

Abstract
Vitronectin and plasminogen activator inhibitor-1 (PAI-1) are important physiological binding partners that work in concert to regulate cellular adhesion, migration, and fibrinolysis. The high affinity binding site for PAI-1 is located within the N-terminal somatomedin B domain of vitronectin; however, several studies have suggested a second PAI-1-binding site within vitronectin. To investigate this secondary site, a vitronectin mutant lacking the somatomedin B domain (rΔsBVN) was engineered. The short deletion had no effect on heparin-binding, integrin-binding, or cellular adhesion. Binding to the urokinase receptor was completely abolished while PAI-1 binding was still observed, albeit with a lower affinity. Analytical ultracentrifugation on the PAI-1-vitronectin complex demonstrated that increasing NaCl concentration favors 1:1 versus 2:1 PAI-1-vitronectin complexes and hampers formation of higher order complexes, pointing to the contribution of charge-charge interactions for PAI-1 binding to the second site. Furthermore, fluorescence resonance energy transfer between differentially labeled PAI-1 molecules confirmed that two independent molecules of PAI-1 are capable of binding to vitronectin. These results support a model for the assembly of higher order PAI-1-vitronectin complexes via two distinct binding sites in both proteins.
First identified as serum-spreading factor, vitronectin was recognized as a protein in serum capable of promoting cellular adhesion and spreading (1). After these initial findings, further roles for vitronectin in maintaining hemostasis, wound healing, angiogenesis, and tumor metastasis have been elucidated based on the diverse number of ligands with which it interacts. Vitronectin works to regulate hemostasis via interactions with heparin and PAI-1.2 Vitronectin competes for heparin binding with both thrombin and antithrombin, effectively reducing the anticoagulant ability of heparin (2). Because PAI-1 inhibits plasminogen activators that convert plasminogen into the protease plasmin, vitronectin acts to prevent fibrinolysis by prolonging the functional lifespan of PAI-1 and localizing it to fibrin clots (3).
A high resolution structure of vitronectin has not been determined, although a model of the domains of vitronectin was proposed based on computer-calculated structure predictions using threading and docking algorithms (4). This model predicted one complete and one incomplete β-propeller for the central and C-terminal domains, respectively, that comprise the majority of vitronectin between amino acids 131 and 456. The structure of the N-terminal somatomedin B domain (residues 1–51) could not be reliably modeled with the threading algorithm. However, direct structural determinations using NMR (5–8) and x-ray crystallography (9) have elucidated the structure of the somatomedin B domain. Although there is some conflict among the structures that is rooted in the identification of disulfide bonds (6, 9–13), in all of the structures on the somatomedin B domain, the most important feature is a single turn α-helix that defines the binding interface with PAI-1. Small-angle x-ray scattering on monomeric vitronectin generated a low resolution model that revealed a bi-lobed structure for vitronectin (14). Assembling the NMR and x-ray scattering data with the structure predicted from threading suggests that the C-terminal domain lies within a separate lobe from the N-terminal somatomedin B domain (14).
PAI-1, the primary inhibitor of the plasminogen activators uPA and tissue plasminogen activator belongs to the serine protease inhibitor (serpin) superfamily. PAI-1 shares the common structural features and mechanism of inhibition characteristic of the other inhibitory members of the serpin family (15, 16). The tertiary structure of PAI-1 is composed of nine α-helices (hA–hI), three β-sheets (A, B, and C), and a solvent-exposed unstructured loop of ∼20 amino acids referred to as the reactive center loop, because it carries the inhibitory reactive peptide bond. The overall scaffold of the molecule is defined by the five-stranded central β-sheet A. PAI-1 spontaneously alters in conformation, converting from the active state to an energetically more favorable inactive latent state (16, 17, 19), where the reactive center loop translocates into the central β-sheet.
The interaction between vitronectin and PAI-1 has important consequences for the regulation of cellular adhesion to the extracellular matrix of the vasculature. Vitronectin binds a number of integrins as well as the GPI-linked urokinase receptor, uPAR, at sites in the region of the somatomedin B domain. When PAI-1 binds the somatomedin B domain of vitronectin, it directly competes for binding with uPAR at an overlapping binding site, thereby inhibiting uPAR-mediated cellular adhesion (20, 21). Integrin-mediated cellular attachment may also be affected by PAI-1 binding to the somatomedin B domain, presumably by blocking the RGD sequence (22), which is found at residues 45–47. PAI-1 has also been shown to detach cells by disrupting the binding of integrin-uPAR complexes to vitronectin, fibronectin, and collagen type-1 (23).
Because of the diverse roles of these proteins, investigating the nature of PAI-1 binding to vitronectin has been an important area of research. Much effort has been focused on defining the PAI-1-binding site within vitronectin. A large body of work has characterized the somatomedin B domain of vitronectin as the principal site of PAI-1 binding (21, 24–26). The PAI-1-binding site within the somatomedin B domain has been restricted to amino acids 24–30 using monoclonal antibodies and mutagenesis (24). Recent crystallographic (9) and NMR (5–7) structural studies have provided details about the PAI-1-binding site in the somatomedin B domain in the vicinity of a single-turn α-helix between residues 26 and 30. Conversely, the complementary vitronectin binding site within PAI-1 has been thoroughly investigated. A region between helices D, E, and F in PAI-1 termed the flexible joint region was proposed to comprise the primary binding site for vitronectin by Lawrence et al. (27) in 1994 and was finally fully mapped to this region by Jensen et al. (28) in 2002. The epitope was later confirmed by x-ray crystallography studies of the PAI-1-somatomedin B complex (9).
However, there is evidence that PAI-1 can bind to a region other than the somatomedin B domain of vitronectin. Competition experiments using synthetic peptides or monoclonal antibodies (29–32) and cleavage of vitronectin with proteases (33) have identified a PAI-1-binding site in the C-terminal region of vitronectin or a site in the connecting region (34). Studying vitronectin-PAI-1 complexes with analytical ultracentrifugation revealed a 2:1 PAI-1-vitronectin binding stoichiometry and the formation of higher order complexes at a PAI-1-vitronectin ratio of 4:2 (32, 35). These reports of alternative PAI-1-binding sites, coupled with the unusual stoichiometry of the vitronectin-PAI-1 complex, have caused us to pursue a deletion mutagenesis approach to address this problem. The studies reported here were designed to evaluate directly whether there is a second site for the binding of PAI-1 to vitronectin that lies outside the well characterized N-terminal site in the somatomedin B domain. A mutant form of vitronectin, rΔsBVN, was engineered omitting the first 40 amino acids, so that the mature peptide begins at lysine 41 upon secretion and removal of the signal sequence. These experiments confirm a second PAI-1-binding site within vitronectin that lies outside of the somatomedin B region.
EXPERIMENTAL PROCEDURES
Materials—Native vitronectin was purified from human plasma using a modified protocol of the method developed by Dahlback and Podack (36, 37). Multimeric vitronectin was prepared by denaturation of the protein in 8 m urea for 2 h at room temperature followed by dialysis into PBS (140 mm NaCl, 3 mm KCl, 10 mm Na2HPO4, 2 mm KH2PO4, pH 7.4). Recombinant human wild-type PAI-1 and recombinant stable PAI-1 mutant, 14-1B (38), used in the ELISA assays were both purchased from Molecular Innovations, Inc. PAI-1 used in the Biacore experiments and latency transition assays was produced according to the method outlined in Jensen et al. (28). The PAI-1 protein preparations were tested for activity using a urokinase inhibition assay; all behaved similarly and fully inhibited urokinase. The numbering used to denote the amino acid positions in PAI-1 throughout the presented work is Ser1-Ala2-Val3-His4-His5, according to Andreasen et al. (39). All other reagents were of analytical reagent grade or better.
Cell Culture—Cultures of Spodoptera frugiperda (Sf9) or Trichoplusnia ni (Hi5) cells (Invitrogen) were routinely grown as monolayers in Corning Ti 75-cm2 culture flasks at 27 °C or as suspension cultures in side-arm spinner flasks with gentle stirring at room temperature. Sf9 cells were grown in serum-free Ex-Cell 420 medium, whereas Hi5 cells were grown in Ex-Cell 401 (both from J. R. H. Biosciences) without the addition of serum. Rabbit smooth muscle cells (the kind gift of Dr. Daniel Lawrence, University of Michigan) were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum. U937 cells (American Type Culture Collection) were grown in RPMI 1640 media supplemented with 10% fetal bovine serum. U937 cells were stimulated for 24 h with 1 ng/ml human transforming growth factor β-1 (Sigma) and 50 nm 1α25-dihydroxyvitamin D3 (Calbiochem) (40).
