

Abstract
Arterial hyperpolarization to ACh reflects co-activation of KCa3.1 (IKCa)-channels and KCa2.3 (SKCa)-channels in the endothelium, that transfers through myoendothelial gap junctions (MEGJs) and diffusible factor(s) to affect smooth muscle relaxation (EDHF response). However, ACh can differentially activate KCa3.1 and KCa2.3-channels and we investigated the mechanisms responsible in rat mesenteric arteries. KCa3.1-channel input to EDHF-hyperpolarization was enhanced by reducing external [Ca2+]o, but blocked either with forskolin to activate PKA or by limiting smooth muscle [Ca2+]i increases stimulated by phenylephrine (PE) depolarization. Imaging [Ca2+]i within the endothelial cell projections forming MEGJs, revealed increases in cytoplasmic [Ca2+]i during endothelial stimulation with ACh that were unaffected by simultaneous increases in muscle [Ca2+]i evoked by PE. If gap junctions were uncoupled, KCa3.1-channels became the predominant input to EDHF-hyperpolarization, and relaxation was inhibited with ouabain implicating a crucial link through Na+/K+-ATPase. There was no evidence for an equivalent link through KCa2.3-channels, nor between these channels and the putative EDHF-pathway involving natriuretic peptide receptor-C (NPR-C). Reconstruction of confocal z-stack images from pressurized arteries revealed KCa2.3-immunostain at endothelial cell borders, including endothelial cell projections, while KCa3.1-channels and Na+/K+-ATPase α2/α3-subunits were highly concentrated in endothelial cell projections and adjacent to MEGJs. Thus, extracellular [Ca2+]o appears to modify KCa3.1-channel activity through a PKA-dependent mechanism independent of changes in endothelial [Ca2+]i. The resulting hyperpolarization links to arterial relaxation largely through Na+/K+-ATPase, possibly reflecting K+ acting as an EDHF. In contrast, KCa2.3-hyperpolarization appears mainly to affect relaxation through MEGJs. Overall, these data suggest that K+ and myoendothelial coupling evoke EDHF-mediated relaxation through distinct, definable pathways.
Keywords: Potassium channel, endothelial cells, hyperpolarization, membrane potential, electrophysiology, vasodilation
Introduction
The importance of the arterial endothelium for relaxation of the subjacent smooth muscle is well established. Whatever the final endothelium-derived effector, a key event is an initial increase in endothelial cell [Ca2+]i. In the case of endothelium-derived hyperpolarizing factor (EDHF, the NO and prostanoid-independent pathway), this increase crucially activates endothelial KCa2.3- and KCa3.1-channels. Activation of these KCa-channels leads on to arterial hyperpolarization and dilation (see review1), and a changing role for each sub-type has been implicated in pathological responses within blood vessels.2,3
Although activation of both KCa2.3- and KCa3.1-channels leads ultimately to vascular dilation, each can provide a variable contribution to EDHF-hyperpolarization. Individual input is influenced by the extent of ongoing arterial (background) constriction. Increasing endothelial cell [Ca2+]i (with ACh) in quiescent rat mesenteric arteries hyperpolarizes the resting membrane potential through KCa2.3-channels alone, but during smooth muscle depolarization and the associated contraction necessary to observe EDHF-relaxation, KCa3.1-channels are also activated.4 This differential activation correlates with a distinct subcellular distribution of the channels. KCa3.1-channels are localized within the endothelial cell projections through the internal elastic lamina (IEL) that form MEGJs with the adjacent smooth muscle. In contrast, KCa2.3-channels are diffusely distributed throughout the plasmalemma of endothelial cells.5 How the different channel types are activated independently is not known, but concentrating KCa3.1-channels within the narrow endothelial cell projections may hinder access of activating Ca2+ from the main body of the endothelial cells, while perhaps facilitating activation by a signal derived from or associated with the contracting smooth muscle. In small arteries and arterioles, Ca2+ and/or a Ca2+ signal has been shown to spread from the muscle to the endothelium.6-8 In the mesenteric artery, this process relies on Ca2+-signalling via MEGJs.7,8 Extracellular Ca2+ levels ([Ca2+]o) may also have a significant influence on endothelial cell signaling. In the mesenteric artery, an extracellular calcium-sensing receptor (CaSR) on the endothelium links to activate KCa3.1-channels selectively, and appears to co-localize with this channel (but not with KCa2.3-channels).21 This raises the possibility that alterations in extracellular Ca2+ concentration may help determine the relative contribution from KCa3.1/2.3-channels. In addition, KCa3.1- but not KCa2.3-channel activity is inhibited by PKA phosphorylation in enteric neurons, indicating regulation may occur independently of intracellular [Ca2+] change.9-11
In the rat mesenteric artery, transfer of endothelial hyperpolarization to the muscle in part reflects K+ ion efflux through endothelial KCa-channels and in part spread of hyperpolarization via MEGJs.12-15 Extracellular K+, mimicking its action as a diffusible EDHF, stimulates smooth muscle hyperpolarization and relaxation primarily by activating Na+/K+-ATPase.16 However, in situ the ability of K+ to act as an EDHF is inversely related to ongoing arterial contraction, because as contraction increases Na+/K+-ATPase activity is swamped by K+ leaving the smooth muscle through BKCa-channels.16-18 In contrast, MEGJs operating in parallel to K+ enable a constant spread of hyperpolarization, and this route to relaxation thus becomes predominant as arterial tone approaches maximum.15 So one possibility is that KCa2.3- and KCa3.1-channels might each separately underlie one of these routes to vasodilation. In support of this possibility, recent evidence from rat mesenteric artery has suggested that KCa2.3-channel activation may be intimately linked to the release of C-type natriuretic peptide, that acts as a diffusible EDHF through natriuretic peptide receptor-C (NPR-C).30
Aims of the present study were therefore to investigate how KCa3.1-channels are independently activated, and second to show if the subcellular distribution of KCa2.3/KCa3.1-channels within rat mesenteric endothelial cells can be correlated with discrete EDHF pathways, with functional consequences for relaxation and of direct relevance to endothelial changes associated with vascular disease.
