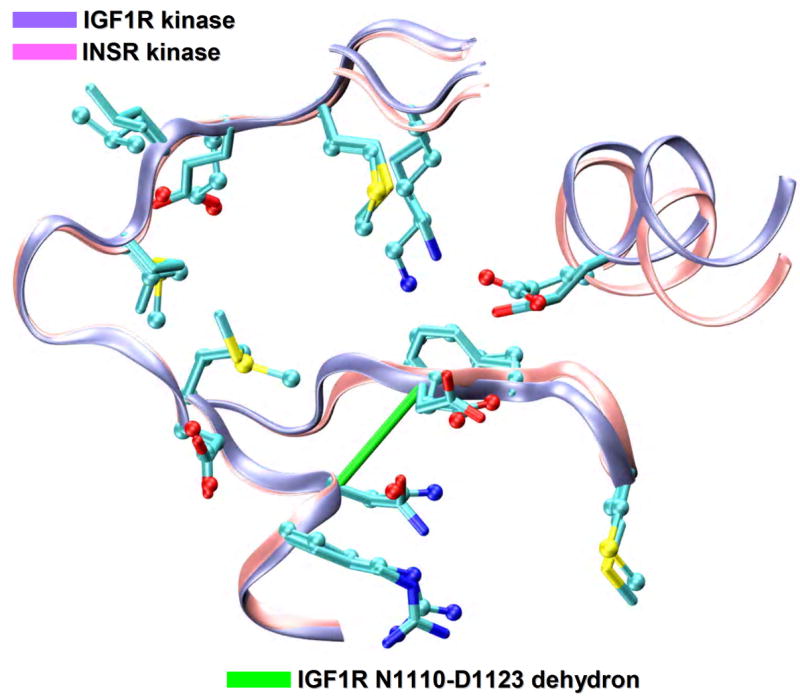
Targeting non-conserved kinase packing defects. (a) Aligned backbones (ribbon representation) of Src (PDB.1BYG, ice blue), CDK2 (PDB.1AQ1, pink), Chk1 (PDB.1NVR, orange) and PDK1 (PDB.1OKY, lime) kinases complexed with staurosporine. The Src Q250-E267 dehydron (green virtual bond joining α-carbons) maps into well-wrapped backbone hydrogen bonds (grey virtual bond joining α-carbons): K65-E81 in CDK2, K69-E85 in Chk1 and K144-S160 in PDK1. Methylation at the position indicated by the yellow arrow would turn the otherwise promiscuous ligand into a wrapper of the nonconserved packing defect. (b) Aligned backbones (ribbon representation) of Abl (PDB.1FPU, ice blue) and c-Kit (PDB.1T46, pink) kinases in the induced-fit conformation generated by co-crystallyzation with imatinib. The nonconserved dehydron C673-G676 (green virtual bond joining α-carbons) in c-Kit, which aligns with the well wrapped M318-G321 hydrogen bond (grey virtual bond joining α-carbons) in Abl, could be targeted by a methylation modification of imatinib in a specific position (yellow arrow) to achieve specificity. (c) Ribbon representation of the c-Kit (PDB.1T46, ice blue) kinases in the induced-fit conformation generated by co-crystallyzation with imatinib. The F811-A814 dehydron (green virtual bond joining α-carbons) present close to the D816V mutation site could be targeted by a methylation modification of imatinib in a specific position (yellow arrow) to overcome drug resistance. (d) Aligned ATP-binding pockets (ribbon representation) of IGF1R (PDB.1K3A, backbone: ice blue, sidechains: licorice representation) and INSR (PDB.1GAG, backbone: pink, sidechains: ball-and-stick representation) kinases. The degree of structure and sequence identity is very high at the ATP binding site. However, the nonconserved dehydron N1110-D1123 (green virtual bond joining α-carbons) in IGF1R, which does not align with any well wrapped hydrogen-bond or dehydron in the INSR, could be targeted to discriminate these closely related paralogs.