Generation of Recombinant Vitronectin—Both full-length vitronectin and a deletion mutant of vitronectin lacking the first 40 amino acids were expressed in the baculovirus FastBac system (Invitrogen) as in the method described earlier (41). Both constructs were subcloned into the pFastBac-1 vector (Invitrogen) and utilized the endogenous vitronectin signal sequence to direct protein secretion. The deletion mutant, rΔsBVN, was generated using the following primers: ΔsB sense, 5′-GGCTGTCGACAAGCCCCAAGTGACTCGCGG-3′ and ΔsB antisense, 5′-GGGGCTTGTCGACAGCCAGAGCAACCCATG-3′. The C termini of both recombinant vitronectin and rΔsBVN have an additional 28 amino acids, containing an myc epitope and a 6× histidine tag. The manufacturer's guidelines were followed to transfect the insect cells for virus generation and subsequent protein expression. Both expressed recombinant vitronectin (rVN) and rΔsBVN were secreted into the medium, and clarified spent medium was passed over Chelating Sepharose-Fast Flow resin (Amersham Biosciences). The column was washed with binding buffer (5 mm imidazole, 0.5 m NaCl, 20 mm Tris, pH 7.9) followed by wash buffer (60 mm imidazole, 0.5 m NaCl, 20 mm Tris, pH 7.9). The bound protein was eluted with elution buffer containing 1 m imidazole. Fractions were analyzed by Western blotting and Coomassie staining. Fractions containing protein were pooled, dialyzed, and concentrated. Protein concentration was determined as described previously (41) by quantitative ELISA and the Pierce BCA Protein Assay. The protein was also subjected to N-terminal protein sequencing, confirming the deletion of the somatomedin B domain as well as cleavage of all but the last amino acid (valine) of the signal sequence. Over the course of these studies, rΔsBVN was expressed from the initial viral stock multiple times. All expressed protein was tested using PAI-1 and heparin binding assays to confirm there was no significant preparation to preparation variability.
Heparin Binding Assays—A direct heparin binding assay was used to measure vitronectin binding to heparin-coated microtiter plates as previously described (41). Briefly, wells were coated with a 1 mg/ml solution of heparin (Sigma) then blocked using 3% casein in PBS. The plates were incubated with serial dilutions of native, multimeric, and recombinant vitronectin. Bound vitronectin was detected using polyclonal antibodies to vitronectin and peroxidase-labeled anti-rabbit secondary antibodies (Vector Laboratories). Kinetic analysis of the rate of inactivation of thrombin by antithrombin in the presence of heparin and the recombinant proteins was measured by continuously monitoring the cleavage of the chromogenic thrombin substrate Chromozym-TH (Roche Applied Science) over time (42). Thrombin was diluted in reaction buffer (PBS with 0.1% polyethylene glycol 8000) to a concentration of 44 nm. A final concentration of 45 nm antithrombin was used in the assay, resulting in 70–80% inhibition of thrombin. Chromozym-TH and heparin (average molecular weight, Mr 6000) were included in the reaction at a final concentration of 0.19 nm and 0.9 μg/ml, respectively. The reactions were monitored at 405 nm for 3 min at 30 °C in 1-ml acrylic cuvettes (Sarstedt). The data were analyzed using the IGOR software package (Wavemetrics) to determine the pseudo-first-order reaction rates.
Cell Adhesion Assays—Two different cell lines, from muscular and monocyte origin, were used to assess the effect of deleting the N-terminal of vitronectin on binding to integrins and uPAR. Adherence of rabbit smooth muscle cells to recombinant vitronectin was determined by a method adapted from Stefansson and Lawrence (22), who showed this interaction to be primarily integrin-mediated. The wells of a 24-well tissue culture plate were coated overnight at 4 °C with a 10 nm solution of vitronectin or recombinant vitronectin diluted in PBS. After rinsing with PBS, the wells were blocked with 500 μl of a 3.5% solution of BSA in PBS at room temperature for 1 h. Rabbit smooth muscle cells were resuspended in serum-free medium containing 1.5% BSA to a concentration of 75,000 cells/well and allowed to adhere for 45 min. Bound cells were quantified by measuring cell surface acid phosphatase. After rinsing the wells to remove any unbound cells, 10 mg/ml p-nitrophenyl phosphate in 0.1 m sodium acetate, pH 5.0, was added to the wells. The reaction was stopped after 1 h with the addition of an equal volume of 1 m Tris, pH 9.0, and the absorbance was measured at 405 nm.
A human lymphoma cell line, U937, was also tested for uPAR-mediated adhesion to wild-type vitronectin and rΔsBVN. For the U937 cell binding assay, 140 nm vitronectin or a recombinant vitronectin derivative was added to the wells of a FluoroNunc plate with Maxisorb Surface microtiter plate (Nunc) suitable for fluorescent applications. Wells were blocked with a solution of 3% BSA in PBS. Stimulated U937 cells were fluorescently labeled by uptake of 10 μm calcein AM (Molecular Probes) for 30 min. Cells were washed two times with serum-free RPMI medium and resuspended at a concentration of 2 × 105 cells/well in serum-free RPMI plus 20 nm uPA (Calbiochem). After an incubation of 1 h at 37 °C in 5% CO2, the plate was rinsed with PBS to remove unbound cells. Cell binding was determined by measuring the fluorescence (ex 485/em 535) of the adhered cells in a Wallac Victor2 1420 Multilabel Counter.
Integrin Binding Assay—Microtiter plates were coated with 100 μl of a 2.5 μg/ml solution of the integrin GPIIbIIIa in binding buffer (50 mm Tris, pH 7.4, 0.1 m NaCl, 1 mm MgCl2, 1 mm CaCl2) according to the protocol described in a previous study (43). Wells were blocked with 3.5% BSA in binding buffer for 1 h at room temperature. Recombinant vitronectin was diluted to 500 nm in binding buffer plus 0.1% Tween and 0.2% BSA and serially diluted down the plate. After incubating for 1 h at 37 °C, the wells were rinsed and anti-vitronectin monoclonal antibody (Quidel, 1:10,000) was added to the wells for 1 h at room temperature. Peroxidase-labeled anti-mouse secondary antibody was added for 1 h at room temperature. The plates were developed using a 0.2 mg/ml solution of 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt in 50 mm sodium citrate, pH 5.5, containing 12 μl of 30% H2O2. Absorbance was read at 405 nm. All the following ELISA assays were developed and read in the same manner.
uPAR Binding Assay—The binding of uPAR to recombinant vitronectin was measured as described by Sidenius et al. (44) with slight modification. The wells of a microtiter plate were coated with multimeric vitronectin and recombinant vitronectin at a concentration of 15 nm in PBS. A 2:1 solution of uPAR (R & D Systems) and uPA (Calbiochem) in PBS containing 0.1% BSA and 0.1% Tween was mixed and allowed to incubate for 1 h at room temperature. Vitronectin-coated wells were blocked with 3% BSA in PBS, and the uPAR:uPA mixture (final concentration per well 60 and 30 nm, respectively) was added to the wells. Following the 1-h incubation, a polyclonal anti-uPAR antibody (Molecular Innovations) was added. The plate was developed as described above.
PAI-1 Binding Assays—Competition experiments were used to compare interactions between PAI-1 and native vitronectin or the recombinant vitronectin samples, as described by Seiffert and Loskutoff (25) with slight modification. Microtiter plates were coated with a 1 μg/ml solution of native vitronectin in PBS overnight at 4 °C. Wells were blocked with 3% BSA in PBS for 1 h at 37 °C then washed. PBS containing 0.1% BSA and 0.1% Tween 20 was used for the washes and for protein dilutions. Vitronectin or recombinant vitronectin was serially diluted on the plate following blocking. A constant concentration of either wild-type or stable PAI-1 (0.4 nm) was added to the wells, and the mixture was incubated at 37 °C for 2 h. The amount of PAI-1 that bound to the immobilized vitronectin was detected using polyclonal anti-PAI-1 antibodies.
Analysis of PAI-1 Binding to rΔsBVN by Surface Plasmon Resonance—All SPR experiments were performed on a BIACORE 3000™ instrument. Recombinant ΔsBVN was coupled directly to the surface of a CM5™ chip by standard amine coupling protocol by applying a 30 μg/ml vitronectin, 10 mm sodium acetate, pH 4.5, until a density of 600 response units accumulated on the chip. Ethanolamine blocked empty flow cells were used as a reference. When necessary, regeneration of the flow cells between runs was achieved by injecting pulses of 10 mm glycine/HCl, pH 2.0, and/or 0.05% SDS in running buffer until baseline levels were acquired. To determine the binding affinity for PAI-1 binding to rΔsBVN, 120-μl samples of active PAI-1 diluted in running buffer were injected in an array of concentrations from 0.25 to 1500 nm at a flow rate of 30 μl/min. The equilibrium dissociation constant (KD) was calculated according to the method outlined by Rich and Myszka (45). The sensorgram responses at equilibrium were plotted against PAI-1 concentration and fit by non-linear least squares to the equation:
 |
(Eq. 1) |
where R is the observed response, Rmax is the maximal response, C is the analyte concentration, and KD is the equilibrium dissociation constant.