Methods
The Online Methods Section gives full details of the experimental techniques used. These included intracellular sharp microelectrode recording and simultaneous tension measurements, and Ca2+ imaging from both smooth muscle and endothelial cells and immunohistochemistry of rat isolated small mesenteric arteries.
Results
Reducing external [Ca2+]o recruits KCa3.1-channel input to EDHF hyperpolarization
Reducing external [Ca2+]o from 2.5 to 1.0 mmol/L slightly reduced smooth muscle resting membrane potential (from -51.7 ± 1.6 mV to -47.8 ± 0.9 mV, n=7 & 12) and depressed the cumulative EDHF-hyperpolarization to ACh (maximal increase 23.9 ± 2.4 mV, n=7 and 20.1 ± 1.7 mV, n=12 respectively Figure 1A). In contrast to the situation in 2.5 mmol/L Ca2+ (and see also ref 4) apamin failed to block EDHF-hyperpolarization in 1 mmol/L Ca2+ (n=4). Furthermore, low [Ca2+]o facilitated the ability of the KCa3.1-channel activator, 1-EBIO (100 and 300 μmol/L) to evoke smooth muscle hyperpolarization Figure 1B. In separate experiments using 1.0 mmol/L [Ca2+]o with apamin present (resting membrane potential: -48.6 ± 1.8 mV, n=8), 1 μmol/L forskolin significantly reduced EDHF-hyperpolarization by 80-100% (Figure 1C) and hyperpolarization to 300 μmol/L 1-EBIO from 8.5 ± 1.9 mV to 2.9 ± 0.9 mV (n=5).
Figure 1.

Summarized data showing the average change in membrane potential (Δ Em) to cumulative increases in [ACh] applied to evoke EDHF-hyperpolarization in mesenteric arteries. A, Reducing [Ca2+]o from 2.5 mmol/L to 1.0 mmol/L depressed the steady state hyperpolarization to ACh (n=7 & 12, respectively). In 1.0 mmol/L Ca2+ and with 50 nmol/L apamin present approximately 50% hyperpolarization remained (n=4), whereas in 2.5 mmol/L [Ca2+]o, apamin fully blocked hyperpolarization (n=13). B, Histogram summarizing the increase in smooth muscle membrane potential following endothelial cell stimulation with 1-EBIO in the presence of 50 nmol/L apamin. In 1.0 mmol/L [Ca2+]o, hyperpolarization to both 100 and 300 μmol/L 1-EBIO was enhanced (n=4&5, respectively). Asterisk indicates significant difference relative to control (2.5 mmol/L [Ca2+]o, P<0.05). C, In 1.0 mmol/L [Ca2+]o Krebs with 50 nmol/L apamin present EDHF-hyperpolarization evoked with ACh was significantly inhibited in the additional presence of 1 μmol/L forskolin (n=8, P<0.05).
Alteration in [Ca2+]o also modified arterial tone (Online Supplementary Figure 1). Increasing [Ca2+]o from 1.0 mmol/L relaxed pre-contracted arteries via endothelial cell KCa3.1-channels, with complete relaxation obtained in the presence of 2.5 mmol/L [Ca2+]o.
Limiting smooth muscle Ca2+ increase modifies KCa2.3- and KCa3.1-channel input to EDHF-dilation in rat mesenteric arteries
In the presence of the voltage-gated Ca2+ channel (VGCC) inhibitor nifedipine (10 μmol/L) and during depolarization to PE (1 μmol/L: 7.2 ± 1.8 mV, n=12), apamin (50 nmol/L) alone effectively abolished EDHF hyperpolarization (23.4 ± 1.3 mV hyperpolarization to 3 μmol/L ACh reduced to 1.9 ± 1.3 mV, n=12) (Figure 2A-C), whereas TRAM-34 (1 μmol/L) had little effect (hyperpolarization to 3 μmol/L ACh 19.6 ± 1.4 mV, n=12) (Figure 2B). Hyperpolarization to the direct KCa3.1-channel activator 1-EBIO (100 or 300 μmol/L) was not altered by nifedipine (control: 3.9 ± 1.0 and 8.8 ± 0.8 mV, n=4; versus nifedipine: 3.5 ± 0.7 and 6.2 ± 2.2 mV, n=5, respectively). In the presence of another VGCC inhibitor diltiazem (10 μmol/L), and PE depolarization (7.0 ± 0.9 mV, n=6), apamin again effectively abolished EDHF hyperpolarization (33.0 ± 1.5 mV hyperpolarization to 3 μmol/L ACh reduced to 5.6 ± 2.2 mV, n=6) (Figure 2C), whereas subsequent addition of the ATP-sensitive K+ (KATP)-channel opener levcromokalim (3 μmol/L) evoked a robust hyperpolarization of 29.4 ± 3.9 mV (n=6). Nifedipine reduced the smooth muscle cell [Ca2+]i and tension increase to PE (Figure 2D-F).
Figure 2.

