

Abstract
Angiotensin-converting enzyme (ACE) has been well-recognized for its role in blood pressure regulation. ACE is made by many tissues, though it is most abundantly expressed on the luminal surface of vascular endothelium. ACE knockout mice show a profound phenotype with low blood pressure, but also with hemopoietic and developmental defects, which complicates understanding the biological functions of ACE in individual tissue types. Using a promoter-swapping strategy, several mouse lines with unique ACE tissue expression patterns were studied. These include mice with ACE expression in the liver (ACE 3/3), the heart (ACE 8/8), and macrophages (ACE 10/10). We also investigated mice with a selective inactivation of either the N- or C-terminal ACE catalytic domain. Our studies indicate that ACE plays a role in many other physiologic processes beyond simple blood pressure control.
Keywords: Angiotensin-converting enzyme, Mice, Gene targeting, Tissue-specific expression, Heart, Macrophages
Introduction
Since the early 1980s, the routine use of angiotensin-converting enzyme (ACE) inhibitors in clinical medicine has established that this enzyme plays an important role in blood pressure control and electrolyte balance, mostly due to the conversion of angiotensin I (Ang I) to angiotensin II (Ang II). Recently, the development of genetic techniques has facilitated new ways to study ACE. The result has been that this old friend continues to provide surprises, as scientists have discovered its many in vivo functions apart from its well-known role in cardiovascular system.
ACE (peptidyl dipeptidase A, CD143, EC 3.4.15.1) is a zinc-dependent dipeptidyl carboxypedtidase. In vertebrates, two isoforms of ACE have been characterized, somatic ACE (sACE) [1, 2] and testis ACE (tACE) [3, 4]. sACE is the isozyme made by endothelium and other somatic tissues. It is composed of two homologous catalytic domains, generally known as the N- and C-domains. In contrast, tACE is the result of testis-specific transcription from a distinct and separate promoter in the ACE gene [5, 6]; it is only composed of the C-domain. Both isoforms of ACE are glycosylated [2, 7]. ACE is a type I ectoenzyme anchored to cell membranes by a hydrophobic C-terminus [8]. While most ACE is bound to cells, the shedding of ACE releases active enzyme into the extracellular milieu.
Zinc-dependent peptidases such as ACE can be traced back to invertebrates [9] and even bacteria [10]. Typically, these proteins are composed of a single catalytic domain. In contrast, the sACE found in vertebrates have two independent catalytic domains with a high conservation of both amino acid and exon structure. This suggests that vertebrate ACE is the result of gene duplication. The principal functional unit of each ACE domain is the M2-type zinc metallopeptidase motif, HEMGH. Although the two domains of sACE share many enzymatic properties, they can be differentiated by substrate and inhibitor preferences, as well as chloride dependence [11].
When assessed by RT-PCR, ACE was found in all 72 human tissues tested [12]. ACE is abundantly expressed on the luminal surface of vascular endothelium [13], where it is well positioned to immediately respond to the fluctuating levels of circulating substrates, such as angiotensin I. ACE is also highly expressed by epithelial cells of many tissues, including kidney and the gastrointestinal tract [14]. In leukocytes, ACE expression is induced during the differentiation of human monocytes into both macrophages and dendritic cells [15, 16]. In the testes, sACE is made by Leydig cells [17], while tACE is made by germinal cells, such as spermatids [6]. There is no tACE equivalent in ovaries, since this tissue only makes sACE which is found in germinal epithelium [18]. ACE is also found in various body fluids as the solubilized form shed from tissues [19]. Therefore, ACE has access to substrates originating from different tissues. As compared to renin, ACE is a relatively non-specific enzyme, and it acts to remove C-terminal dipeptides from a variety of naturally occurring substrates. These substrates are short peptides, which vary in length from protected tripeptides (such as chemotactic peptide) to tridecapeptides (such as neurotensin). Large and folded substrates are not generally susceptible to ACE-mediated cleavage [20]. The wide spectrum of substrates for ACE and its ubiquitous distribution throughout the body already subtly suggest that this enzyme may be involved in physiologic and pathologic processes other than blood pressure control. This is further supported by the finding of functional ACE-like enzymes in invertebrates, which lack an identifiable renin-angiotensin system.