Analytical Ultracentrifugation—Sedimentation velocity experiments were performed in a Beckman Optima XL-I ultracentrifuge with interference optics. For evaluating the effects of increasing salt concentrations on the vitronectin-PAI-1 complexes, vitronectin and wild-type PAI-1 were mixed (3.2 μm) and dialyzed against PBS with varying concentrations of NaCl (150, 250, and 500 mm). The samples were dialyzed for 2 h at 4 °C. The samples were allowed to equilibrate to room temperature before loading into the centerpieces. For the analytical ultracentrifuge experiments using the 14-1B PAI-1 variant, vitronectin, and 14-1B PAI-1 were dialyzed into PBS. Vitronectin and PAI-1 were mixed in a 1:1 molar ratio and incubated at 25 °C for 1 h. In both experiments, the samples were loaded (400 μl) into double-sector charcoal-filled Epon centerpieces. Sedimentation velocity was performed at 50,000 rpm at 25 °C in an An50 Ti eight-holed rotor. Interference scans were collected at 1-min intervals. Analysis of sedimentation velocity data on PAI-1-vitronectin complexes was performed using a Lamm equation distribution model that calculates the concentration of species of a given sedimentation coefficient (c(s)) with the program Sedfit (46). c(s) distributions were calculated over a range in sedimentation coefficients of 3–30, and an integration tool was used to evaluate the amount of signal (total fringes) corresponding to higher order PAI-1/vitronectin complexes (s values between 14 and 26).
Fluorescence Resonance Energy Transfer Measurements—FRET fluorescence measurements were made using a Varian Cary Eclipse spectrofluorometer at 25 °C. Individual spectra were collected in semi-micro quartz cuvettes (0.5 × 1.0 cm) as averages of 5 emission scans using bandwidths of 5 nm and 10 nm for the excitation and emission beams, respectively. All emission spectra were recorded with 0.1-s integration over a 1.0 nm step resolution. For the 5-iodoacetamidofluorescein (5-IAF) probe on the PAI-1P1′-FL variant, the excitation wavelength was 490 nm, and emission spectra were collected between 500 and 700 nm. Energy transfer between the 5-IAF probe on PAI-1P1′-FL and the tetramethylrhodamine-5-iodoacetamide (5-TMRIA) probe on PAI-1P1′-TMR was measured as the decrease in donor fluorescence intensity, because the signal obtained for the loss of donor fluorescence is typically greater than the re-emitted fluorescence after subtraction of the appropriate control spectra. Donor-acceptor fluorescence (FD-A), which corresponds to the fluorescence of the donor PAI-1P1′-FL in the presence of acceptor PAI-1P1′-TMR, was measured as the emission spectra of a 1:1 mixture of PAI-1P1′-FL (20 nm) and PAI-1P1′-TMR (20 nm) after incubation with monomeric vitronectin (20 nm). The raw fluorescence spectra were normalized for the contribution of non-FRET acceptor by subtraction of spectra from equivalent binding experiments containing a 1:1 mixture of labeled acceptor and unlabeled donor (wild-type PA1-1). The fluorescence of the donor probe only (FD) was resolved by measuring the fluorescence of binding reactions containing a 1:1 mixture of PAI-1P1′-FL and unlabeled wild-type PAI-1 after incubation at 25 °C with monomeric vitronectin. All fluorescence spectra for FRET measurements are averages of two to four independent experimental acquisitions.
The efficiency of FRET (E) between the 5-IAF and 5-TMRIA probes was calculated using the following expression (47, 48).
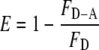 |
(Eq. 2) |
The distance between the two fluorophores coupled to PAI-1P1′-FL and PAI-1P1′-TMR, which participate in FRET, is then described by Equation 3,
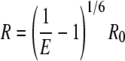 |
(Eq. 3) |
where R is the distance between the donor and acceptor probes, Ro is the distance at which there is a 50% transfer of the fluorescence energy from the donor probe to the acceptor probe, and E is the FRET efficiency defined by Equation 2, above (47, 48). The value of Ro is defined by the expression in Equation 4 (48, 49),
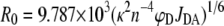 |
(Eq. 4) |
and is such that it is dependent on κ2, the orientation factor, which is routinely given a value of 2/3 indicating that both donor and acceptor fluorophores exhibit isotropic motion (47–49), n the experimentally determined refractive index of 1.3364 (using a Carl Zeiss ABBE-type refractometer), ϕD the experimental quantum yield of the donor fluorophore, and JDA the overlap integral for the donor and acceptor fluorophore pair, which was previously determined to be 3.06 × 10–13 m–1 cm3 (49). The quantum yield was measured by comparison of the emission spectrum of PAI-1P1′-FL to that of a fluorescein reference in 0.1 m NaOH (ϕ = 0.95) and evaluated as 0.143 by Equation 5,
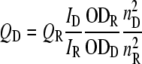 |
(Eq. 5) |
where QD and QR are the quantum yields of the donor and reference, respectively, ID and IR are the respective integrated fluorescence emission intensities over the 500 to 600 nm spectrum, ODD and ODR are the optical densities at the excitation wavelength of 490 nm, and the values n2D and n2R represent the refractive indices (determined as 1.3364 for both donor and reference samples).
RESULTS
A deletion mutant of vitronectin, rΔsBVN, was constructed to evaluate whether there is a second site for the binding of PAI-1 in vitronectin, which lies outside the well characterized N-terminal site in the somatomedin B domain. The first 40 amino acids of vitronectin were deleted, removing essentially all of the well structured disulfide cross-linked fold of the somatomedin B domain (typically delimited as residues 1–44), but retaining the RGD integrin-binding sequence found within a more flexible region at residues 45–47. The recombinant protein was produced using the baculovirus expression system as previously described (41). N-terminal sequencing confirmed the absence of the somatomedin B domain and cleavage of all but the final amino acid of the native signal sequence (valine). Gel-filtration chromatography of rΔsBVN showed that it was oligomeric (data not shown), as observed previously with recombinant full-length and C-terminal truncated vitronectin expressed without a tag in the baculovirus system (41). The presence of the His6 tag had no effect on oligomerization, because rΔsBVN expressed without the tag was also oligomeric (data not shown). Therefore, in assessing the functions of rΔsBVN, the deletion mutant was compared both to multimeric and monomeric plasma vitronectin.
Heparin Binding Activity of the Deletion Mutant Is Comparable to That of Multimeric Vitronectin—As a first step, the recombinant proteins were tested for the ability to bind heparin. The results shown in Fig. 1A demonstrate that the recombinant vitronectin and rΔsBVN bound equally well to immobilized heparin in a solid-phase binding assay. Binding of both the recombinant forms of vitronectin is similar to that of multimeric and much greater than that of native vitronectin, demonstrating the increased binding of the multimeric forms of vitronectin to heparin. This difference in the heparin-binding ability of monomeric and multimeric vitronectin has been attributed to the multivalent binding capacity of the multimeric form of vitronectin (50, 51).
FIGURE 1.

Recombinant forms of vitronectin bind to heparin. In a direct heparin-binding assay (A), a fixed concentration of heparin was immobilized in microtiter wells and blocked with casein. Varying concentrations of rVN (▴), rΔsBVN (□), native vitronectin (♦), and multimeric vitronectin (○) were incubated in the heparin-bound wells for 1 h. After rinsing the wells, bound vitronectin was detected using polyclonal anti-vitronectin antibodies and a peroxidase-labeled secondary antibody. Neutralization of the anticoagulant activity of heparin is followed in B. Heparin activity was measured by a decrease in thrombin activity due to inhibition by antithrombin. Thrombin activity was continuously monitored over time by measuring the hydrolysis of the chromogenic substrate Chromozym-TH. Variations in the Kapp were measured at various concentrations of vitronectin. Kapp rates were determined using the IGOR pro software and fitting the data using least-squares analysis. Kapp values were standardized to the reaction rate of heparin-catalyzed inhibition in the absence of vitronectin. Data points are shown at varying concentrations of multimeric vitronectin (○), rVN (▴), and rΔsBVN (□).
A solution-phase heparin neutralization assay was also performed to compare the heparin binding ability of the recombinant proteins. This assay measures the activity of thrombin in the presence of its inhibitor (antithrombin) and heparin (41, 42). Heparin acts as a catalyst in the reaction, bringing thrombin and antithrombin together more rapidly, resulting in increased inhibition of thrombin. When vitronectin is added to the mixture, it competes with both thrombin and antithrombin for heparin binding, reducing the rate of interaction between thrombin and antithrombin (the Kapp). The effects of adding varying concentrations of native vitronectin and recombinant vitronectin are shown in Fig. 1B. The concentrations of rVN and rΔsBVN needed to inhibit heparin activity by 50% were 0.04 and 0.2 μm, respectively. The corresponding value for multimeric vitronectin was 0.16 μm, consistent with previous measurements using this assay (41, 50). The Kapp of 0.04 μm for rVN was comparable to other recombinant forms of vitronectin previously expressed in the baculovirus system, which demonstrated 50% inhibition at 0.05 μm (41). The Kapp of the multimeric forms of vitronectin are all tighter than that of monomeric vitronectin with a Kapp of 0.6 μm (50). The results of the solid-phase and solution-phase heparin binding assays indicate that both rVN and rΔsBVN bind heparin with the same high valency interaction characteristic of the multimeric form of vitronectin.