ACh-mediated stimulation of KCa3.1-channels is prevented by blocking VGCC in rat mesenteric arteries. A, Original traces demonstrating concentration-dependent EDHF-induced smooth muscle hyperpolarization evoked by ACh in the presence of nifedipine (10 μmol/L) and prior depolarization to PE. Hyperpolarization overshot the resting membrane potential (dashed line), and was abolished by apamin (50 nmol/L). B, Summarized data showing the average change in membrane potential (Δ Em) to cumulative increases in [ACh] in arteries stimulated with PE in the presence of nifedipine (10 μmol/L, n=4-10), or in the additional presence of either apamin (50 nmol/L, n=4-12) or TRAM-34 (1 μmol/L, n=7-14), or both inhibitors together (n=5-8). C, Summarized data showing the average change in membrane potential (Δ Em) to cumulative increases in [ACh] in arteries stimulated with PE in the presence of diltiazem (10 μmol/L, n=4-6), or in the additional presence of apamin (50 nmol/L, n=5-7). D, Smooth muscle Ca2+ signals in the absence (upper traces) and presence of nifedipine (10 μmol/L, lower traces) colour-coded to the field of interest shown in the image in E. Applying 0.6 or 1 μmol/L PE stimulated a marked increase in the global average level of [Ca2+] (black line), representing asynchronous Ca2+ increases within individual muscle cells. Both the average increase and the majority of the oscillations were abolished in the presence of nifedipine (see Online Video 1 for movie corresponding to these data). F, Summarized data showing inhibition of the increase in [Ca2+]i stimulated by 1 μmol/L PE (left panel, control n=6, nifedipine n=3) and the associated block of arterial contraction (right panel, paired data). Asterisk indicates significant difference relative to control baseline (P<0.05).
Imaging both global and subcellular increases in endothelial cell [Ca2+] to ACh, failed to reveal any significant difference between ACh applied under resting conditions (baseline) compared to during smooth muscle stimulation with PE (Figure 3A & B). In Figure 3A, the measurement regions could be limited to the holes through the IEL, where a Ca+ increase was clearly observed within endothelial cell projections.
Figure 3.
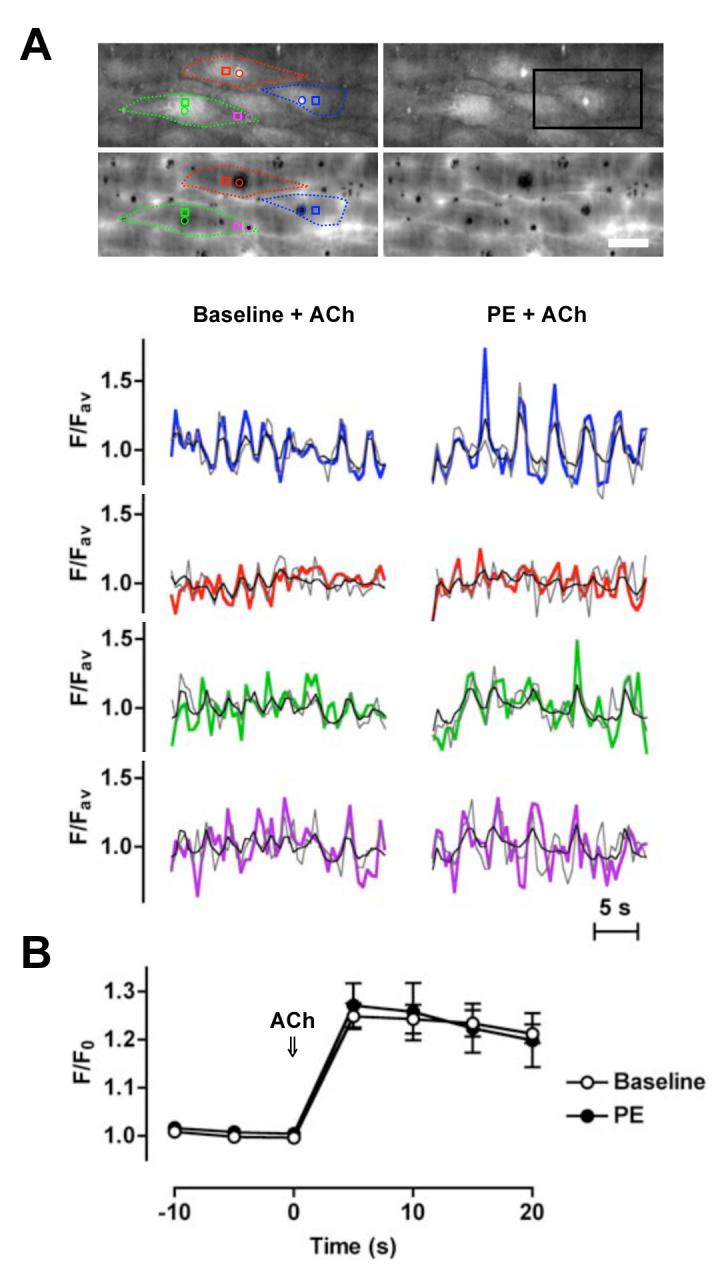
Similar increases in endothelial cell [Ca2+]i evoked with ACh alone and applied during smooth muscle stimulation with PE. A, Mesenteric arteries were mounted in a pressure myograph and changes in endothelial cell [Ca2+]i assessed at the interface between the endothelium and the IEL. Upper micrographs show loaded endothelial cells (average of 20 sec). Note bright spots (endothelial cell projections) that correspond to holes in the IEL (lower micrographs). Bar = 20 μm. Lower panels show the time course of fluorescence changes in individual endothelial cells in response to ACh (1 μmol/L) added under baseline conditions (left) and in the presence of PE (0. 3 - 0.6 μmol/L, right). Regions were placed over (colour) and adjacent to (grey) the endothelial cell projections, and around the whole cell (black). The colours relate to subcellular regions within the micrographs (see Online Video 2 for movie corresponding to these data from the region indicated by the black box). Representative of 3 experiments. B, Mesenteric arteries were mounted in a wire myograph and changes in (whole) endothelial cell [Ca2+]i measured before and during ACh (1 μmol/L) either from the baseline or in the presence of PE (0.3 - 0.6 μM, n=3, paired data). Note that endothelial cell projections were not loaded in these experiments (see Online Methods Supplement for details).
