

Abstract
Hypothesis: Bile and pancreatic juice exclusion from gut activates acinar stress kinases and exacerbates gallstone pancreatitis as evidenced by ameliorating effects of replacement therapy in an experimental model of duct ligation-induced acute pancreatitis. In early stages of gallstone pancreatitis, bile-pancreatic juice cannot enter the gut. Enteral exclusion worsens pancreatitis by causing feedback hyperstimulation of the exocrine pancreas that activates acinar cell stress kinases. Investigations using a unique surgical model, the Donor Rat Model, showed that duodenal replacement of bile-pancreatic juice in rats with duct ligation attenuates pancreatic stress kinase activation, reduces pancreatic cytokine production, and ameliorates pancreatic morphologic changes. These findings suggest that exclusion-induced acinar hyperstimulation, in the presence of duct obstruction, exacerbates acute pancreatitis via stress kinase activation. Although acinar hyperstimulation has often been implicated in acute pancreatitis pathogenesis, the lack of supporting evidence remains a conspicuous lacuna. The proposed hypothesis draws on fresh evidence to present a new paradigm that re-examines the role of exocrine pancreatic hyperstimulation in gallstone pancreatitis pathogenesis.
Rationale
The most common cause of acute pancreatitis worldwide is gallstone impaction of the distal common bile-pancreatic duct1. As acute pancreatitis has already progressed considerably at the time of clinical presentation, experimental models are required to investigate mechanisms of disease pathogenesis. Duct ligation-induced acute pancreatitis in rats is a valuable experimental model of gallstone pancreatitis to study early events in disease pathogenesis. Duct ligation excludes bile-pancreatic juice from the gut. In the presence of an obstructed duct, bile-pancreatic juice exclusion-induced hyperstimulation of pancreatic digestive enzyme secretion imposes excessive stress on the acinar cell. Excess stress activates acinar stress kinase pathways that induce acinar production of acute inflammatory mediators. Once initiated, acute inflammation progresses by activation of several parallel series of pro-inflammatory signaling cascades and leukocyte responses.
Identity crisis
Several theories attempt to explain the pathogenesis of acute pancreatitis. The very existence of multiple theories for one disease provides prima facie evidence that little is understood about the pathogenesis. In terms of disease pathogenesis, is acute pancreatitis really one entity or does it comprise a group of distinct pathogenic entities, each the manifestation of a different etiology? From a pathogenic perspective, acute pancreatitis suffers from an identity crisis.
Final “Common” Pathway may not be that common
One school of thought suggests that, irrespective of the etiology that underlies an attack of acute pancreatitis, there exists a final “common” pathway within the pancreatic acinar cell that triggers acute pancreatic inflammation. However, the nature of this hypothetical pathway still eludes us. A fundamental problem with this approach is that it presumes the existence of a final “common” pathway in all forms of acute pancreatitis. What if a final “common” pathway does not actually even exist?
Leave no stone unturned
On the other hand, if we begin with the assumption that each etiology of acute pancreatitis has a different pathogenic pathway and investigate each separately, the approach may provide us a systematic way of looking at key events in disease pathogenesis. Applying this logic, the pathogenesis of gallstone-induced acute pancreatitis should be investigated separately from the pathogenesis of alcohol-induced acute pancreatitis. If a final “common” pathway does exist it would eventually be elucidated during the course of investigating each etiology separately. The proposed hypothesis pertains to the pathogenesis of gallstone pancreatitis.
The other side of the stone
In view of insufficient evidence to support the “hypersecretion” theory, the conventional approach has a bias towards events on the pancreatic side of the obstructing stone. “Bile reflux” into the pancreatic duct and “duct obstruction” have been implicated. However, pancreatic duct ligation in opossums – where bile reflux does not occur – is associated with necrotizing pancreatitis, signifying that bile reflux may after all have little relevance in disease pathogenesis1. The proposed hypothesis addresses several unanswered questions. Are there events on the duodenal side of the stone that are pertinent to disease pathogenesis? Does bile-pancreatic juice exclusion from gut and the consequent feedback hyperstimulation influence disease severity? Does bile exclusion interact with pancreatic juice exclusion to amplify the enteral response to exclusion and thus worsen pancreatitis? Do neural and hormonal pathways interact to impact acinar cell responses to hyperstimulation? Are duct obstruction and hyperstimulation dependent on each other for development of more severe acute pancreatitis?