The Recombinant Vitronectin, rΔsBVN, Binds Integrins—To further address the functionality of rΔsBVN, the ability of the protein to bind integrins and support cell adhesion was tested. The deletion mutant, rΔsBVN, was designed to ablate the N-terminal PAI-1-binding site, while leaving intact the integrin-binding RGD sequence between residues 45 and 47. Whereas some adhesive proteins, such as fibronectin, contain secondary integrin binding sites, it has been shown that the RGD sequence is the mediator of integrin binding to vitronectin (52).
To test the binding of integrins to the recombinant vitronectins, a solid-phase assay using purified integrins was performed (Fig. 2A). The integrin used in this assay was GPIIbIIIa, which is known to mediate the binding of activated platelets to vitronectin (53). Varied concentrations of the recombinant proteins were incubated with immobilized GPIIbIIIa, and the amount of bound vitronectin was measured. Both rVN and rΔsBVN bound GPIIbIIIa efficiently over a wide concentration range. Fig. 2B shows the results of the cell adhesion assay. Rabbit smooth muscle cells were allowed to attach to wells coated with equal molar concentrations of immobilized multimeric vitronectin, rVN, or rΔsBVN. After thoroughly washing the unbound cells from the wells, substrate was added to detect the number of bound cells using acid phosphatase as a surface marker. The cells bound equally well to rVN, rΔsBVN, and multimeric vitronectin. Taken together, these results demonstrate that the RGD site of rΔsBVN is exposed and available for integrin binding.
FIGURE 2.

Recombinant ΔsBVN binds integrins, but not uPAR. The recombinant ΔsBVN bound to immobilized integrin GPIIbIIIa (A) and supported the binding of rabbit smooth muscle cells (B). A, varying concentrations of recombinant vitronectin (▴) and recombinant ΔsBVN (□) were incubated for 1 h in GPIIbIIIa-coated wells. Wells were washed, and bound vitronectin was detected using a monoclonal anti-vitronectin antibody and a peroxidase-labeled secondary antibody. The binding of rabbit smooth muscle cells to vitronectin is shown in B. Equal molar amounts (8 nm) of multimeric vitronectin (solid bar), rVN (open bar), and rΔsBVN (shaded bar) were immobilized in the wells of a tissue culture plate. Following blocking of the wells with a solution of 3.5% BSA in PBS, rabbit smooth muscle cells were added and allowed to adhere for 1 h. The amount of cell binding was determined by following the conversion of ρ-nitrophenyl phosphate to ρ-nitrophenol by alkaline phosphatase on the cell at an absorbance of 405 nm. The binding of smooth muscle cells to blocked wells alone was negligible and was subtracted as background. The recombinant ΔsBVN does not bind to soluble uPAR (C) or support U937 cell binding (D). C, an equal molar concentration of multimeric vitronectin (solid bar), rΔsBVN (open bar), or rVN (shaded bar) was used to coat the wells of a microtiter plate. The wells were blocked with 3% BSA in PBS before incubation with soluble uPAR. The amount of bound uPAR after washing was determined using polyclonal anti-uPAR antibodies and a peroxidase-labeled secondary antibody. D, stimulated U937 cells were stained with calcein, resuspended in serum-free medium with 15 nm uPA, and added to the wells of a microtiter plate that had been coated with equal molar concentrations of multimeric vitronectin (solid bar), rΔsBVN (open bar), or rVN (shaded bar). Cell binding was determined by measuring the fluorescent intensity of the calcein-labeled cells after washing with PBS. U937 cell binding to BSA-blocked wells accounted for 4% (∼850,000 units) of the binding to multimeric vitronectin and was subtracted as background. In both panels, the percent binding was calculated relative to multimeric vitronectin.
Recombinant Vitronectin Lacking the Somatomedin B Domain Does Not Bind to uPAR—The adhesive functions of vitronectin are attributed not only to integrin binding but also to interactions with the GPI-linked urokinase receptor, uPAR. Because binding sites for PAI-1 and uPAR extensively overlap in the N-terminal domain, the deletion mutant likely eliminates vitronectin binding to this receptor. Again, this activity was tested with purified receptors and intact cells. Testing the binding of vitronectin to purified soluble uPAR (Fig. 2C), multimeric and recombinant full-length vitronectin bound to uPAR equally whereas rΔsBVN binding was negligible. The results using purified uPAR corroborate the findings with U937 cells. U937 cells adhered to vitronectin upon activation at which time uPAR was expressed and localized to the cell membrane. The cell binding data in Fig. 2D show that activated U937 cells adhered strongly to both multimeric vitronectin (solid bar) and rVN (shaded bar). However, there was essentially no binding of the U937 cells to rΔsBVN.
Wild-type and Stable PAI-1 Exhibit Differences in Binding to rΔsBVN—The results presented thus far demonstrate the suitability of using a truncated vitronectin expressed using the baculovirus system to study the function of the somatomedin B domain. One prominent goal of this project was to investigate the PAI-1-binding ability of rΔsBVN. A competitive binding assay was used in which varying concentrations of native vitronectin, multimeric vitronectin, rVN, and rΔsBVN were incubated with a fixed concentration of PAI-1; competition for binding of PAI-1 to solid-phase monomeric vitronectin was measured. Fig. 3A clearly shows binding of wild-type PAI-1 to rΔsBVN. Native and multimeric vitronectin as well as rVN effectively compete for wild-type PAI-1 binding, almost completely inhibiting PAI-1 binding to immobilized vitronectin.
FIGURE 3.

The recombinant ΔsBVN retains PAI-1-binding activity. Equal molar concentrations of native vitronectin (♦), multimeric vitronectin (○), rVN (▴), and rΔsBVN (□) were tested for binding to wild-type PAI-1 (A) or the 14-1B mutant form of PAI-1 (B) in a competitive assay where native vitronectin is immobilized in the wells of a microtiter plate. A fixed concentration of PAI-1 was added to wells in the presence of increasing concentrations of competing vitronectin. Bound PAI-1 was detected using a polyclonal anti-PAI-1 antibody. Percent binding inhibition was calculated using the equation, [(Ax – A0)/A0]*100, where A0 is the absorbance in the absence of competing vitronectin, and Ax is the absorbance at a given concentration of vitronectin.
A different vitronectin-binding behavior is observed with the stable PAI-1 mutant, 14-1B, compared with wild-type PAI-1 in the competitive binding assay. A unique property of PAI-1 among other serpins is its propensity to convert to a latent, inactive form that can no longer inhibit plasminogen activators. The binding of vitronectin to PAI-1 decreases the rate at which PAI-1 converts to this latent form. Stable forms of PAI-1 that do not collapse to the latent form at an appreciable rate have been engineered by incorporating amino acid replacements at a variety of loci, including the core β-sheet (16, 27, 38, 54, 55). The stable variant of PAI-1 that has been best characterized and has yielded the crystallographic structure of active PAI-1 (55) is the 14-1B mutant with the mutations N152H, K156T, Q321L, and M356I (16, 38). Although most functions are indistinguishable for wild-type and the stable 14-1B PAI-1, recent work suggests that there are subtle differences in structure and function (35, 56). For this reason, the stable 14-1B variant was compared with native PAI-1 in binding of full-length vitronectin and the deletion mutant, rΔsBVN. The results of the competitive PAI-1 binding using stable 14-1B PAI-1 are shown in Fig. 3B. Multimeric vitronectin, native vitronectin, and rVN inhibit the binding of stable PAI-1 to immobilized monomeric vitronectin. However, rΔsBVN is an ineffective competitor for PAI-1 binding in this assay, indicating a difference in affinity for 14-1B versus wild-type PAI-1 binding to rΔsBVN.
Affinity of Binding at the Secondary Site Is Measured by Equilibrium Analysis Using SPR—Initial SPR experiments confirmed the competitive PAI-1 binding ELISA data, showing binding of wild-type PAI-1 to rΔsBVN. To determine the equilibrium dissociation constant for PAI-1 binding to the second site of vitronectin only, varying concentrations of PAI-1 were injected across the captured rΔsBVN to produce the sensorgrams shown in Fig. 4A. The responses from the PAI-1 injections were concentration-dependent, and all reached equilibrium within ∼1 min. To extract a binding constant for this interaction, the responses at equilibrium were plotted against PAI-1 concentration (Fig. 4B). The data were fit to a simple 1:1 binding isotherm (Equation 1) and yielded a KD of 29 ± 3 nm.
FIGURE 4.

PAI-1 binds to the second site on vitronectin with lower affinity than the SMB domain. Representative sensorgrams obtained from the injection of varying concentrations of PAI-1 (1, 5, 10, 25, 50, 75, 250, and 500 nm) over immobilized rΔsBVN in a BIACORE 3000™ are shown in A. A duplicate series of sensorgram responses were normalized and plotted versus PAI-1 concentration (B). The points were fit to the 1:1 interaction model (Equation 1) to yield the KD of 29 ± 3 nm with an R2 value of 0.997.