KCa3.1-channel or Na+/K+-ATPase block abolishes EDHF-dilation when a gap-junction uncoupler is present
In unstimulated arteries, 30-60 minutes exposure to the gap-junction uncoupler carbenoxolone (100 μmol/L) did not alter either the smooth muscle (-50 ± 1.7 mV and -50.6 ± 0.9 mV, n=11 and 6, respectively) or the endothelial cell resting potential (-52.4 ± 1.3 mV and -51.9 ± 1.3 mV, n=11 and 6). With ACh, carbenoxolone reduced smooth muscle (EDHF)-hyperpolarization from a maximum of 20.9 ± 1.2 mV (to -69.7 ± 1.7 mV, n=9) to 9.9 ± 3 mV (to -60.5 ± 2.7 mV, n=6) (Figure 4A-B), but did not reduce (direct) endothelial cell hyperpolarization to 1 μmol/L ACh (8.9 ± 1.4 mV and 8.1 ± 1.7 mV, n=5) (Figure 4C). However, it reversibly blocked the indirect hyperpolarization recorded from endothelial cells, where hyperpolarization was induced by stimulating KATP channels in the smooth muscle (5 μmol/L levcromakalim: control 19.8 ± 2.1 mV, n=5; plus carbenoxolone, 0.4 ± 0.4 mV, n=3; after washout, 19.7 ± 2.0 mV, n= 3) (Figure 4C).
Figure 4.

Gap junctions facilitate the contribution of KCa2.3-channels to EDHF-hyperpolarization in rat mesenteric artery. A, Original traces demonstrating ACh-mediated concentration-dependent increases in smooth muscle cell membrane potential from the resting membrane potential (dashed lines). Carbenoxolone (100 μmol/L) did not alter the resting membrane potential, but reduced the hyperpolarization to ACh. B, Summarized data showing the average change in membrane potential (Δ Em) to cumulative increases in [ACh] to evoke EDHF-hyperpolarization in arteries from resting membrane potential (left, n=6-11). In the presence of PE (right panels), carbenoxolone (100 μmol/L) depressed EDHF hyperpolarization (n=5-9) but not relaxation to ACh (n=8-14). The EDHF-mediated relaxation was reduced by the addition of apamin (50 nmol/L, n=9-13), and almost abolished by TRAM-34 (1 μmol/L, n=5-12) or ouabain (100 μmol/L, n=3-9), each applied when carbenoxolone was present. Asterisk indicates significant difference relative to control (P<0.05).
Carbenoxolone (100 μmol/L) did not alter contraction to either submaximal or maximal concentrations of PE, or the subsequent EDHF relaxation to ACh during submaximal contraction. However, in arteries maximally contracted with PE the EDHF-mediated relaxation, which under these stimulation conditions relies solely on MEGJs,15 was abolished even though levcromakalim (1 μmol/L) was able to stimulate maximal relaxation (97.1 ± 1.0%, n=6, Table 1).
Table 1.
Carbenoxolone (Carb, 100 μmol/L) only blocks EDHF-relaxation to ACh in mesenteric arteries maximally contracted with PE. Average relaxation to ACh obtained in arteries undergoing submaximal (low) contraction or maximal (high) contraction to PE. Asterisk indicates significant difference relative to control (P<0.05), paired data for each level of tension
| Tension (mN) | [PE] (μmol/L) | % Relaxation | pD2 | n | |
|---|---|---|---|---|---|
| Low Contraction | |||||
| Control | 10.8 ± 0.6 | 0.4 ± 0.1 | 96.4 ± 1.2 | 7.2 ± 0.1 | 5 |
| + Carb | 8.9 ± 0.7 | 0.6 ± 0.1 | 92.9 ± 3.2 | 6.9 ± 0.1 | 5 |
| High Contraction | |||||
| Control | 19.0 ± 2.5 | 10.0 ± 0.0 | 95.4 ± 0.6 | 7.0 ± 0.1 | 6 |
| + Carb | 18.6 ± 1.8 | 10.0 ± 0.0 | 10.3 ± 7.7* | 7.2 ± 0.5 | 6 |
In arteries submaximally stimulated with PE, depolarization and contraction did not differ from control (control 9.2 ± 1.0 mV, and 6.3 ± 0.4 mN; with carbenoxolone 9.2 ± 1.6 mV, and 6.9 ± 0.6 mN, n=8), but the ACh-evoked increase in membrane potential was reduced by around 10 mV (Figure 4B), increasing to -63.4 ± 4.5 mV (n=7) compared with -70.9 ± 2.4 mV (n=8) without carbenoxolone. However, this reduction in maximum hyperpolarization was not sufficient to modify the associated relaxation. In contrast, in the additional presence of 1 μmol/L TRAM-34 relaxation to ACh was almost abolished (92.5 ± 2.8 %, n=5 reduced to 26.1 ± 6.7%, n=5, Figure 4B). Ouabain (100 μmol/L) had a similar inhibitory effect to TRAM-34, depressing relaxation to ACh (from 98.9 ± 1.0 %) to only 23.5 ± 5.7 % in the presence of carbenoxolone (n=3). In contrast, under similar conditions apamin had only a slight inhibitory effect (Figure 4B).
Possible link between KCa2.3-channels and C-type natriuretic peptide (CNP)?
Use of the selective natriuretic peptide receptor-C (NPR-C) antagonist, M372049 (100 nmol/L) and comparison of EDHF reponses (to ACh) with the action of exogenous CNP did not suggest any link between KCa2.3-channel activation and the subsequent activation of NPR-C leading to hyperpolarization and relaxation, Online Supplementary Figure 2.
Dimensional localization of Na+/K+-ATPase and K-channels within the rat mesenteric artery
Immunoflourescence indicating Na+/K+-ATPase α2 (and similarly for α3)-subunits was distributed throughout the cells within pressurized rat mesenteric arteries (Figure 5). Particularly intense, punctate fluorescence signal was discretely aligned with holes through the IEL and above the plane of focus of the endothelium, at the interface between the IEL and the smooth muscle (noted to be an endothelial cell projection19). This corresponds with regions of MEGJ formation where we have previously demonstrated co-localization of KCa3.1-channels, connexin (Cx)40 and Cx37.5,15 In contrast, staining for Kir2.1-channels was homogeneous within endothelial cells, and that for KCa2.3-channels very clear at inter-endothelial cell borders, similar to staining for Cx37 and Cx40.5,20 There was also strong staining for both Kir2.1- and KCa3.1-channels in endothelial cell projections, whereas staining for KCa3.1-channels and Na+/K+-ATPase was not evident (or at least markedly less) in other regions of the endothelial cells. No immunostaining was observed in arteries where the primary antibody was omitted during preparation, or following prior incubation with blocking peptide.