Rolling stones gather trouble
Using observations from rats with ligation-induced acute pancreatitis2–4, let us attempt to describe a sequence of altered events from the point at which a stone rolls out of the gallbladder into the distal biliary tract and until the onset of acute pancreatic inflammation. A gallstone migrates from the gallbladder, rolls down the biliary tract, impacts at the ampulla of Vater, and occludes the distal common bile-pancreatic duct. At the time of duct occlusion, two important events occur: duct obstruction proximal to the point of occlusion and bile-pancreatic juice exclusion from the gut distal to the point of occlusion. Enteral exclusion of bile-pancreatic juice initiates feedback hyperstimulation of the exocrine pancreas via neuro-hormonal pathways. Exclusion-induced acinar hyperstimulation, in the presence of duct obstruction, imposes excessive stress on the acinar cell. The stress of this combined hyperstimulation-obstruction activates acinar cell stress kinase pathways that induce acinar cell production of acute inflammatory mediators. This rolling stone and its gathering storm, every so often, leaves behind a smoldering cauldron of cytokines, chemokines, oxygen-derived free-radicals, and inflammatory cells that conspire to demolish these revolting groups of acini (pancreatic necrosis) and damage innocent distant organs (systemic inflammation and multi-organ failure), with the possible ultimate outcome of taking the life of the very organism that harbors them (mortality).
Excess stress generates sparks
To illustrate the extreme stress created by the contradictory and opposing forces of hyperstimulation and obstruction, let us envision a car that is forced to accelerate at full-throttle against a sturdy garage door that refuses to open (Figure 1). The door does not budge, the vehicle does not move, all cylinders in full-revolution soon overheat from too much stress, sparks begin to fly, and the engine bursts into flames. Sparks - the first cytokines that initiate pancreatitis. Flame - the root word of “inflammation”.
Figure 1. Hyperstimulation against obstruction.

A car engine over-heated from forceful acceleration against an unyielding garage door illustrates acinar cell stress from hyperstimulation in the presence of an obstructed duct. In this diagrammatic representation of acinar hyperstimulation in gallstone pancreatitis, the engine going up in flames exemplifies acute inflammation of the pancreas.
Paradigm lost
For several decades, atropine – an inhibitor of exocrine pancreatic secretion – was routinely used in the initial treatment of acute pancreatitis. In 1979, a landmark study showed no improvement in outcomes and the practice was abandoned in view of the undesirable effects of atropinization5. Studies of cholecystokinin-A (CCK-A) receptor antagonists in experimental models of acute pancreatitis showed contradictory results6,7. The concept that dampening exocrine pancreatic stimulation could be beneficial in acute pancreatitis is clearly a well established idea, but few believe in it any longer. Consequently, the “hyperstimulation theory” was shelved based on the assumption that cell surface receptor antagonism is efficient even in the presence of acute inflammation.