Measurement of FRET between Differentially Labeled PAI-1 Molecules Indicates Binding to Two Sites on the Surface of Monomeric Vitronectin—Whereas the structure of a recombinant somatomedin B domain bound to PAI-1 has been solved, there are as yet no available experimental structures describing the relative orientation of the second PAI-1-binding site relative to the primary site in the somatomedin B domain within full-length vitronectin. Small-angle x-ray scattering shows that the C-terminal domain of vitronectin lies within a separate lobe from the N-terminal somatomedin B domain (14). FRET was chosen as a reasonable approach to test the relative proximity of the PAI-1-binding sites within the bi-lobed structure. For this approach, we reasoned that placement of the donor and acceptor fluorophores on the P1′ residue of the reactive center loop of PAI-1 would create a labeled variant that is known to be fully active (57) and yet provides the best arrangement in which the tethered probe will remain solvent-exposed and free from interactions with the bound vitronectin (58). To this end, we engineered a derivatizable cysteine residue at the P1′ position of the reactive center loop (M349C) and labeled the resulting PAI-1 variant with either 5-IAF or 5-TMRIA to act as a fluorescence donors (PAI-1P1-FL) or acceptors (PAI-1P1′-TMR), respectively.
Equal amounts of PAI-1P1-FL and PAI-1P1′-TMR were incubated with monomeric vitronectin at the requisite 2:1 ratio of PAI-1-vitronectin to ensure the formation of complexes in the sample that have both the high affinity and low affinity binding sites on vitronectin saturated, because conditions favoring formation of complexes that are not saturated at both sites would effectively decrease the concentration of available FRET pairs. Reaction mixtures were incubated at 25 °C for at least 5 min prior to initiating the acquisition of spectra to allow the formation of complexes to proceed to completion, based on our measured rates above. The observation of FRET between molecules of PAI-1P1′-FL and PAI-1P1′-TMR is evident from comparison of the fluorescence emission spectra obtained in the presence and absence of the acceptor PAI-1P1′-TMR, which demonstrated a 19.3% quench in donor fluorescence in the presence of acceptor (Fig. 5). Total donor-acceptor fluorescence (FD-A) and donor only fluorescence (FD) were calculated from integrations of the total peak fluorescence between 500 and 700 nm for each respective spectrum and subsequent evaluation of Equation 2 indicated an efficiency of energy transfer of 0.19 for the PAI-1P1′-FL/PAI-1P1′-TMR FRET pair co-localized on the surface of monomeric vitronectin. A calculated Ro of 45 Å for this donor and acceptor pair was determined from Equation 4 using an experimentally determined quantum yield of 0.143 for the PAI-1P1′-FL donor (Equation 5). The standard assumption that both the donor and acceptor probes exhibited isotropic motion was invoked, and therefore a value of 2/3 was valid for the orientation factor κ2 (47–49). This value, in combination with the above determination for the efficiency of FRET, was used to calculate the intermolecular distance between the fluorescence labels on PAI-1P1′-FL and PAI-1P1′-TMR when in a complex with monomeric vitronectin by evaluation of Equation 3, yielding a calculated distance of 57 Å.
FIGURE 5.

FRET reveals the co-localization of two PAI-1 molecules on plasma vitronectin. Fluorescence donor PAI-1P1′-FL (20 nm) was reacted with vitronectin (20 nm) in the presence of “cold” wild-type PAI-1 (20 nm, dashed line) or with acceptor PAI-1P1′-TMR (20 nm, solid line) to yield a final 2:1 ratio (PAI-1-vitronectin). Emission spectra were recorded following excitation at 490 nm and corrected for dilution and the contribution of residual acceptor PAI-1P1′-TMR fluorescence when excited at 490 nm.
Under the conditions of this experimental design (i.e. a 1:1 ratio of donor and acceptor fluorophores) and assuming two binding sites for PAI-1 on vitronectin, the statistical expectation is that only 50% of the binding events will have resulted in productive donor-acceptor FRET pairs. The remaining 50% of the binding events will presumably be equally distributed between donor-donor and acceptor-acceptor pairs. Whereas any acceptor-acceptor pairs would have a negligible contribution to the recorded fluorescence spectra, a donor-donor pair of PAI-1P1′-FL/PAI-1P1′-FL will result in fluorescence quenching due to homo-FRET between the two 5-IAF probes on account of the small Stokes shift found with the fluorescein fluorophore. The net result would be a reduction in the integrated intensity of the observed fluorescence spectra. Nonetheless, the magnitude of this effect on the spectra obtained in the presence and absence of acceptor, as well as the calculated values for FD-A and FD, should be equivalent for both conditions using this experimental design. Therefore, homo-FRET should not have any significant influence on the calculated intermolecular distance between donor and acceptor probes, because the calculation of energy transfer efficiency (Equation 2), which is used in the distance evaluation of Equation 3, is dependent on the relative ratio of FD-A/FD fluorescence.
Ionic Interactions Mediate the Secondary Vitronectin-PAI-1 Interaction—Analytical ultracentrifugation was performed to evaluate conditions favorable for formation of higher order PAI-1-vitronectin complexes. These experiments were adapted from the published analyses using sedimentation velocity experiments, which demonstrate that binding of PAI-1 in a 2:1 stoichiometry promotes association to higher order forms (35). The analyses of the data utilize a global fit to all scans taken during the course of the sedimentation velocity experiment and are plotted as a c(s) function (35, 46, 59), which is the distribution of species over a range of sedimentation coefficients, showing the concentration of the individual species. Thus, the binding of PAI-1 to both the somatomedin B domain and the secondary site appears to be required for higher order associations to ensue. For this study, sedimentation velocity experiments were conducted using varying concentrations of NaCl (Fig. 6). Equimolar mixtures of vitronectin and PAI-1 at a physiological salt concentration (150 mm) form a 2:1 PAI-1-vitronectin complex (∼6.5 S (35)). Raising the NaCl concentration favors the formation of 1:1 PAI-1-vitronectin complexes. At the highest salt concentration (500 mm), the PAI-1 and vitronectin complexes were solely 1:1 (∼5.5 S), whereas the 250 mm sample was a mixture of 1:1 and 2:1 PAI-1-vitronectin complexes. The sensitivity of the observed formation of higher order complexes indicates that occupancy of the second site on vitronectin is dependent on charge-charge interactions between vitronectin and PAI-1.
FIGURE 6.

Sedimentation velocity analysis of equimolar mixtures of vitronectin and PAI-1 at varying NaCl concentrations. Mixtures of 3.2 μm vitronectin and PAI-1 were evaluated by analytical ultracentrifugation using the sedimentation velocity method. Data were analyzed using the c(s) method using SEDFIT (46). Plots showing the distribution of species in each sample in the range of sedimentation coefficients from 4 to 10 S are shown for samples in 20 mm sodium phosphate containing 150 mm (solid line), 250 mm (hatched line), and 500 mm (dashed line) NaCl.
Active PAI-1 and the 14-1B Mutant Differ in Their Ability to Form Higher Order Complexes—Using a different approach, an evaluation was made of the need for occupation of both the somatomedin B domain and secondary site with PAI-1 for association to higher order complexes. Recalling that our binding assays show a weaker affinity of the rΔsBVN for the stable 14-1B PAI-1 mutant (Fig. 3B), it was reasoned that higher order complexes would form to a lesser extent upon binding to the stable mutant. The 14-1B mutant has been commonly used to mimic wild-type PAI-1, although there is accumulating evidence that the 14-1B mutant does not altogether recapitulate the structure and function of wild-type PAI-1 (56). Once again, sedimentation velocity analysis was used. Fig. 7A shows a typical analysis of the sedimentation equilibrium data on a mixture of vitronectin and wild-type PAI-1 using the continuous c(s) method to display the multiple species in the mixture (35, 46, 59). Consistently with our previous work (35), it is apparent from these data that wild-type PAI-1 promotes the formation of higher order complexes in addition to stoichiometric 1:1 and 1:2 vitronectin-PAI-1 complexes (35). Continuous c(s) distributions for mixtures of vitronectin with wild-type PAI-1 or the variant 14-1B in the region of s-values corresponding to these higher order species are shown in Fig. 7B. From this comparison, it is clear that the higher order species are more abundant in mixtures containing the native form of PAI-1 rather than the 14-1B mutant. The increase in species with s-values between 14 and 26 for both forms of PAI-1 is quantified in Fig. 7C. Not only is there a lower total concentration of these higher order complexes with the stable mutant form, but also the relative distribution across the range in s values is skewed so that there are diminishing amounts of complexes at higher s values with the stable mutant versus native PAI-1 (Fig. 7B).
FIGURE 7.