Curbing imatinib cross-reactivity and side effects through wrapping-based imatinib redesign
Undesired side effects [3,8] of drug treatment can sometimes be traced to the inhibitory impact on the primary target, as in the reported cardiotoxicity of imatinib, attributed to its impact on Bcr-Abl [9]. This constitutively active chimeric kinase, arising from aberrant chromosomal translocation, is the primary target in the treatment of chronic myeloid leukemia (CML) [3,39,40]. Using the drug-as-wrapper concept, we recently reported [36] a modified version of imatinib that reduces its impact on Bcr-Abl, while retaining anticancer activity through inhibition of c-Kit kinase, a primary target to treat GIST (gastrointestinal stromal tumors) [41]. The structural alignment of imatinb-bound Abl (PDB.1FPU) and c-Kit (PDB.1T46) reveals the nonconserved dehydron C673-G676 in c-Kit which aligns with the well-wrapped M318-G321 hydrogen bond in Abl (Figure 3b). This difference in the pattern of packing defects in the catalytic loop prompted us to develop a methylated variant of imatinib which hampers Abl inhibition while re-focusing the impact on c-Kit kinase. Thus, the therapeutic profile of the re-optimized wrapping variant is different from that of imatinib. The wrapping ligand is intended for GIST treatment, while being less cardiotoxic than the parental compound. We delineated the molecular basis for this target discrimination through in-vitro kinetics assays and high-throughput screening [6]. Thus, while imatinib binds to both c-Kit and Bcr-Abl, the wrapping variant only binds to c-Kit, as evidenced by the experimentally obtained dissociation constants: KdAbl=50nM, Kdc-Kit=55nM for imatinib, and KdAbl=11μM, Kdc-Kit=43nM for the wrapping variant [36]. We further demonstrated controlled inhibitory impact in vivo by assaying for antitumor activity on different cell lines and finally established the therapeutic impact of the optimized compound in a novel GIST animal model, corroborating a significant reduction in cardiotoxicity [36].
Overcoming imatinib resistance in c-Kit kinase through wrapping-based imatinib redesign
Kinases are moving targets since the cell develops mechanisms of drug resistance, mainly mutations, which hamper drug association [42]. The development of drug-resistant mutations of targeted proteins poses a further challenge to inhibitor design [42–44]. The c-Kit kinase is inhibited by imatinib [41], but in malignancies like systemic mastocytosis, the kinase develops the mutation D816V in the activation loop, promoting imatinib resistance [42–44]. Thus, we redesigned imatinib so that it inhibits both the wild-type kinase and the imatinib-resistant mutant [37]. The prototype was re-engineered to be a better stabilizer of the induced-fit conformation of the activation loop. The wrapping analysis performed in the c-Kit kinase (PDB.1T46) revealed a nonconserved dehydron (F811-A814) close to the mutation site (D816V) (Figure 3c). Molecular modeling led us to introduce another specific methylation in imatinib to promote a tighter grip on the activation loop of c-Kit kinase and overcome the destabilizing effect of the mutation. The dual inhibitory effect of the prototype was confirmed through in vitro spectroscopic kinase assays [37]: while imatinib only inhibits the wild-type kinase, the wrapping prototype inhibits both the wild-type and the D816V mutant (Table 1). The focused effect of the prototype over a vast cross-section of the human kinome was corroborated by high-throughput screening [6], confirming the prototype’s selective impact (Table 1) [37]. Finally, cell proliferation assays on lines that express wild-type and imatinib-resistant kinase confirmed the dual anticancer activity of the prototype, in contrast with the efficacy of the parental compound [37].
Table 1.
Comparison of binding properties of imatinib and wrapping prototype.