Paradigm regained
If this assumption turned out to be inaccurate, it would imply a giant leap in logic. Receptor blockade efficiency elucidated in physiologic models (no acute inflammation) was extrapolated and applied in pathologic models (acute inflammation) without evidence that the antagonists are as effective in the latter setting. Indeed, there is evidence to the contrary. Using amylase activity in zymogen fractions of pancreas as an index of hyperstimulation, experiments have shown that combined CCK-A and cholinergic muscarinic receptor blockade completely reverses pancreatic hyperstimulation in rats with free external diversion via a bile-pancreatic fistula (physiologic state) but only shows a minimal inhibitory effect in duct ligation-induced acute pancreatitis4. Interestingly, in the same study, duodenal bile-pancreatic juice replacement using the Donor Rat Model completely reversed pancreatic hyperstimulation in both the physiologic and the pathologic state. In a separate study using the Donor Rat Model, duodenal bile-pancreatic juice replacement was associated with substantially greater amelioration of pancreatic morphological changes in duct ligation-induced acute pancreatitis compared to the marginal amelioration seen with combined CCK-A and cholinergic antagonism7. These experiments using the Donor Rat Model provide new evidence to propose a novel paradigm to explain the role of exocrine pancreatic hyperstimulation in the pathogenesis of ligation-induced acute pancreatitis in rats – the hyperstimulation-obstruction paradigm of sequential biological interactions.
Evolution of the Donor Rat Model
The initial experiment evaluated the role of bile exclusion in disease pathogenesis8. A surgical shunt between the proximal bile duct and duodenum in rats with duct ligation resulted in marginal improvement in pancreatic morphological changes and hypercholecystokininemia. The continued elevation in plasma CCK concentration and substantial residual pancreatic inflammatory changes in the shunted group were attributable to ongoing pancreatic juice exclusion in the presence of duct obstruction. Elegant physiological studies by Japanese investigators established a profound synergy between pancreatic juice exclusion and bile exclusion in feedback hyperstimulation of the exocrine pancreas9. In conscious rats without acute pancreatitis, they showed that combined external diversion of both bile and pancreatic juice has a synergistic rather than additive effect on hypercholecystokininemia and exocrine pancreatic protein secretion9. In the rat model of duct ligation-induced acute pancreatitis, it is not practically possible to shunt pancreatic juice into the duodenum without negating the physical effects of duct obstruction. The unique surgical model, the Donor Rat Model (Figure 2), circumvents these logistical obstacles2. The model provides a tool to test the hypothesis that combined replacement of both bile and pancreatic juice ameliorates ligation-induced acute pancreatitis to a greater extent than either bile replacement alone or pancreatic juice replacement alone2.
Figure 2. The Donor Rat Model.

Each donor rat was prepared with a bile-pancreatic fistula and a duodenal fistula, allowing external diversion and duodenal re-circulation of bile and pancreatic juice. Enteral feeding was instituted via a gastric fistula during a three-day post-laparotomy recovery period. When experiments were begun, bile and pancreatic juice were donated to an experimental (recipient) rat via a duodenal fistula beginning immediately before duct ligation. In an additional set of donor rats, a separate bile fistula and pancreatic juice fistula was created allowing experimental rats with duct ligation to receive either bile replacement alone or pancreatic juice replacement alone2 (not shown here).
The hyperstimulation-obstruction paradigm of sequential biological interactions
Experiments suggest that three biological interactions sequentially exacerbate ligation-induced acute pancreatitis in rats (Figure 3):
Figure 3. Paradigm of sequential biological interactions.