Analytical ultracentrifugation experiments demonstrate differences in higher order species formed by vitronectin with wild-type PAI-1 versus 14-1B PAI-1 variant. Mixtures of vitronectin (0.5 mg/ml) in a 1:1 molar ratio with wild-type PAI-1 or 14-1B PAI-1 were analyzed by sedimentation velocity (50,000 rpm) in a Beckman XL-I analytical ultracentrifuge. Data were analyzed using a continuous c(s) distribution model with the program SEDFIT (46). A, continuous c(s) distribution of complexes formed upon mixing of vitronectin with wild-type PAI-1. B, distribution profiles of higher order species formed between vitronectin and wild-type (solid line) or 14-1B stable mutant PAI-1 (dotted line). C, bar graph comparing the relative amounts of higher order species (s = 14–26) formed when vitronectin is mixed with either wild-type or 14-1B PAI-1. Peaks in the c(s) distribution in s = 14–26 range were integrated, and results were normalized relative to the total amount of protein loaded (concentrations are given in fringes). Error bars represent root mean square deviation values normalized to total loading concentration (fringes).
DISCUSSION
A Mutant Form of Vitronectin Lacking the Somatomedin B Domain Exhibits Normal Heparin- and Cell-binding Functions—Prokaryotic expression systems have been used extensively in the study of vitronectin function (9, 26, 60–62). The lack of post-translational modifications is one of the potential drawbacks in using these systems, especially with a protein like vitronectin that has numerous post-translational modifications, including disulfide bonds. The baculovirus system was chosen for expression of the vitronectin deletion mutant lacking the somatomedin B domain, because this system has been used successfully to express vitronectin (41, 52, 63, 64). Characterization of full-length vitronectin produced by the baculovirus cells determined it to be functionally similar to multimeric vitronectin (41). Significantly, the recombinant protein reacts with conformation-specific monoclonal antibodies and is generally considered to be a reasonable mimic of the form of vitronectin found in the extracellular matrix (41, 65, 66).
rΔsBVN was compared with monomeric and multimeric vitronectin using functional assays that span a broad range of activities. Based on the findings in two independent assays, there were no significant differences in the ability of rΔsBVN, rVN, and multimeric vitronectin to bind heparin. The RGD sequence important for integrin binding was accessible in the rΔsBVN, as concluded by the cell binding and integrin binding assays. For the cell binding assay, there was no appreciable difference observed for any of the vitronectin samples. Therefore, the determinants necessary to promote cellular adhesion are contained outside the somatomedin B domain and are accessible even in an oligomeric state. The integrin binding ability of the recombinant proteins was also investigated using an established vitronectin binding integrin, GPIIbIIIa (also referred to as αIIbβ3). This integrin, expressed on the surface of platelets, binds vitronectin and facilitates their adhesion to sites of injury during thrombus formation (67). Both of the recombinant proteins, rVN and rΔsBVN, exhibited similar binding to immobilized GPIIbIIIa. The results of these experiments indicate that the cell binding sequence (RGD) necessary for integrin adhesion is exposed in both rΔsBVN and rVN.
Cells attach to vitronectin in the extracellular matrix via their interactions with not only integrins but with uPAR as well. The binding of uPA to uPAR causes a conformational change in uPAR that increases its affinity for vitronectin (68). Because uPA is responsible for the generation of plasmin, this system is important in controlling extracellular proteolysis and cellular adhesion. The uPAR-binding site within vitronectin has been localized to the somatomedin B domain (21, 26, 69). Our results support this conclusion. There was no significant binding of either purified uPAR or activated U937 cells to rΔsBVN lacking the somatomedin B domain.
The Mutant Form of Vitronectin Missing the Somatomedin B Domain Exhibits Residual PAI-1 Binding—A high affinity binding site for PAI-1 has been localized to the somatomedin B domain. However, there are numerous reports that allude to PAI-1-binding sites in the connecting region (34, 70) or the heparin-binding domain (29–31, 33). These studies coupled with results from our laboratory that show two PAI-1 molecules will bind to one vitronectin molecule (32, 35) suggest there is a second PAI-1-binding site in vitronectin outside of the somatomedin B domain. By using a deletion mutant of vitronectin lacking the high affinity PAI-1-binding site, the somatomedin B domain, we were able to address the issue more directly. The result from a competitive PAI-1 binding assay (Fig. 3A) clearly demonstrates that wild-type PAI-1 binds rΔsBVN, because the rΔsBVN deletion mutant competes with native plasma vitronectin for binding to wild-type PAI-1. At saturating concentrations, the rΔsBVN deletion mutant inhibits binding of PAI-1 to full-length vitronectin by roughly 50%, consistent with PAI-1 binding to only one of two sites that is retained in the deletion mutant. The interaction between active wild-type PAI-1 and rΔsBVN is appreciable, as evidenced by a KD of 30 nm (∼2 orders of magnitude less than full-length vitronectin with the somatomedin B domain intact, which is ∼0.3 nm (26, 71–74)). The deletion mutant is multimeric; therefore, avidity may affect the PAI-1 binding and the KD may appear tighter due to multivalent display of these secondary PAI-1-binding sites. (The effect of multimeric vitronectin yielding apparent tighter binding to heparin has been attributed to this effect (51).)
The observation of two separable binding sites on vitronectin and PAI-1 was further substantiated by the use of FRET to demonstrate the binding to two molecules of PAI-1 at different binding sites on vitronectin. For this experiment, PAI-1 molecules were labeled with separate fluorophores that can serve as a donor and acceptor and the labeled PAI-1 molecules were mixed with vitronectin to test for FRET, which is distance-dependent and will occur if two PAI-1 are bound to the same molecule of vitronectin. These experiments were performed at concentrations of vitronectin and PAI-1 that are well below those that lead to assembly into higher order complexes to prevent any possibility that FRET could be attributed to intermolecular association and formation of a multimeric vitronectin scaffold to which PAI-1 is bound. Fluorescence measurements on the complexes demonstrated energy transfer, and the FRET signal was concentration-dependent, so that increasing the ratio of vitronectin to PAI-1 decreased the FRET signal, in effect sequestering PAI-1 on separate vitronectin molecules by binding to the high affinity site in the somatomedin B domain. The FRET measurements indicate the binding sites to be separated by ∼57 Å on the surface of vitronectin, which has an ellipsoid shape with a length of ∼110 Å (14).
Stable PAI-1 (14-1B) Displays an Altered Affinity Compared with Wild-type PAI-1 for the Somatomedin B Deletion Mutant—The proposal of two binding sites for PAI-1 on vitronectin has been controversial. Our original work in the ultracentrifuge that demonstrated a 2:1 stoichiometry for the PAI-1-vitronectin complex also contained data using two different monoclonal antibodies, mAB153 and mAB8E6, where each inhibited binding to vitronectin only by ∼50% (32). This result provided some of our first evidence for two sites, which were recognized as separate epitopes by the two monoclonal antibodies (32). However, this interpretation has been questioned by others who instead observed full inhibition of binding using either of the two antibodies; their suggestion involved mAB8E6 exerting steric effects that perturb the primary site in the somatomedin B domain, rather than defining a second PAI-1-binding site on vitronectin (26). However, these contrary experiments used different reagents than the ones we have employed in this study. Notably, the other experiments used denatured vitronectin, along with the stable mutant of PAI-1 (14-1B) (16, 38), which is often used by experimentalists and deemed to be equivalent to wild-type PAI-1. As seen in Figs. 3 and 7 our experiments have established differences in the binding affinities of the 14-1B stable mutant to the separate PAI-1-binding sites within vitronectin as evidenced in the competitive PAI-1 binding assay using vitronectin and rΔsBVN where full-length vitronectin binds 14-1B PAI-1 and the somatomedin B deletion mutant binds very weakly to 14-1B PAI-1. These results using this stable PAI-1 mutant suggest that it is the negligible affinity of 14-1B for the second site on vitronectin, outside of the somatomedin B domain, that accounts for the inconsistent results with antibodies noted in the experiment above.
These findings add to accumulating evidence that the well characterized 14-1B stable mutant of PAI-1 does not altogether recapitulate the structure and function of wild-type PAI-1. The differences noted between wild-type PAI-1 and the 14-1B mutant may be based on structural differences. In an elegant study that was recently published using donor-donor energy migration and dimer formation by BODIPY fluorophores, the reactive center loop of PAI-1 in solution was shown to be closely associated with the central β-sheet or partially inserted into β-sheet A rather than extended as suggested in crystallographic studies using the 14-1B mutant (56). The different conformations of the wild-type and the 14-1B stable PAI-1 mutant may be responsible for the differences in binding events described above and in the reported PAI-1 binding abilities in the literature.
Two Binding Sites on Vitronectin Support Stepwise Binding and Assembly of Complexes—Over the last several years, work from our laboratory has addressed the hypothesis that higher order PAI-1-vitronectin complexes form in a stepwise and concentration-dependent fashion via 1:1 and 2:1 intermediates, with the 2:1 complex serving a key role in assembly of higher order complexes. A variety of approaches has been used to test this idea, including sedimentation velocity experiments in the analytical ultracentrifuge that suggested that PAI-1 and vitronectin assemble into higher order forms via an ordered pathway that is triggered upon saturation of the two PAI-1-binding sites of vitronectin to form the 2:1 complex (35). This 2:1 PAI-1-vitronectin complex appeared to be an obligatory intermediate in the assembly of higher order complexes. The demonstration that 14-1B PAI-1 exhibits weaker binding to the second site that lies outside the somatomedin B domain agrees with this model, because this mutant promotes association to higher order complexes to a lesser extent compared with wild-type PAI-1.