| Kd c-Kit (exp) | Kd D816V (exp) | High-throughput screening targets | Therapeutic use | |
|---|---|---|---|---|
| Imatinib | 25 nM | 11 μM | ABL, KIT, LCK, PDGFR | CML, GIST |
| Wrapping prototype | 21 nM | 39 nM | KIT, D816V, LCK, PDGFR | GIST, imatinib-resistant (KIT: D816V) GIST |
Future challenge: discriminating IGF1R and INSR kinases
The ability of IGF1R to regulate both proliferative and anti-apoptotic signaling suggests that selective inhibition of its kinase domain may yield a promising cancer therapy [11,12]. Cross reactivities of IGF1R kinase inhibitors due to a simultaneous inhibition of the INSR kinase, a very close paralog, may be life-threatening, as inhibiting the latter would lead to diabetic-related stress in erythrocyte glucose uptake [45,46]. The degree of structural similarity between the two paralogs is staggeringly high (RMSD=1.2Å, IGF1R: PDB.1K3A, INSR: PDB.1GAG, Figure 1), as expected from the 80% sequence identity, which approaches 100% for the ATP binding site (Figure 3d). Researchers at Novartis have recently disclosed the IGF1R inhibitory properties of a class of pyrrolopyrimidines [47,48] found by high-throughput screening. In particular NVP-ADW742 and NVP-AEW541 have been used preclinically to delineate their efficacy as IGF1R kinase inhibitors, with approximately 16- and 26-fold more potent than the activity on INSR, respectively (NVP-ADW742: IC50IGF1R=170nM, IC50INSR=2.8μM and NVP-AEW541: IC50IGF1R=86nM, IC50INSR=2.3μM). These inhibitors seem to have some selectivity, but the issue of how much inhibition of the INSR is acceptable remains unaddressed. In light of these findings, a wrapping design emerges as a possible alternative tool to maximize selectivity among these two related targets. Contrary to the vast majority of kinases that have a threonine in the gatekeeper position, both IGF1R and INSR kinases have a methionine gatekeeper residue, increasing the likelihood of cross reactivity. Nevertheless, differences arise in their dehydron patterns: there is a dehydron in IGF1R (N1110-D1123) that is not conserved in the INSR kinase (Figure 3d). This dehydron involves the highly conserved aspartate from the DFG catalytic triad located in the activation loop and does not align with any dehydron or well-wrapped hydrogen bond in the INSR kinase (D1150). Thus, to gain selectivity, our wrapping design dictates that a purported ligand should target the N1110-D1123 dehydron in IGF1R kinase.
Conclusions
Wrapping design holds promise as a new paradigm for rational development of drugs that exclude water around accessible hydrogen bonds of the protein target. This concept represents a departure from the standard approach which focuses on promoting pairwise intermolecular interactions. Not only uses wrapping design a novel modality of ligand association, but also guides modifications to existing ligands, in contrast with a combinatorial search strategy.
Using commercially available ligands and drug inhibitors as lead compounds, we are able to introduce a variety of wrapping modifications and test them on cell lines and animal models for enhanced specificity and anticancer activity. Based on a trustworthy structural context, the wrapping re-design should be focused on lead compounds that fulfil the following two conditions:
cross reactivity must have been independently assessed by profiling the inhibitor for its affinity against a significant number of kinases;
the complexation of the inhibitor with a selected target protein is structurally reported in the PDB, so that the nonconserved packing defects in the target may be identified with full certainty.
A shortcoming in engineering drugs that wrap protein packing defects arises because a reliable identification of such defects requires three-dimensional structures of the protein target. This is a difficult problem for kinases, since they possess flexible motifs around the ATP-pocket [16], prone to adopt induced fits that can only be safely determined from spectroscopic data. Thus, to design drugs that wrap dehydrons within the protein flexible regions, a representative ensemble of crystal structures for kinase/ligand induced-fits is required [13].
Future developments in drug design will arise as we harness the novel experimental techniques developed by high-throughput screening pioneers David Lockhart and Patrick Zarrinkar [6]. These techniques allow an unprecedented level of drug-affinity profiling against hundreds of kinases, representing a major advance that will revolutionize drug discovery since it will enable a broad assessment of cross reactivity. We have already undertaken first steps to combine our wrapping design with the novel screening tool in order to improve the modulation of the inhibitory impact.
Drugs designed using the wrapping concept have already been tested in vitro and in animal models, and the wrapping modification of imatinib that curbs its cardiotoxicity will soon enter into the clinical-trial phase. Thus, the foundational steps for a molecular drug therapy based on targeting packing defects have been undertaken and every indication heralds its promising future as a translational platform to drug design that can minimize or control side effects.