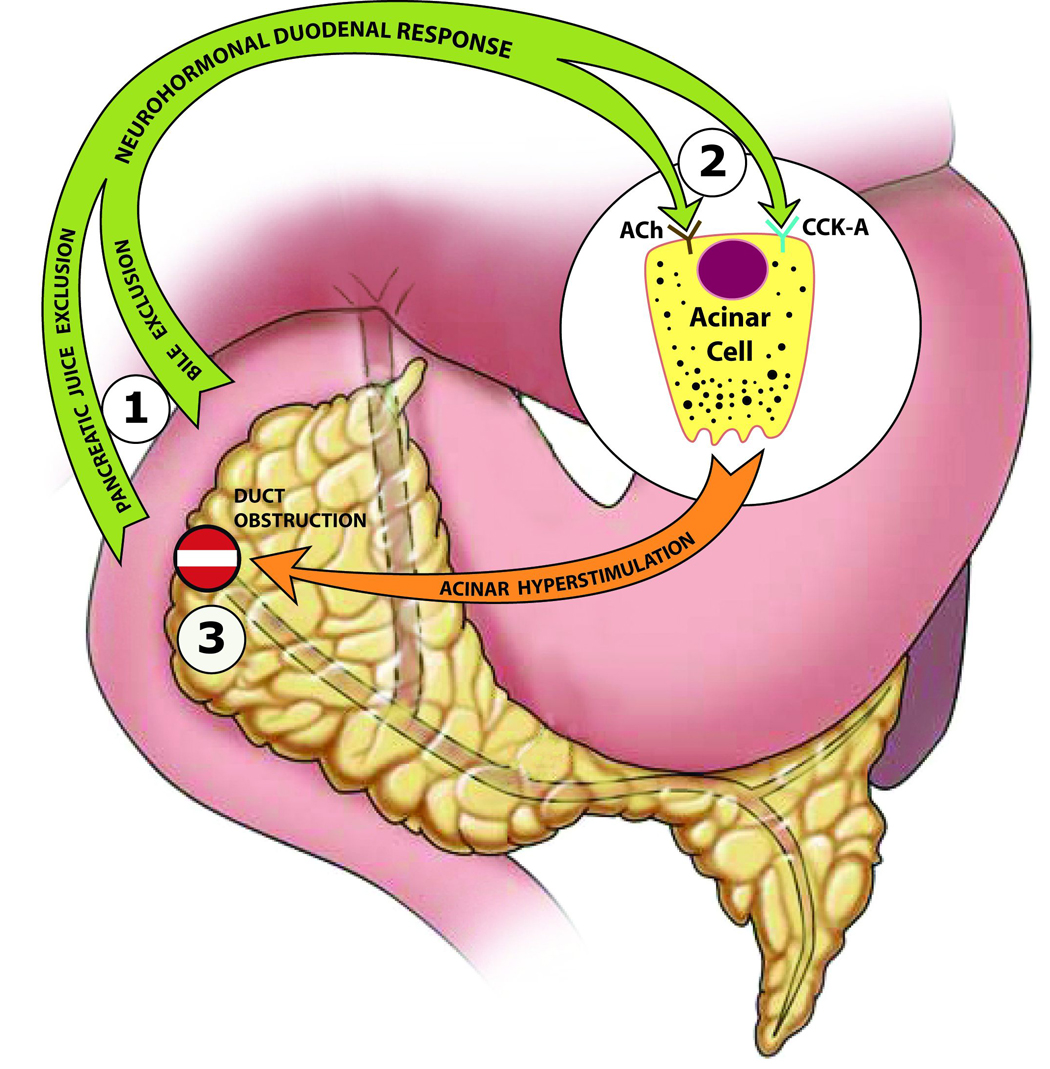
This is a diagrammatic representation of a paradigm for early events in the pathogenesis of duct ligation-induced acute pancreatitis in rats. Three sequential biological interactions (circled numbers) involving hyperstimulation delineate salient extracellular events that affect the pancreatic acinar cell environment in the rat model: 1) A synergistic interaction between pancreatic juice exclusion and bile exclusion augments the neuro-hormonal duodenal response to bile-pancreatic juice exclusion, 2) An interaction between CCK-A receptor stimulation and cholinergic (ACh) muscarinic receptor stimulation further increases acinar hyperstimulation, and 3) An interaction between acinar hyperstimulation and duct obstruction (“No Entry” sign) imposes excess stress on the acinar cell, activating stress kinases that induce production of acute inflammatory mediators.
The First Interaction: Using the Donor Rat Model, combined replacement of bile and pancreatic juice was associated with greater attenuation of hypercholecystokininemia and amelioration of pancreatic morphological changes after duct ligation than with replacement of either bile or pancreatic juice alone2. This indicates that a synergistic interaction between bile exclusion and pancreatic juice exclusion exacerbates ligation-induced acute pancreatitis.