ACE knockout mice
One of the first insights into the many physiologic roles of ACE was the analysis of “null” or knockout mice created via targeted homologous recombination in embryonic stem cells. Our group analyzed ACE knockout mice (termed ‘ACE 1/1’) and found many changes from normal animals. The loss of ACE resulted in a marked reduction of blood pressure, abnormalities of blood electrolytes, structural lesions of the kidney, the inability to concentrate urine, anemia, and male reproductive defects [21, 22]. Although a knockout mouse is an extremely important tool, the great advantage of the knockout model—the total lack of a particular protein from conception until the death of the animal—is also a major limitation. An animal null for a protein represents an extreme phenotype, which is quite different from a human treated with a pharmacological inhibitor. Moreover, in the setting of a complex phenotype such as that seen in an ACE knockout mouse, it is easy to imagine that one physiologic defect (for example low blood pressure) may induce secondary physiologic consequences.
Consider another example: ACE knockout mice are anemic. It is difficult to know whether the anemia is a secondary consequence of the renal abnormalities present in ACE null mice or is due to a direct effect of ACE on erythropoiesis. Thus, examining knockout mice alone is not fully satisfying in understanding the role of this enzyme in an organ-by-organ fashion. To facilitate more a detailed analysis, our group has generated mouse models in which ACE expression is altered either by selective inactivation of one of the two ACE catalytic domains or by promoter ‘swapping’ so that ACE is made only in limited tissue types. We introduce these models below and show how each contributes to the idea that the physiologic roles of ACE are more diverse than previously understood.
Mice with the inactivation of one catalytic domain of ACE
Although ACE catalysis has been studied extensively in vitro, the exact physiologic relevance of each of the two catalytic domains of ACE is unknown. To study whether there are true physiologic differences in the function between the ACE N- and C-domains, we created mouse models in which point mutations were introduced into the ACE gene inactivating only one of the sACE catalytic domains. This was achieved by selectively replacing the histidines in one of the zinc-binding motifs with lysines, rendering that domain catalytically inactive. Mouse models lacking either the N- or C-domains of sACE are termed ACE 7/7 [23] and ACE 13/13 [24] (Fig. 1). The introduction of point mutations does not change the expression profile of sACE, as determined by Western blot analysis of ACE expression by individual tissues. Both ACE 7/7 (N-domain knockout) and ACE 13/13 (C-domain knockout) have a normal blood pressure. However, the manner in which each of these two models manages to maintain normal blood pressure is different. In the plasma from ACE 7/7 mice, the concentrations of Ang I, Ang II, and renin are not different from that of wild-type (WT) mice. This suggests that the C-domain alone (the active domain in ACE 7/7 mice) plays the major role in normal blood pressure control. In contrast, ACE 13/13 mice maintain normal plasma levels of angiotensin II and a normal blood pressure by increasing plasma levels of Ang I by about 7-fold. Thus, ACE 13/13 mice, a model lacking C-domain ACE activity, can only maintain a normal blood pressure by up-regulating renin concentrations (3-fold), markedly up-regulating plasma Ang I and thus, forcing normal concentrations of plasma Ang II. This conclusion was supported by experiments in which Ang I was infused intravenously while monitoring acute blood pressure changes. ACE 7/7 mice (which maintain normal function of the ACE C-domain) respond to Ang I infusion similar to WT mice. In contrast, ACE 13/13 mice (which possess N-domain activity) respond with a slower and much less vigorous conversion of angiotensin I to angiotensin II, and with subsequently less elevation in acute blood pressure.
Fig. 1.