Figure 5.

Endothelial cell immunohistochemical expression pattern for Na+/K+-ATPase, Kir2.1-, KCa3.1- and KCa2.3-channels in rat mesenteric arteries. (A-D, top panels) Confocal images of the wall of pressurized arteries showing a single z-axis plane through the IEL (green) and simultaneously acquired corresponding expression of protein (red). (A-D, bottom panels) Reconstruction of the confocal z-axis multi-plane stack through the wall of the artery corresponding to a line drawn through the images in the upper panels at the positions indicated by arrows. Note the presence of protein staining within the holes through the IEL in both the upper and lower panels. Both the Na+/K+-ATPase α2-subunit and KCa3.1-channels are highly expressed within the holes, whereas Kir2.1- and KCa2.3-channels are also highly expressed within endothelial cells and at endothelial cell borders, respectively. In these images, the Alexa Fluor 488 secondary antibody was only applied to the luminal side of the artery, and the confocal laser and PMT settings were identical for each z-stack. Bar indicates 20 μm in the x-axis in all images. Representative of at least 3 arteries. E, Quantification of protein expression within the holes through the IEL (n=3-5).
Discussion
These data show that the ability of KCa3.1-channels to contribute to EDHF-hyperpolarization depends on extracellular [Ca2+]o, possibly linked to the extent of basal stimulation of the extracellular CaSRs known to colocalize with these channels in the mesenteric artery endothelium. Recruitment of KCa3.1-channels to EDHF-hyperpolarization (and relaxation) did not appear to reflect Ca2+ or a Ca2+-release signal passing from the smooth muscle into the endothelial projections that form MEGJs. However, it may reflect a local depletion of extracellular [Ca2+]o around the endothelial projections containing KCa3.1 as [Ca2+]o enters the smooth muscle through VGCC, because recruitment was reduced in the presence of VGCC blockers to limit the ability of PE to raise muscle [Ca2+]i. Finally, KCa3.1-channel input may be enhanced by the K+ ions acting on the same, or closely adjacent cells via the Na+/K+-ATPase (and possibly Kir-channels). This suggests that K+ efflux through KCa-channels discretely localized in endothelial projections may underlie its action as a ‘diffusible’ EDHF, while hyperpolarization due to KCa-channels expressed near inter-endothelial cell borders (KCa2.3) relies primarily on extant MEGJs for effective spread into the media.
In the rat mesenteric artery, we have recently shown that KCa3.1-channels are concentrated in the head region of endothelial cell projections protruding through the IEL to form MEGJs.5 In the endothelial cells, strong punctate signal for KCa3.1-channels co-localized with Cx37 and Cx40 at the interface with the smooth muscle. Furthermore, at an ultrastructural level KCa3.1-channels and both Cx37 and Cx40 conjugated gold-label was associated with endothelial cell projections forming MEGJs. In contrast, although KCa2.3-channels were localized in close proximity to Cx37, Cx40 and Cx43, they appeared to be mainly adjacent to endothelial-endothelial cell gap junctions.5,15 These observations correlate with electrophysiological and immunoprecipitation data. In the former, the contribution from KCa2.3- and KCa3.1-channels to ACh-mediated hyperpolarization could be separated. Endothelial KCa3.1-channel activity was only apparent when the arterial smooth muscle was depolarized and contracted (leading to EDHF-repolarization to close to resting potential).4 In the latter, KCa2.3- and KCa3.1-channels were associated with caveolin-rich and -poor fractions of endothelial cell membrane, respectively.21 The present experiments subtly extend our morphological data, and provide novel insight into the control of endothelial KCa3.1-channel activation and therefore their contribution to EDHF responses. Reconstruction of confocal z-stack images from the wall of mesenteric arteries, obtained in a physiological orientation (as opposed to perfusion fixation and flat mounting for microscopy),5 confirmed the restriction of KCa3.1-channels within endothelial cell projections, but also revealed a co-localization of KCa2.3.
With regard to the mechanism responsible for recruiting the KCa3.1-channels within endothelial cell projections, this was shown to be sensitive to [Ca2+]o around the outside of the projections. Reducing [Ca2+]o recruited KCa3.1-channels to EDHF-hyperpolarization. These observations are consistent with recent evidence in lean Zucker rats, where both KCa2.3- and KCa3.1-channels contributed to EDHF-hyperpolarization in arteries bathed in Krebs containing 1.6 mmol/L [Ca2+].2 A reasonable explanation in both studies is that lowering [Ca2+]o reduces basal stimulation of an extracellular G-protein linked CaSR, receptors known to co-localize with KCa3.1-channels in ‘non-caveolin’ membrane fractions of mesenteric endothelial cells.21 Thus, lowering [Ca+]o from 2.5 to 1 mmol/L reduced extracellular CaSR saturation and effectively increased availability of membrane KCa3.1-channels for activation by increases in [Ca2+]i within the endothelial projections.