The Second Interaction: Combined CCK-A and muscarinic receptor antagonism was associated with a significant amelioration of pancreatic morphologic changes after duct ligation, while CCK-A receptor antagonism alone or muscarinic receptor antagonism alone had relatively minimal effects7. This indicates that an interaction between CCK-A receptor stimulation and muscarinic receptor stimulation exacerbates ligation-induced acute pancreatitis.
The Third Interaction: Exclusion alone (free external diversion of bile-pancreatic juice without duct obstruction) does not cause acute pancreatitis, obstruction alone (duct ligation with bile-pancreatic juice replacement from a donor rat) is associated with only mild pancreatitis, while combined duct obstruction and bile-pancreatic juice exclusion is associated with acute pancreatitis of greater severity2,4. This indicates that an interaction between exclusion-induced acinar hyperstimulation and duct obstruction exacerbates acute pancreatitis2,4,7.
Genesis of the central hypothesis
When considered in sequence, the three biological interactions afford a logical progression of certain salient events in early stages of disease pathogenesis and provide a map for further investigation to explore these uncharted waters. Bile exclusion when combined with pancreatic juice exclusion augments the duodenal neurohormonal response to exclusion. Exclusion-induced acinar hyperstimulation is further enhanced by interactions between neural and hormonal G protein-coupled receptors (GPCRs) (e.g., CCK-A, muscarinic, serotonin, and secretin receptors). Acinar hyperstimulation, in the presence of duct obstruction, imposes excessive stress on the acinar cell and activates stress kinases such as p38, c-Jun N-terminal Kinase (JNK), and Extracellular signal-Regulated Kinase (ERK)3,10. The stress kinases activate nuclear transcription factors (e.g., nuclear factor kappa-B or NF-kB) that induce production of pro-inflammatory cytokines. Cytokines such as tumor necrosis factor-alpha and interleukin-1-beta initiate, maintain and propagate acute inflammation in the exocrine pancreas and eventually even in distant organs. Stress kinase inhibitors attenuate caerulein-stimulated overproduction of cytokines and chemokines by pancreatic fragments in vitro10. In ligation-induced acute pancreatitis, progressive activation of pancreatic stress kinases, activation and nuclear translocation of NF-kB, and overproduction of cytokines and chemokines is ameliorated by duodenal bile-pancreatic juice replacement from a donor rat. Taken together, these findings allow us to generate a central hypothesis for the early stages of pathogenesis of ligation-induced acute pancreatitis in the rat model: Enteral bile and pancreatic juice exclusion-induced exocrine pancreatic hyperstimulation activates acinar cell stress kinases that exacerbate duct occlusion-induced acute pancreatitis.
Luminal messengers of the enteral response to exclusion
In rats, a luminal CCK-releasing peptide is degraded by trypsin digestion when trypsin enters the enteral lumen in sufficient quantities from a normally secreting pancreas11. When luminal trypsin levels fall, as when the bile-pancreatic duct is ligated, the levels of the CCK-releasing peptide increase in the enteral lumen and stimulate I-cells within the duodenal mucosa to secrete CCK. Gut hormones other than CCK (e.g., serotonin, secretin), neurotransmitters other than acetylcholine (e.g., vasoactive intestinal peptide, gastrin-releasing peptide, substance P, CCK, neurotensin), vago-vagal reflexes, neural-hormonal interactions and hormonal-hormonal interactions may all contribute to stimulation of the exocrine pancreas7,12. Duodenal absence of bile salts has an indirect effect on the response by destabilizing the trypsin molecule and a direct effect via cholinergic mechanisms2. This explains the synergistic interaction between pancreatic juice exclusion and bile exclusion in feedback hyperstimulation of the exocrine pancreas. Consistent with this explanation, combined replacement of trypsin and Na-taurocholate showed similar improvement of pancreatic morphological changes as that achieved by combined bile-pancreatic juice replacement from a donor rat, while replacement of trypsin alone or Na-taurocholate alone failed to achieve statistically significant improvement7.