Generation of ACE 7/7 mice lacking N-terminal ACE catalytic activity. Arrows indicate both the somatic and testis ACE promoters. Black boxes represent exons 1–25 of sACE. A testis-specific exon is indicated in the gray box located between exons 12 and 13. Exons 8 and 20 (patterned boxes) include the N- and C-catalytic domains of sACE, respectively. Site-directed mutagenesis was used to convert the zinc-binding domain within the eighth exon (N-domain) from HEMGH to KEMGK. A neomycin resistance cassette (neoR), used for positive selection, was inserted into the seventh intron of the ACE gene. A thymidine kinase (TK) cassette, used for negative selection, was inserted at the 5’-end of the construction. The resulting allele contains both mutated N-domain of ACE and neo gene (“targeted allele”). Excision of the neo gene by Cre recombinase resulted in the final mutated allele with a residual 34-base pair loxP sequence present in the seventh intron. To create ACE 13/13 mice, the same approach was used except for this line, we targeted that C-terminal catalytic site present in exon 20. ACE 13/13 mice have an N-terminal domain equivalent to WT mice
Two other conclusions are important in considering ACE 7/7 and ACE 13/13 mice. First, both models showed normal plasma levels of bradykinin 1–7 and bradykinin 1–9, suggesting no particular domain preference in the degradation of bradykinin. With the ACE 7/7 mice (N-domain knockout) we also examined the plasma levels of AcSDKP (N-domain-specific substrate) and its role in erythropoiesis. AcSDKP has been suggested as an erythrocyte lineage bone marrow-suppressive peptide [25]. The elevation of AcSDKP has been suggested as a possible reason for anemia in ACE null mice (ACE 1/1). Not surprisingly, ACE 7/7 mice have an elevated AcSDKP level in both plasma and urine, but this had no influence on basal hematocrit measurements or the ability of the mice to recover after phenylhydrazine-induced anemia. This experiment strongly suggests that the anemia induced in ACE null mice is almost certainly due to factors other than AcSDKP. Further, it calls into question whether AcSDKP is a bone-marrow-suppressive peptide in vivo.
Tissue-restricted expression of ACE
To investigate the role of ACE in an organ-by-organ fashion, we have genetically modified the ACE locus to disassociate ACE expression from the somatic ACE promoter and place it under the control of different tissue-specific promoters (Fig. 2). In each of these models, the expression profile of ACE is different from that of a WT mouse since now ACE is strictly regulated by the tissue-specific expression pattern of the new promoter, which is typically different from that of the endogenous somatic ACE promoter. One example is ACE 3/3 mice, in which an albumin promoter directs expression of ACE so that most ACE in the body is made by hepatocytes [26]. The importance of this model centers on the question of the relative importance of systemic versus local expression of ACE. In WT mice, localized expression of ACE on the surface of vascular endothelium is thought responsible for the local delivery of Ang II to vascular smooth muscle. ACE 3/3 mice have approximately 80% normal plasma ACE activity, probably due to shedding of ACE by hepatocytes. However, these mice totally lack ACE expression by vascular endothelium. Thus, the ACE 3/3 mice represent a unique way of assessing whether ACE expression by vascular endothelium is obligatory for normal blood pressure control. ACE 3/3 mice have a normal blood pressure. They respond to fluid restriction by concentrating urine in a fashion indistinguishable from WT mice. Their renal histology is unremarkable. And they have a hematocrit that is normal. Thus, this model shows that sufficient compensatory activity exists in the renin–angiotensin system to totally adjust for lack of ACE expression by vascular endothelium.
Fig. 2.