Limiting increases in smooth muscle [Ca2+]i were also linked to the ability of endothelial KCa3.1-channels to provide significant input to EDHF-hyperpolarization. Nifedipine or diltiazem reduced voltage-dependent Ca2+ entry into the smooth muscle, associated with a loss of KCa3.1-channel input to the EDHF response (evoked by ACh) and, as a consequence, sole reliance on the activation of apamin-sensitive KCa2.3-channels. One possible explanation is that during ‘normal’ increases in smooth muscle [Ca2+]i, KCa3.1-channel input to the EDHF-response is enabled because the increase in smooth muscle [Ca2+]i following depolarization indirectly facilitates Ca2+ signaling in the endothelial cell projections and activates the channels. Direct measurement of endothelial cell [Ca2+]i levels in both resistance arteries and arterioles has shown that smooth muscle stimulation with PE leads on to [Ca2+]i increases in the endothelium.6-8 In isolated mesenteric arteries from the rat, endothelial cell subpopulations displayed an increase in Ca2+ during muscle stimulation with PE, apparently reflecting spread of IP3 from the muscle to the endothelium.7 However, the additional movement of Ca2+ into endothelial cell projections (and throughout the cells)8,22 could not be discounted by these experiments. But signalling via this route does appear to explain our data. We were unable to detect any difference in [Ca2+]i increases to ACh within the endothelial cell projection (or whole cell) during simultaneous muscle stimulation with PE. The caveat here is that it remains possible the resolution and limitations of our system prevented detection of very small and localized changes within endothelial cell projections or discrete changes in [Ca2+]i within the projections during changes in diameter.
Alternatively, Ca2+ entry into the contracting muscle itself may be sufficient normally to reduce the [Ca2+]o in the restricted extracellular space round the endothelial cell projection. As such, this would reduce ‘basal’ CaSR stimulation and facilitate linked KCa3.1-channel availability, enabling activation of the latter by the increased [Ca2+]i in the projections evoked by ACh. In support of this scenario, during synchronous neuronal activity the opening of VGCCs is known to substantially deplete (circa 1 mmol/L) extracellular [Ca2+]o in the limited volume of fluid surrounding the cells.23-25 Whatever the precise explanation, within endothelial cell projections KCa3.1-channel activity may be suppressed through the action of PKA. In enteric neurons, PKA appears to maintain the KCa3.1-currents underlying slow, post-spike, after-hyperpolarization in a closed state.9 In our experiments, the ability of forskolin to inhibit EDHF-hyperpolarization due to KCa3.1-channel activity suggests a similar mechanism operates in mesenteric endothelial cells.
One possible confounding influence in our experiments with VGCC blockers, is the potential for nifedipine directly to block KCa3.1-channels. Dihydropyridines were initially shown to block the Gardos channel in erythrocytes,26,27 and more recently Jiang et al28 reported block of EDHF-hyperpolarization in guinea-pig cochlear artery, where in contrast to the mesenteric artery hyperpolarization appears to depend on KCa3.1-channels alone. However, this did not appear to be a significant consideration in our experiments because 1) diltiazem, which unlike the dihydropyridines does not block KCa3.1-channels,26,28 had a similar effect to nifedipine, and 2) nifedipine did not alter hyperpolarization to the KCa3.1-channel activator, 1-EBIO. The apparent inability of nifedipine to block KCa3.1-channels may relate to the experimental conditions. For example, block of the Gardos channel with nifedipine decreases with increasing extracellular K+, a situation which would be predicted in our experiments in the presence of PE.26 Or it may simply be that although KCa3.1-channel activation is reduced, the reduction is not sufficient to suppress the overall EDHF-hyperpolarization in intact arteries.
Another important observation in our study was the ability of either ouabain or TRAM-34 to block EDHF responses in the presence of the gap junction uncoupler carbenoxolone. Although carbenoxolone did reduce EDHF hyperpolarization, this did not modify the associated arterial relaxation. However, once ouabain was added along with carbenoxolone, EDHF-relaxation was abolished. Although this concentration of ouabain blocks smooth muscle hyperpolarization and relaxation to exogenous K+,16 it has only a small inhibitory effect against the overall EDHF hyperpolarization and relaxation in submaximally contracted arteries, because the EDHF response is sustained by MEGJs as explained in the Introduction.12,15 However, once the MEGJ pathway is compromised, in this case with carbenoxolone, blocking the action of endogenous K+ significantly impacts on EDHF-relaxation. The fact that a gap-junction uncoupler (carbenoxolone) did not alter EDHF responses in submaximally contracted arteries, but abolished them in maximally contracted arteries although not altering relaxation to levcromakalim, supports this scheme. Interestingly, with carbenoxolone present in submaximally contracted arteries, TRAM-34 (but not apamin) caused similar block to ouabain. This suggests that endogenous K+ originating from the KCa3.1-channels in close proximity to focused clusters of Na+/K+-ATPase, underlies the ability of K+ to act as EDHF. In the absence of carbenoxolone, block of both KCa3.1- and KCa2.3-channels, with TRAM-34 and apamin together, is necessary to block EDHF effects.4 The concentration of carbenoxolone we used has been shown not to alter the [Ca2+]i increase evoked in the endothelium with ACh,15 and blocks the spread of calcein from the endothelium into the smooth muscle.8 Furthermore, we now show that carbenoxolone does not modify the resting potential in either the endothelial or smooth muscle cells, nor the endothelial cell hyperpolarization evoked with ACh. However, notably it did block hyperpolarization to levcromakalim from spreading through MEGJs to the endothelium. In this vessel, KATP channels are found only on the smooth muscle cells not the endothelium.29 So although it remains possible carbenoxolone has effects in addition to uncoupling gap-junctions, this agent clearly and effectively blocks the spread of EDHF-hyperpolarization through MEGJs, and without disrupting the key events responsible for initiating an EDHF-response viz Ca2+ handling and KCa-channel activation within the endothelium.
Finally, in rat mesenteric arteries KCa2.3-channel activation has recently been suggested to be somehow linked to the release of CNP, with CNP then acting as an EDHF to hyperpolarize and relax the adjacent smooth muscle.30 Furthermore, in this study the NPR-C selective antagonist M372049 was found to act synergistically with ouabain to block EDHF responses, leading to the suggestion that CNP acts alongside a KCa3.1-dependent signal, the two ‘arms’ underpinning the EDHF-response in mesenteric arteries. While a link between KCa3.1-channels and Na+/K+-ATPase activation is consistent with our data, we were unable to provide any evidence to suggest CNP plays a significant role in the EDHF response. While M372049 clearly blocked arterial relaxation to exogenous CNP, it did not increase the inhibitory action of ouabain against EDHF-hyperpolarization and relaxation, and it failed to inhibit the apamin-sensitive (KCa2.3-channel) component of EDHF-hyperpolarization. The fact that exogenous CNP only evoked a modest relaxation without any hyperpolarization, and that M372049 did not modify EDHF responses to ACh, also argues strongly against a significant role for CNP in the EDHF-response.