Acknowledgments
The research of A. F. is supported by NIH grant R01-GM072614, by the John and Ann Doerr Fund for Computational Biomedicine (Program GC4R 2005) and by the Rice Computational Research Cluster funded by NSF under Grant CNS-0421109.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dancey J, Sausville EA. Issues and progress with protein kinase inhibitors for cancer treatment. Nat Rev Drug Discov. 2003;2:296–313. doi: 10.1038/nrd1066. [DOI] [PubMed] [Google Scholar]
- 2.Levitski A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 3.Donato NJ, Talpaz M. Clinical use of tyrosine kinase inhibitors: Therapy for chronic myelogenous leukemia and other cancers. Clin Cancer Res. 2000;6:2965–2966. [PubMed] [Google Scholar]
- 4.Tibes R, et al. Tyrosine kinase inhibitors and the dawn of molecular cancer therapeutics. Annu Rev Pharmacol Toxicol. 2005;45:357–384. doi: 10.1146/annurev.pharmtox.45.120403.100124. [DOI] [PubMed] [Google Scholar]
- 5.Gibbs J, Oliff A. Pharmaceutical research in molecular oncology. Cell. 1994;79:193–198. doi: 10.1016/0092-8674(94)90189-9. [DOI] [PubMed] [Google Scholar]
- 6.Fabian MA, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nature Biotechnology. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 7.Chen JP, et al. Molecular basis for specificity in the druggable kinome: sequence-based analysis. Bioinformatics. 2007;23:563–572. doi: 10.1093/bioinformatics/btl666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chemistry & Biology. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 9.Kerkela R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nature Medicine. 2006;12:908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 10.Druker BJ. Molecularly targeted therapy: have floodgates opened? Oncologist. 2004;9:357–360. doi: 10.1634/theoncologist.9-4-357. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, et al. Screening for small molecule inhibitors of insulin-like growth factor receptor (IGF-1R) kinase: comparison of homogeneous time-resolved fluorescence and 33P-ATP plate assay formats. J Exp Ther Oncol. 2004;4:111–119. [PubMed] [Google Scholar]
- 12.Wang Y, et al. Inhibition of insulin-like growth factor-I receptor (IGF-IR) signaling and tumor cell growth by a fully human neutralizing anti-IGF-IR antibody. Mol Cancer Ther. 2005;4:1214–1221. doi: 10.1158/1535-7163.MCT-05-0048. [DOI] [PubMed] [Google Scholar]
- 13.Fedorov O, et al. Insights for the development of specific kinase inhibitors by targeted structural genomics. Drug Discovery Today. 2007;12:365–372. doi: 10.1016/j.drudis.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Rellos P, et al. Structure and regulation of the human NEK2 centrosomal kinase. J Biol Chem. 2007;282:6833–6842. doi: 10.1074/jbc.M609721200. [DOI] [PubMed] [Google Scholar]
- 15.Shen K, et al. Protein kinase structure and function analysis with chemical tools. Biochim, Biophys Acta. 2005;1754:65–78. doi: 10.1016/j.bbapap.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 16.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 18.Noble MEM, et al. Protein kinase inhibitors: Insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 19.Bogoyevitch MA, Fairlie DP. A new paradigm for protein kinase inhibition: blocking phosphorylation without directly targeting ATP binding. Drug Discovery Today. 2007;12:622–633. doi: 10.1016/j.drudis.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Ohren JF, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–1197. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- 21.Barnett SF, et al. Identification and characterization of pleck-strin homology domain dependent and isozyme specific Akt inhibitors. Biochem J. 2004;385:399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma B, et al. Protein-protein interactions: Structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc Natl Acad Sci USA. 2003;100:5772–5777. doi: 10.1073/pnas.1030237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernández A, Maddipati S. A priori inference of cross reactivity for drug-targeted kinases. J Med Chem. 2006;49:3092–3100. doi: 10.1021/jm060163j. [DOI] [PubMed] [Google Scholar]
- 24.Fernández A, Scheraga HA. Insufficiently dehydrated hydrogen bonds as determinats of protein interactions. Proc Natl Acad Sci USA. 2003;100:113–118. doi: 10.1073/pnas.0136888100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernández A, Scott RL. Adherence of packing defects in soluble proteins. Phys Rev Lett. 2003;91:018102. doi: 10.1103/PhysRevLett.91.018102. [DOI] [PubMed] [Google Scholar]