Implications, ramifications, extrapolations
The hyperstimulation-obstruction paradigm proposed here justifies current protocols for gallstone pancreatitis that recommend endoscopic extraction of stones impacted at the ampulla. Stone extraction relieves duct obstruction and facilitates flow of natural bile and pancreatic juice to the enteral lumen. Patients in whom the stone has already passed could potentially still have occlusive ampullary edema that may aggravate pancreatic inflammation by perpetuating obstruction and exclusion. Furthermore, acute pancreatitis triggered by a gallstone or any other cause is associated with subdued acinar cell function (secretory block), raising the intriguing possibility that intra-cellular obstruction and pancreatic enzyme exclusion may form part of a final “common” pathway that continues to activate stress kinases and thus fans the flames of acute pancreatic inflammation. An extrapolation of this logic raises the question whether enteral replacement therapy may be beneficial in patients with acute pancreatitis initiated by gallstones, and possibly even other etiologies. After all, replacement therapy has improved symptoms in selected patients with chronic pancreatitis, suggesting that some parallels may be drawn in the pathogenic mechanisms involved in the two entities.
Criticism and controversy related to the proposed hypothesis
What about bile reflux, trypsinogen activation, and pancreatic autodigestion?
Since the early twentieth century, conventional thinking has maintained that bile reflux and pancreatic autodigestion by trypsin are central to the pathogenesis of gallstone pancreatitis. A century later, is it rational to examine the possibility that gallstone pancreatitis can develop without reflux of bile into the pancreatic duct? Is trypsinogen activation an effect rather than the cause of pancreatitis? Is active trypsin essential for development of acute pancreatitis or is it merely a secondary factor that exacerbates pancreatitis?
How “human” are rats?
The Achilles' heel of the proposed hypothesis is the potential dissimilarity between rodents and humans in species-specific physiology and behavior. For instance, if rats were more human they would have a gallbladder, create gallstones, consume alcohol in company or in solitude, and become inflicted with acute hemorrhagic necrotizing mesmerizing pancreatitis. To the best of our knowledge, rats neither have a gallbladder nor voluntarily get intoxicated. Conversely, how different are humans from rodents with regards to regulation of exocrine pancreatic secretion? Interestingly, there is accumulating evidence that human pancreatic acinar cells do not harbor CCK-A receptors – an observation that could potentially cast a pessimistic shroud of doubt over the proposed hypothesis. On the other hand, there is also evidence that CCK-A receptors in humans may actually regulate acinar digestive enzyme secretion in a surrogate manner via cholinergic pathways rather than directly12. Furthermore, GPCRs other than the CCK-A receptor may play the predominant role in enteral exclusion-induced acinar hyperstimulation in humans. In other words, hyperstimulation can be implicated in disease pathogenesis irrespective of the specific cell surface receptors incriminated. Therefore, it is reasonable to advocate further experimental and clinical studies to evaluate the central hypothesis “Acinar cell stress associated with enteral exclusion-induced acinar hyperstimulation in the presence of duct obstruction exacerbates acute pancreatic inflammation during the early stages of gallstone-induced acute pancreatitis.”
ACKNOWLEDGMENTS
Professor Michael J. McMahon, PhD, ChM, FRCS, General Infirmary at Leeds, Leeds University, Leeds, UK, who stimulated my interest in acute pancreatitis research; who, along with other surgical colleagues, opened the doors of academic surgery for me in 1987. Dr. David L. Nahrwold and Dr. Raymond J. Joehl for providing me the opportunity and mentoring me during my Research Fellowship in acute pancreatitis basic science studies at the Department of Surgery, Northwestern University Feinberg School of Medicine, Chicago, IL, 1991–1994. Dr. Richard A. Prinz for nurturing my enthusiasm for acute pancreatitis research and providing me general surgery training at the Department of Surgery, Rush University Medical Center, Chicago, IL, 1994–1999.