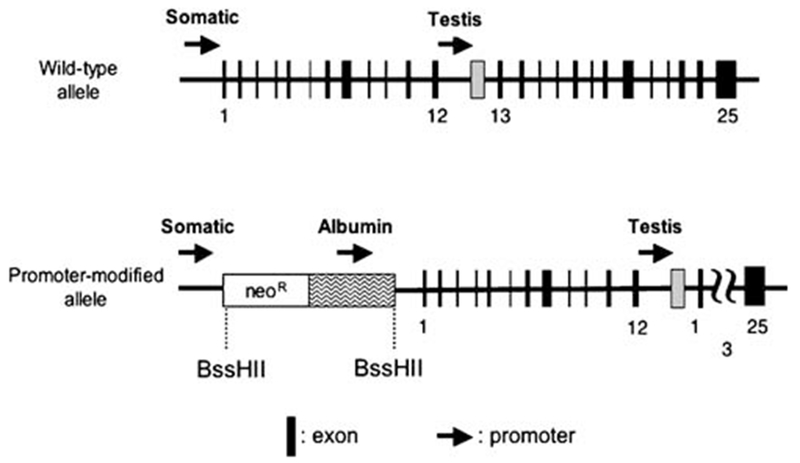
Promoter manipulation as a means of selective expression of ACE. In the ACE 3/3 gene, homologous recombination was used to position a neomycin resistance cassette (neoR) and an albumin promoter cassette between the somatic ACE promoter and exon 1. The neoR cassette works for both positive selection and to block the somatic ACE promoter, such that the structural portion of the ACE gene is now under the control of the albumin promoter
More intriguing discoveries came when other mouse lines were developed. One line is ACE 8/8, in which ACE expression is directed by the cardiac-specific α-myosin heavy chain promoter [27]. This model was developed to test the widely accepted idea that elevated cardiac levels of Ang II are deleterious. In the heart, Ang II was implicated in regulating cardiac remodeling and promoting cardiac hypertrophy [28, 29]. Hence, we believed that creating a mouse model in which ACE was selectively expressed in cardiac tissue would result in elevated local production of Ang II. ACE 8/8 mice express between 50- and 100-fold normal cardiac ACE levels, with ACE expression found on the surface of cardiac myocytes. Cardiac Ang II peptide levels are approximately 4-fold those of WT mice, while plasma Ang II levels are comparable to those of WT mice. In this model, some ACE is also made by tissues of the lung. ACE 8/8 mice have a systolic blood pressure of about 102 mm Hg (WT have approximately 111 mm Hg), normal ventricular size and normal left ventricular contraction velocity. ACE 8/8 mice show no evidence of heart failure, pericardial effusion or increased ventricular fibrosis. However, ACE 8/8 mice are not normal, as indicated by a markedly increased mortality observed after weaning. Examination of this model documented increased atrial size and, perhaps more important, a variety of cardiac electrical abnormalities, including various degrees of atrioventricular heart block and a marked reduction of cardiac electrical amplitude. These mice are prone to ventricular tachycardia. The electrical abnormalities reflect a major reduction of cardiac connexin 40 and connexin 43, the gap junction proteins responsible for cell–cell electrical coupling [30].
While the basic defects of the ACE 8/8 mice are relatively clear, the explanation for the biochemical abnormalities is more complex. This conclusion is the result of studying compound heterozygous mice created by breeding ACE 8/8 mice with ACE 1/1 mice. The compound mice, designated ACE 1/8, have one ACE null allele (‘1’) and one ACE allele targeting expression to the heart (‘8’) [31]. Because of this, these mice express less cardiac ACE than ACE 8/8 mice but still have most of their ACE expression restricted to heart tissues. Even though compound heterozygous mice have elevated levels of cardiac Ang II (similar to ACE 8/8 levels), they do not show the structural or electrical defects observed in ACE 8/8 mice. In addition, aortic banding generated equivalent levels of cardiac hypertrophy in ACE 1/8 mice as compared to WT controls, suggesting that the local increase of cardiac Ang II present in ACE 1/8 mice did not augment cardiac hypertrophy beyond the hemodynamic changes induced by banding. The study of ACE 1/8 mice, as well as ACE 8/8 mice genetically lacking the production of angiotensinogen, strongly suggests that abnormal cardiac levels of Ang II are not responsible for the pathologies observed in the ACE 8/8 mice. Interestingly, a recent review has discussed the role of Ang II as a mediator of left ventricular hypertrophy, apart from its role in elevating blood pressure. This concluded that most evidence in mice does not support a direct role of cardiac Ang II in the induction of ventricular hypertrophy [32].