The present work, which substantiates the idea of a second-PAI-1-binding site in vitronectin, suggests that a complementary second site should reside in PAI-1. Other work from our laboratory extends these results and defines the complementary binding site within PAI-1, a necessary feature that must exist for an ordered mechanism of binding and assembly of higher order complexes to be operable.3 Furthermore, we have used use pre-steady state kinetics to evaluate role of these dual sites in the mechanism of binding.4 Results from the kinetic measurements demonstrate two-step binding of PAI-1 to vitronectin. Significantly, this kinetic work demonstrates that the two-phase binding mechanism mediates the formation of stoichiometric 1:1 complexes of vitronectin and PAI-1; it is not limited to assembly of higher order complexes.
The verification of a second PAI-1-binding site on vitronectin apart from the N-terminal somatomedin B domain is important in constructing a unified picture of the interplay between PAI-1 and vitronectin that can account for their multiple roles in the circulation and the extracellular matrix. Both PAI-1 and vitronectin have different activities depending upon their localization. The anti-protease activity of PAI-1 in the circulation is targeted specifically at pericellular proteolysis when localized to the extracellular matrix or cell/matrix boundary. A paradox has emerged from studies in animal models that have shown dose-dependent effects of PAI-1 in regulating tumor-growth and angiogenesis (18, 75), with low and high concentrations giving opposite effects. There is currently no mechanism to explain why low concentrations of PAI-1 have anti-adhesive and anti-tumor effects, whereas high levels of PAI-1 are proadhesive and pro-tumorigenic. The availability of two binding sites for PAI-1 on vitronectin may help rectify this paradox. Thus, the dose-dependent PAI-1 effects may arise from the assembly of oligomeric vitronectin-PAI-1 that occurs when both binding sites are occupied. Clearly, further work is needed to test this hypothesis.
This work was supported by NHLBI, National Institutes of Health Grant HL50676. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PAI-1, plasminogen activator inhibitor type 1; uPA, urokinase-type plasminogen activator; uPAR, urokinase receptor; rVN, recombinant full-length vitronectin; rΔsBVN, recombinant vitronectin lacking the somatomedin B domain due to deletion of residues 1–40; mVN, multimeric vitronectin; PBS, phosphate-buffered saline; ELISA, enzyme-linked immunosorbent assay; BSA, bovine serum albumin; SPR, surface plasmon resonance; P1–P1′, Schecter and Berger nomenclature for the reactive center loop residues of PAI-1, where P1, P2, P3, P4,... and P1′, P2′, P3′, P4′,... denote those residues on the N-terminal and C-terminal sides of the scissile bond, respectively; 5-IAF, 5-iodoacetamidofluorescein; TMRIA, tetramethylrhodamine-5-iodoacetamide; PAI-1P1′-FL, PAI-1 labeled at the P1′ residue with 5-IAF; PAI-1P1′-TMR, PAI-1 labeled at the P1′ residue with TMRIA; GPI, glycosylphosphatidylinositol.
C. R. Schar, J. K. Jensen, A. Christenson, G. E. Blouse, P. A. Andreason, and C. B. Peterson, submitted for publication.
G. E. Blouse, D. M. Dupont, C. R. Schar, J. K. Jensen, K. H. Monor, J. Y. Anagli, H. Gardsvoll, M. Ploug, C. B. Peterson, and P. A. Andreasen, submitted for publication.
References
- 1.Holmes, R. (1967) J. Cell Biol. 32 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Preissner, K. T., and Muller-Berghaus, G. (1986) Eur. J. Biochem. 156 645–650 [DOI] [PubMed] [Google Scholar]
- 3.Podor, T. J., Campbell, S., Chindemi, P., Foulon, D. M., Farrell, D. H., Walton, P. D., Weitz, J. I., and Peterson, C. B. (2002) J. Biol. Chem. 277 7520–7528 [DOI] [PubMed] [Google Scholar]
- 4.Xu, D., Baburaj, K., Peterson, C. B., and Xu, Y. (2001) Proteins 44 312–320 [DOI] [PubMed] [Google Scholar]
- 5.Mayasundari, A., Whittemore, N. A., Serpersu, E. H., and Peterson, C. B. (2004) J. Biol. Chem. 279 29359–29366 [DOI] [PubMed] [Google Scholar]
- 6.Kamikubo, Y., De Guzman, R., Kroon, G., Curriden, S., Neels, J. G., Churchill, M. J., Dawson, P., Oldziej, S., Jagielska, A., Scheraga, H. A., Loskutoff, D. J., and Dyson, H. J. (2004) Biochemistry 43 6519–6534 [DOI] [PubMed] [Google Scholar]
- 7.Kamikubo, Y., Kroon, G., Curriden, S. A., Dyson, H. J., and Loskutoff, D. J. (2006) Biochemistry 45 3297–3306 [DOI] [PubMed] [Google Scholar]
- 8.Kjaergaard, M., Gardsvoll, H., Hirschberg, D., Nielbo, S., Mayasundari, A., Peterson, C. B., Jansson, A., Jørgensen, T. J. D., Poulsen, F., and Ploug, M. (2007) Prot. Sci. 16 1934–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou, A., Huntington, J. A., Pannu, N. S., Carrell, R. W., and Read, R. J. (2003) Nat. Struct. Biol. 10 541–544 [DOI] [PubMed] [Google Scholar]
- 10.Kamikubo, Y., Okumura, Y., and Loskutoff, D. J. (2002) J. Biol. Chem. 277 27109–27119 [DOI] [PubMed] [Google Scholar]
- 11.Horn, N. A., Hurst, G. B., Mayasundari, A., Whittemore, N. A., Serpersu, E. H., and Peterson, C. B. (2004) J. Biol. Chem. 279 35867–35878 [DOI] [PubMed] [Google Scholar]
- 12.Li, X., Zou, G., Yuan, W., and Lu, W. (2007) J. Biol. Chem. 282 5318–5326 [DOI] [PubMed] [Google Scholar]
- 13.Zhou, A. (2007) Prot. Sci. 16 1502–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lynn, G. W., Heller, W. T., Mayasundari, A., Minor, K. H., and Peterson, C. B. (2005) Biochemistry 44 565–574 [DOI] [PubMed] [Google Scholar]
- 15.Huber, R., and Carrell, R. W. (1989) Biochemistry 28 8951–8966 [DOI] [PubMed] [Google Scholar]
- 16.Stout, T. J., Graham, H., Buckley, D. I., and Matthews, D. J. (2000) Biochemistry 39 8460–8469 [DOI] [PubMed] [Google Scholar]
- 17.Mottonen, J., Strand, A., Symersky, J., Sweet, R. M., Danley, D. E., Geoghegan, K. F., Gerard, R. D., and Goldsmith, E. J. (1992) Nature 355 270–273 [DOI] [PubMed] [Google Scholar]
- 18.Devy, L., Blacher, S., Grignet-Debrus, C., Bajou, K., Masson, V., Gerard, R. D., Gils, A., Carmeliet, G., Carmeliet, P., Declerck, P. J., Noel, A., and Foidart, J. M. (2002) FASEB J. 16 147–154 [DOI] [PubMed] [Google Scholar]
- 19.Baker, D., and Agard, D. A. (1994) Biochemistry 33 7505–7509 [DOI] [PubMed] [Google Scholar]
- 20.Waltz, D. A., Natkin, L. R., Fujita, R. M., Wei, Y., and Chapman, H. A. (1997) J. Clin. Invest. 100 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng, G., Curriden, S. A., Wang, S., Rosenberg, S., and Loskutoff, D. J. (1996) J. Cell Biol. 134 1563–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stefansson, S., and Lawrence, D. A. (1996) Nature 383 441–443 [DOI] [PubMed] [Google Scholar]
- 23.Czekay, R. P., Aertgeerts, K., Curriden, S. A., and Loskutoff, D. J. (2003) J. Cell Biol. 160 781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Royle, G., Deng, G., Seiffert, D., and Loskutoff, D. J. (2001) Anal. Biochem. 296 245–253 [DOI] [PubMed] [Google Scholar]
- 25.Seiffert, D., and Loskutoff, D. J. (1991) J. Biol. Chem. 266 2824–2830 [PubMed] [Google Scholar]