- 26.Fernández A, Scott RL. Dehydron: A structurally encoded signal for protein interaction. Biophys J. 2003;85:1914–1928. doi: 10.1016/S0006-3495(03)74619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernández A. Keeping dry and crossing membranes. Nature Biotechnology. 2004;22:1081–1084. doi: 10.1038/nbt0904-1081. [DOI] [PubMed] [Google Scholar]
- 28.Fernández A, Berry RS. Molecular dimension explored in evolution to promote proteomic complexity. Proc Natl Acad Sci USA. 2004;101:13460–13465. doi: 10.1073/pnas.0405585101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernández A, et al. Structural defects and the diagnosis of amyloidogenic propensity. Proc Natl Acad Sci USA. 2003;100:6446–6451. doi: 10.1073/pnas.0731893100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernández A, Berry RS. Proteins with hydrogen-bond packing defects are highly interactive with lipid bilayers: Implications for amyloidogenesis. Proc Natl Acad Sci USA. 2003;100:2391–2396. doi: 10.1073/pnas.0335642100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deremble C, Lavery R. Macromolecular recognition. Curr Opin Struc Biol. 2005;15:171–175. doi: 10.1016/j.sbi.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 32.Ma B, et al. Comparison of the protein-protein interfaces in the p53-DNA crystal structures: Towards elucidation of the biological interface. Proc Natl Acad Sci USA. 2005;102:3988–3993. doi: 10.1073/pnas.0500215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajamani D, et al. Anchor residues in protein-protein interactions. Proc Natl Acad Sci USA. 2004;101:11287–11292. doi: 10.1073/pnas.0401942101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernández A, et al. Inhibitor design by wrapping packing defects in HIV-1 proteins. Proc Natl Acad Sci USA. 2004;101:11640–11645. doi: 10.1073/pnas.0404641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernández A. Incomplete protein packaging as a selectivity filter in drug design. Structure. 2005;13:1829–1836. doi: 10.1016/j.str.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernández A, et al. An anticancer c-Kit kinase inhibitor is re-engineered to make it more active and less cardiotoxic. J Clin Invest. 2007 doi: 10.1172/JCI32373. In press: publication date: December 3, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernández A, et al. Rational drug redesign to overcome drug resistance in cancer therapy: imatinib moving target. Cancer Res. 2007;67:4028–4033. doi: 10.1158/0008-5472.CAN-07-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pietrosemoli N, et al. Dehydration propensity of order-disorder intermediate regions in soluble proteins. J Proteome Res. 2007;6:3519–3526. doi: 10.1021/pr070208k. [DOI] [PubMed] [Google Scholar]
- 39.Schindler T, et al. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 40.Gambacorti-Passerini C, et al. Inhibition of the ABL kinase activity blocks the proliferation of BCR/ABL+ leukemic cells and induces apoptosis. Blood Cells Mol Dis. 1997;23:380–394. doi: 10.1006/bcmd.1997.0155. [DOI] [PubMed] [Google Scholar]
- 41.Attoub S, et al. The c-Kit tyrosine kinase inhibitor STI-571 for colorectal cancer therapy. Cancer Res. 2002;62:4879–4883. [PubMed] [Google Scholar]
- 42.Druker BJ. Circumventing resistance to kinase-inhibitor therapy. N Engl J Med. 2006;354:2594–2596. doi: 10.1056/NEJMe068073. [DOI] [PubMed] [Google Scholar]
- 43.Shah NP, et al. Dasatinib (BMS-354825) inhibits KITD816V, an imatinib-resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis. Blood. 2006;108:286–291. doi: 10.1182/blood-2005-10-3969. [DOI] [PubMed] [Google Scholar]
- 44.Schittenhelm MM, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66:473–481. doi: 10.1158/0008-5472.CAN-05-2050. [DOI] [PubMed] [Google Scholar]
- 45.White MF. Insulin Signaling in Health and Disease. Science. 2003;302:1710–1711. doi: 10.1126/science.1092952. [DOI] [PubMed] [Google Scholar]
- 46.Plum L, et al. Central insulin action in energy and glucose homeostasis. J Clin Invest. 2006;116:1761–1766. doi: 10.1172/JCI29063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitsiades CS, et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hemato-logic malignancies, and solid tumors. Cancer Cell. 2004;5:221–230. doi: 10.1016/s1535-6108(04)00050-9. [DOI] [PubMed] [Google Scholar]
- 48.Garcia-Echeverria C, et al. In vivo antitumor activity of NVP-AEW541- A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5:231–239. doi: 10.1016/s1535-6108(04)00051-0. [DOI] [PubMed] [Google Scholar]