Financial support The author received research awards from: National Pancreas Foundation, Boston, MA, Research Grant, 2003; American College of Surgeons, Chicago, IL, Faculty Research Fellowship Award 2003–2005; National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, Clinical Scientist Career Development Award (Grant #K08-DK062805), 2003–2007.
ILLUSTRATIONS All diagrams were drawn by the departmental medical illustrator, Mr. Thomas Weinzerl.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Steer ML. Pathobiology of Experimental Acute Pancreatitis. The Yale Journal of Biology and Medicine. 1992;65:421–430. [PMC free article] [PubMed] [Google Scholar]
- 2.Samuel I, Toriumi Y, Wilcockson DP, Turkelson CM, Solomon TE, Joehl RJ. Bile and pancreatic juice replacement ameliorates early ligation- induced acute pancreatitis in rats. Am. J. Surg. 1995;169(4):391–399. doi: 10.1016/s0002-9610(99)80183-4. [DOI] [PubMed] [Google Scholar]
- 3.Samuel I, Zaheer S, Zaheer A. Bile-pancreatic juice exclusion increases p38MAPK activation and TNF-alpha production in ligation-induced acute pancreatitis in rats. Pancreatology. 2005;5(1):20–26. doi: 10.1159/000084486. [DOI] [PubMed] [Google Scholar]
- 4.Samuel I, Joehl RJ. Bile-pancreatic juice replacement, not cholinergic and cholecystokinin-receptor blockade, reverses acinar cell hyperstimulation after bile-pancreatic duct ligation. Am. J. Surg. 1996;171(1):207–211. doi: 10.1016/S0002-9610(99)80101-9. [DOI] [PubMed] [Google Scholar]
- 5.Cameron JL, Mehigan D, Zuidema GD. Evaluation of atropine in acute pancreatitis. Surg Gynecol Obstet. 1979;148:206–208. [PubMed] [Google Scholar]
- 6.Gomez G, Townsend CM, Green D, Rajaraman S, Uchida T, Thompson JC. Involvement of cholecystokinin receptors in the adverse effect of glucocorticoids on diet-induced necrotizing pancreatitis. Surgery. 1989;106(2):230–238. [PubMed] [Google Scholar]
- 7.Samuel I, Toriumi Y, Zaheer A, Joehl RJ. Mechanism of acute pancreatitis exacerbation by enteral bile-pancreatic juice exclusion. Pancreatology. 2004;4(6):527–532. doi: 10.1159/000080527. [DOI] [PubMed] [Google Scholar]
- 8.Toriumi Y, Samuel I, Wilcockson DP, Turkelson CM, Solomon TE, Joehl RJ. Increased circulating CCK in obstruction-induced acute pancreatitis: II - Pancreatic duct obstruction with and without bile duct obstruction. J Surg Res. 1993;54:132–135. doi: 10.1006/jsre.1993.1020. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura R, Miyasaka K, Funakoshi A, Kitani K. Interactions between bile and pancreatic juice diversions on cholecystokinin release and pancreas in conscious rats. Proc Soc Exp Biol Med. 1989;192:182–186. doi: 10.3181/00379727-192-42976. [DOI] [PubMed] [Google Scholar]
- 10.Samuel I, Zaheer A, Fisher RA. In Vitro Evidence for Role of ERK, p38, and JNK in Exocrine Pancreatic Cytokine Production. J. Gastrointest. Surg. 2006;10(10):1376–1383. doi: 10.1016/j.gassur.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Miyasaka K, Funakoshi A. Luminal feedback regulation, monitor peptide, CCK-releasing peptide, and CCK receptors. Pancreas. 1998;16(3):277–283. doi: 10.1097/00006676-199804000-00012. [DOI] [PubMed] [Google Scholar]
- 12.Owyang C, Logsdon CD. New insights into neurohormonal regulation of pancreatic secretion. Gastroenterology. 2004;127(3):957–969. doi: 10.1053/j.gastro.2004.05.002. [DOI] [PubMed] [Google Scholar]