Overall, our data indicate a clear correlation between the discrete localization of KCa3.1-channels within the endothelial cell projections that form MEGJs, and their respective activation during smooth muscle stimulation. A suggested outline mechanism is presented in Figure 6. The complex dynamics of [Ca2+]o change within the very restricted intercellular space between muscle and endothelial cell projections may provide the key to explaining why KCa3.1-channels are able to contribute to EDHF hyperpolarization and relaxation during muscle contraction, dropping out as membrane potential and tension (reflecting smooth muscle [Ca2+]i) returns to resting levels, while KCa2.3-channels still continue to increase membrane potential to close to EK. In addition, spatial clustering of KCa3.1-channels and Na+/K+-ATPase in a concentrated microdomain within the endothelial cell projections, may serve to focus K+ efflux for optimal stimulation of the pump thus amplifying and/or initiating EDHF-hyperpolarization in the smooth muscle cells.
Figure 6.

Suggested key steps underlying endothelial cell KCa-channel activation and subsequent transfer of hyperpolarization to the mesenteric smooth muscle. Step 1. At rest in 2.5 mmol/L [Ca2+]o Krebs solution, the endothelial cell Ca2+ sensing receptor (CaSR) is maximally stimulated and KCa3.1-channels are in some way inactivated, possibly via PKA-phosphorylation. Step 2. Stimulation of the endothelium with ACh raises cytoplasmic [Ca2+]i, activating apamin-sensitive KCa2.1-channels causing hyperpolarization which can spread through homocellular (endothelial-endothelial) and heterocellular (myoendothelial, MEGJs) gap junctions. KCa3.1 are not available for agonist-induced [Ca2+]i activation. Step 3. Activation of smooth muscle VDCC (with PE) causes a local ‘sink’ of [Ca2+]o in the vicinity of the endothelial projections. This reduction reduces stimulation of the CaSR and phosphorylation of KCa3.1-channels in the projections. Step 4. At this point, ACh-stimulation of the endothelium raises cytoplasmic [Ca2+]i, now activating ‘available’ KCa3.1 as well as KCa2.3. As in Step 2, the resulting hyperpolarization spreads through endothelial-endothelial and MEGJs gap junctions, but now reflects input from both KCa2.3 and KCa3.1. Facilitated by ongoing muscle depolarization, endothelial K+ efflux through KCa-channels is sufficient to stimulate adjacent Na+/K+-ATPase enhancing hyperpolarization. Further amplification may also occur through KIR-channel activation within this microdomain. As smooth muscle cells repolarize to resting levels, VGCC open-probability decreases, removing the Ca2+ ‘sink’, local [Ca2+]o increases towards 2.5 mmol/L and KCa3.1-channel activity is again removed from the control of intracellular [Ca2+]i. Grey, intercellular space; Purple, gap junctions; red, KCa3.1-channels; yellow, KCa2.3-channels; blue, VGCCs; green, Na+/K+-ATPase; and cyan, CaSR.
Supplementary Material
Acknowledgements
We are very grateful to Drs Lorraine McEvoy and Michaela Spitaler for carrying out some of the control experiments with carbenoxolone and the immunohistochemistry, respectively, and Polina Iarova for technical support.
Sources of funding:
Supported by the Wellcome Trust and British Heart Foundation.
Footnotes
Subject codes: [95] Endothelium/vascular type/nitric oxide [97] Other vascular biology (isolated artery electrophysiology) [152] Ion channels/membrane transport
Disclosures:
None
Publisher's Disclaimer: This is an un-copyedited author manuscript accepted for publication in Circulation Research, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the “Fair Use of Copyrighted Materials” (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at http://circres.ahajournals.org/. The American Heart Association disclaims any version derived from it by the National Institutes of Health or other parties.