- 26.Okumura, Y., Kamikubo, Y., Curriden, S. A., Wang, J., Kiwada, T., Futaki, S., Kitagawa, K., and Loskutoff, D. J. (2002) J. Biol. Chem. 277 9395–9404 [DOI] [PubMed] [Google Scholar]
- 27.Lawrence, D. A., Berkenpas, M. B., Palaniappan, S., and Ginsburg, D. (1994) J. Biol. Chem. 269 15223–15228 [PubMed] [Google Scholar]
- 28.Jensen, J. K., Wind, T., and Andreasen, P. A. (2002) FEBS Lett. 521 91–94 [DOI] [PubMed] [Google Scholar]
- 29.Gechtman, Z., Sharma, R., Kreizman, T., Fridkin, M., and Shaltiel, S. (1993) FEBS Lett. 315 293–297 [DOI] [PubMed] [Google Scholar]
- 30.Kost, C., Stuber, W., Ehrlich, H. J., Pannekoek, H., and Preissner, K. T. (1992) J. Biol. Chem. 267 12098–12105 [PubMed] [Google Scholar]
- 31.Preissner, K. T., Grulich-Henn, J., Ehrlich, H. J., Declerck, P., Justus, C., Collen, D., Pannekoek, H., and Muller-Berghaus, G. (1990) J. Biol. Chem. 265 18490–18498 [PubMed] [Google Scholar]
- 32.Podor, T. J., Shaughnessy, S. G., Blackburn, M. N., and Peterson, C. B. (2000) J. Biol. Chem. 275 25402–25410 [DOI] [PubMed] [Google Scholar]
- 33.Chain, D., Kreizman, T., Shapira, H., and Shaltiel, S. (1991) FEBS Lett. 285 251–256 [DOI] [PubMed] [Google Scholar]
- 34.Mimuro, J., Muramatsu, S., Kurano, Y., Uchida, Y., Ikadai, H., Watanabe, S., and Sakata, Y. (1993) Biochemistry 32 2314–2320 [DOI] [PubMed] [Google Scholar]
- 35.Minor, K. H., Schar, C. R., Blouse, G. E., Shore, J. D., Lawrence, D. A., Schuck, P., and Peterson, C. B. (2005) J. Biol. Chem. 280 28711–28720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bittorf, S. V., Williams, E. C., and Mosher, D. F. (1993) J. Biol. Chem. 268 24838–24846 [PubMed] [Google Scholar]
- 37.Dahlback, B., and Podack, E. R. (1985) Biochemistry 24 2368–2374 [DOI] [PubMed] [Google Scholar]
- 38.Berkenpas, M. B., Lawrence, D. A., and Ginsburg, D. (1995) EMBO J. 14 2969–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andreasen, P. A., Riccio, A., Welinder, K. G., Douglas, R., Sartorio, R., Nielsen, L. S., Oppenheimer, C., Blasi, F., and Dano, K. (1986) FEBS Lett. 209 213–218 [DOI] [PubMed] [Google Scholar]
- 40.Waltz, D. A., and Chapman, H. A. (1994) J. Biol. Chem. 269 14746–14750 [PubMed] [Google Scholar]
- 41.Gibson, A. D., and Peterson, C. B. (2001) Biochim. Biophys. Acta 1545 289–304 [DOI] [PubMed] [Google Scholar]
- 42.Peterson, C. B., Morgan, W. T., and Blackburn, M. N. (1987) J. Biol. Chem. 262 7567–7574 [PubMed] [Google Scholar]
- 43.Minor, K. H., and Peterson, C. B. (2002) J. Biol. Chem. 277 10337–10345 [DOI] [PubMed] [Google Scholar]
- 44.Sidenius, N., Andolfo, A., Fesce, R., and Blasi, F. (2002) J. Biol. Chem. 277 27982–27990 [DOI] [PubMed] [Google Scholar]
- 45.Rich, R. L., and Myszka, D. G. (2001) J. Mol. Recognit. 14 223–228 [DOI] [PubMed] [Google Scholar]
- 46.Schuck, P. (2000) Biophys. J. 78 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sivasothy, P., Dafforn, T. R., Gettins, P. G., and Lomas, D. A. (2000) J. Biol. Chem. 275 33663–33668 [DOI] [PubMed] [Google Scholar]
- 48.Lakowicz, J. R. (1999) Principles of Fluorescence Spectroscopy, Second Ed., pp. 367–378, Plenum, New York
- 49.Stratikos, E., and Gettins, P. G. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 453–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhuang, P., Chen, A. I., and Peterson, C. B. (1997) J. Biol. Chem. 272 6858–6867 [DOI] [PubMed] [Google Scholar]
- 51.Peterson, C. B. (1998) Trends Cardiovasc. Med. 8 124–131 [DOI] [PubMed] [Google Scholar]
- 52.Zhao, Y., and Sane, D. C. (1993) Arch. Biochem. Biophys. 304 434–442 [DOI] [PubMed] [Google Scholar]
- 53.Phillips, D. R., Charo, I. F., Parise, L. V., and Fitzgerald, L. A. (1988) Blood 71 831–843 [PubMed] [Google Scholar]
- 54.Stoop, A. A., Eldering, E., Dafforn, T. R., Read, R. J., and Pannekoek, H. (2001) J. Mol. Biol. 305 773–783 [DOI] [PubMed] [Google Scholar]
- 55.Sharp, A. M., Stein, P. E., Pannu, N. S., Carrell, R. W., Berkenpas, M. B., Ginsburg, D., Lawrence, D. A., and Read, R. J. (1999) Struct. Fold Des. 7 111–118 [DOI] [PubMed] [Google Scholar]
- 56.Hagglof, P., Bergstrom, F., Wilczynska, M., Johansson, L. B., and Ny, T. (2004) J. Mol. Biol. 335 823–832 [DOI] [PubMed] [Google Scholar]
- 57.Olson, S. T., Swanson, R., Day, D., Verhamme, I., Kvassman, J., and Shore, J. D. (2001) Biochemistry 40 11742–11756 [DOI] [PubMed] [Google Scholar]
- 58.Gibson, A., Baburaj, K., Day, D. E., Verhamme, I., Shore, J. D., and Peterson, C. B. (1997) J. Biol. Chem. 272 5112–5121 [DOI] [PubMed] [Google Scholar]
- 59.Schuck, P., Perugini, M. A., Gonzales, N. R., Howlett, G. J., and Schubert, D. (2002) Biophys. J. 82 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schvartz, I., Seger, D., Maik-Rachline, G., Kreizman, T., and Shaltiel, S. (2002) Biochem. Biophys. Res. Commun. 290 682–689 [DOI] [PubMed] [Google Scholar]
- 61.Yoneda, A., Ogawa, H., Kojima, K., and Matsumoto, I. (1998) Biochemistry 37 6351–6360 [DOI] [PubMed] [Google Scholar]
- 62.Seiffert, D., Ciambrone, G., Wagner, N. V., Binder, B. R., and Loskutoff, D. J. (1994) J. Biol. Chem. 269 2659–2666 [PubMed] [Google Scholar]
- 63.Seger, D., Gechtman, Z., and Shaltiel, S. (1998) J. Biol. Chem. 273 24805–24813 [DOI] [PubMed] [Google Scholar]
- 64.Wilkins-Port, C. E., Sanderson, R. D., Tominna-Sebald, E., and McKeown-Longo, P. J. (2003) Cell Commun. Adhes. 10 85–103 [DOI] [PubMed] [Google Scholar]
- 65.Seiffert, D., and Loskutoff, D. J. (1996) J. Biol. Chem. 271 29644–29651 [DOI] [PubMed] [Google Scholar]
- 66.Stockmann, A., Hess, S., Declerck, P., Timpl, R., and Preissner, K. T. (1993) J. Biol. Chem. 268 22874–22882 [PubMed] [Google Scholar]
- 67.Thiagarajan, P., and Kelly, K. L. (1988) J. Biol. Chem. 263 3035–3038 [PubMed] [Google Scholar]
- 68.Mondino, A., Resnati, M., and Blasi, F. (1999) Thromb. Haemost. 82 Suppl. 1, 19–22 [PubMed] [Google Scholar]
- 69.Kanse, S. M., Kost, C., Wilhelm, O. G., Andreasen, P. A., and Preissner, K. T. (1996) Exp. Cell Res. 224 344–353 [DOI] [PubMed] [Google Scholar]
- 70.Izumi, M., Shimo-Oka, T., Morishita, N., Ii, I., and Hayashi, M. (1988) Cell Struct. Funct. 13 217–225 [DOI] [PubMed] [Google Scholar]
- 71.Deng, G., Royle, G., Wang, S., Crain, K., and Loskutoff, D. J. (1996) J. Biol. Chem. 271 12716–12723 [DOI] [PubMed] [Google Scholar]
- 72.Seiffert, D., and Loskutoff, D. J. (1991) Biochim. Biophys. Acta 1078 23–30 [DOI] [PubMed] [Google Scholar]
- 73.Lawrence, D. A., Palaniappan, S., Stefansson, S., Olson, S. T., Francis-Chmura, A. M., Shore, J. D., and Ginsburg, D. (1997) J. Biol. Chem. 272 7676–7680 [DOI] [PubMed] [Google Scholar]
- 74.Jensen, J. K., Durand, M. K., Skeldal, S., Dupont, D. M., Bodker, J. S., Wind, T., and Andreasen, P. A. (2004) FEBS Lett. 556 175–179 [DOI] [PubMed] [Google Scholar]
- 75.Lambert, V., Munaut, C., Carmeliet, P., Gerard, R. D., Declerck, P. J., Gils, A., Claes, C., Foidart, J. M., Noel, A., and Rakic, J. M. (2003) Invest. Ophthalmol. Vis. Sci. 44 2791–2797 [DOI] [PubMed] [Google Scholar]