References
- 1.Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- 2.Burnham MP, Johnson IT, Weston AH. Impaired small-conductance Ca2+-activated K+ channel-dependent EDHF responses in Type II diabetic ZDF rats. Br J Pharmacol. 2006;148:434–441. doi: 10.1038/sj.bjp.0706748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohler R, Wulff H, Eichler I, Kneifel M, Neumann D, Knorr A, Grgic I, Kampfe D, Si H, Wibawa J, Real R, Borner K, Brakemeier S, Orzechowski HD, Reusch HP, Paul M, Chandy KG, Hoyer J. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation. 2003;108:1119–1125. doi: 10.1161/01.CIR.0000086464.04719.DD. [DOI] [PubMed] [Google Scholar]
- 4.Crane GJ, Gallagher NT, Dora KA, Garland CJ. Small and intermediate calcium-dependent K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J Physiol. 2003;553:183–189. doi: 10.1113/jphysiol.2003.051896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandow SL, Neylon CB, Chen MX, Garland CJ. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (K(Ca)) and connexins: possible relationship to vasodilator function? J Anat. 2006;209:689–698. doi: 10.1111/j.1469-7580.2006.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc Natl Acad Sci USA. 1997;94:6529–6534. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamboley M, Pittet P, Koenigsberger M, Sauser R, Beny JL, Meister JJ. Evidence for signaling via gap junctions from smooth muscle to endothelial cells in rat mesenteric arteries: possible implication of a second messenger. Cell Calcium. 2005;37:311–320. doi: 10.1016/j.ceca.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Kansui Y, Garland CJ, Dora KA. Enhanced spontaneous Ca2+ events in endothelial cells reflects signalling through myoendothelial gap junctions in pressurized mesenteric arteries. Cell Calcium. 2008 doi: 10.1016/j.ceca.2007.11.012. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogalis F, Harvey JR, Furness JB. PKA-mediated inhibition of a novel K+ channel underlies the slow after-hyperpolarization in enteric AH neurons. J Physiol. 2003;548:801–814. doi: 10.1113/jphysiol.2002.037325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neylon CB, D’Souza T, Reinhart PH. Protein kinase A inhibits intermediate conductance Ca2+-activated K+ channels expressed in Xenopus oocytes. Pflugers Arch. 2004;448:613–620. doi: 10.1007/s00424-004-1302-5. [DOI] [PubMed] [Google Scholar]
- 11.Neylon CB, Fowler CJ, Furness JB. Regulation of the slow afterhyperpolarization in enteric neurons by protein kinase A. Auton Neurosci. 2006;126-127:258–263. doi: 10.1016/j.autneu.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 12.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 13.Edwards G, Feletou M, Gardener MJ, Thollon C, Vanhoutte PM, Weston AH. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br J Pharmacol. 1999;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ Res. 2002;90:1108–1113. doi: 10.1161/01.res.0000019756.88731.83. [DOI] [PubMed] [Google Scholar]
- 15.Mather S, Dora KA, Sandow SL, Winter P, Garland CJ. Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res. 2005;97:399–407. doi: 10.1161/01.RES.0000178008.46759.d0. [DOI] [PubMed] [Google Scholar]
- 16.Dora KA, Garland CJ. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am J Physiol Heart Circ Physiol. 2001;280:H2424–H2429. doi: 10.1152/ajpheart.2001.280.6.H2424. [DOI] [PubMed] [Google Scholar]
- 17.Richards GR, Weston AH, Burnham MP, Feletou M, Vanhoutte PM, Edwards G. Suppression of K+-induced hyperpolarization by phenylephrine in rat mesenteric artery: relevance to studies of endothelium-derived hyperpolarizing factor. Br J Pharmacol. 2001;134:1–5. doi: 10.1038/sj.bjp.0704256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dora KA, Ings NT, Garland CJ. KCa channel blockers reveal hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am J Physiol Heart Circ Physiol. 2002;283:H606–H614. doi: 10.1152/ajpheart.01016.2001. [DOI] [PubMed] [Google Scholar]
- 19.Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ Res. 2000;86:341–346. doi: 10.1161/01.res.86.3.341. [DOI] [PubMed] [Google Scholar]
- 20.Kansui Y, Fujii K, Nakamura K, Goto K, Oniki H, Abe I, Shibata Y, Iida M. Angiotensin II receptor blockade corrects altered expression of gap junctions in endothelial cells from hypertensive rats. Am J Physiol Heart Circ Physiol. 2004;287:H216–H224. doi: 10.1152/ajpheart.00915.2003. [DOI] [PubMed] [Google Scholar]
- 21.Weston AH, Absi M, Ward DT, Ohanian J, Dodd RH, Dauban P, Petrel C, Ruat M, Edwards G. Evidence in favor of a calcium-sensing receptor in arterial endothelial cells: studies with calindol and Calhex 231. Circ Res. 2005;97:391–398. doi: 10.1161/01.RES.0000178787.59594.a0. [DOI] [PubMed] [Google Scholar]
- 22.Isakson BE, Ramos SI, Duling BR. Ca2+ and inositol 1,4,5-trisphosphate-mediated signaling across the myoendothelial junction. Circ Res. 2007;100:246–254. doi: 10.1161/01.RES.0000257744.23795.93. [DOI] [PubMed] [Google Scholar]
- 23.Hofer AM, Brown EM. Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol. 2003;4:530–538. doi: 10.1038/nrm1154. [DOI] [PubMed] [Google Scholar]
- 24.Keicher E, Bilbaut A, Maggio K, Hernandez-Nicaise ML, Nicaise G. The desheathed periphery of aplysia giant neuron. Fine structure and measurement of [Ca2+]o fluctuations with calcium-selective microelectrodes. Eur J Neurosci. 1991;3:10–17. doi: 10.1111/j.1460-9568.1991.tb00806.x. [DOI] [PubMed] [Google Scholar]
- 25.Rusakov DA, Fine A. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron. 2003;37:287–297. doi: 10.1016/s0896-6273(03)00025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaji DM. Nifedipine inhibits calcium-activated K transport in human erythrocytes. Am J Physiol. 1990;259:C332–339. doi: 10.1152/ajpcell.1990.259.2.C332. [DOI] [PubMed] [Google Scholar]
- 27.Ellory JC, Kirk K, Culliford SJ, Nash GB, Stuart J. Nitrendipine is a potent inhibitor of the Ca2+-activated K+ channel of human erythrocytes. FEBS Lett. 1992;296:219–221. doi: 10.1016/0014-5793(92)80383-r. [DOI] [PubMed] [Google Scholar]
- 28.Jiang ZG, Shi XR, Guan BC, Zhao H, Yang YQ. Dihydropyridines inhibit acetylcholine-induced hyperpolarization in cochlear artery via blockade of intermediate-conductance calcium-activated potassium channels. J Pharmacol Exp Ther. 2007;320:544–551. doi: 10.1124/jpet.106.115212. [DOI] [PubMed] [Google Scholar]
- 29.Takano H, Dora KA, Spitaler MM, Garland CJ. Spreading dilatation in rat mesenteric arteries associated with calcium-independent endothelial cell hyperpolarization. J Physiol. 2004;556:887–903. doi: 10.1113/jphysiol.2003.060343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villar IC, Panayiotou CM, Sheraz A, Madhani M, Scotland RS, Nobles M, Kemp-Harper B, Ahluwalia A, Hobbs AJ. Definitive role for natriuretic peptide receptor-C in mediating the vasorelaxant activity of C-type natriuretic peptide and endothelium-derived hyperpolarising factor. Cardiovasc Res. 2007;74:515–525. doi: 10.1016/j.cardiores.2007.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.