ACE expression in macrophages
Recently, we reported another mouse line called ACE 10/10, in which ACE is overexpressed by monocyte and macrophage-lineage cells [33]. In this model, ACE gene expression is regulated by the c-fms promoter. ACE 10/10 mice have normal plasma ACE activity and normal blood pressure, without any anatomic defects. Recent studies by other groups have suggested that Ang II and the receptor for Ang II may be involved in tumor growth, angiogenesis, or metastasis [34–36]. Because of this, we challenged ACE 10/10 mice with mouse models of melanoma or lymphoma. Surprisingly, the ACE 10/10 mice showed an enhanced inflammatory response that markedly limited tumor growth. These findings were reversed by treating ACE 10/10 mice with ACE inhibitors, indicating that it is the presence of ACE, and not its absence from endothelium, that is responsible for the phenotype. Investigation of the immune response showed that tumor challenge of ACE 10/10 mice resulted in increased numbers of tumor-specific CD8 T cells, as compared to WT mice challenged in an equivalent fashion. Enhanced tumor resistance was transferable to WT mice by bone marrow transplantation from ACE 10/10 mice. Macrophages from tumor-bearing ACE 10/10 mice had an increased pro-inflammatory profile characterized by increased interleukin (IL)-12 and nitric oxide expression but a decrease of IL-10. These effects do not appear mediated by Ang II since both pharmacologic inhibition of the AT1 receptor or genetic elimination of angiotensinogen production did not affect enhanced tumor resistance. In contrast, the elimination of CD8 T cells converted ACE 10/10 mice to fully tumor susceptible.
Our group is actively pursuing the mechanisms for the increased immune response observed in ACE 10/10 mice. A possible explanation incorporating the changed phenotype of macrophages with increased CD8 T cell numbers is that ACE overexpression by macrophages may enhance processing of some peptides by antigen-presenting cells. However, there seems little doubt that the changes noted in ACE 10/10 mice are complex, as indicated by experiments where naive ACE 10/10 macrophages were stimulated with lipopolysaccharide (LPS) in vitro. Cells from ACE 10/10 mice secreted far more pro-inflammatory cytokines (such as TNF-α and IL-12) than WT cells. These results show that even without the interplay between T cells and antigen-presenting cells, the ACE overexpressing macrophages from ACE 10/10 mice are intrinsically different from similar WT cells.
Conclusion
ACE and the renin–angiotensin system contribute to blood pressure regulation. Further, there is overwhelming evidence showing an important role of ACE inhibition in treating heart failure. What recent studies show is that ACE probably plays a role in many other physiologic processes. Indeed, the manipulation of ACE expression levels, as present in ACE 10/10 mice, suggests a means by which ACE expression levels may be regulated as a strategy for immune modulation, potentially impacting a variety of common human diseases. Whether such approaches ultimately contribute to an ability to enhance human resistance to tumors or even to chronic infections remains to be seen, but such studies suggest that the role of the peptidase ACE is significantly more complicated than just the conversion of Ang I to Ang II.
Acknowledgments
This work was supported by grants R01 DK039777 and R01 DK051445 from the National Institutes of Health. H.D.X. is supported by Scientist Development Grant 0530136N from the American Heart Association. S.F. is supported by a Beginning Grant-in-Aid 0665176B0 from the American Heart Association and a Pathway to Independence Award from NIH/NHLBI, K99 HL088000.
References
- 1.Bernstein KE, Martin BM, Bernstein EA, Linton J, Striker L, Striker G (1988) The isolation of angiotensin-converting enzyme cDNA. J Biol Chem 263:11021–11024 [PubMed] [Google Scholar]
- 2.Soubrier F, Alhenc-Gelas F, Hubert C, Allegrini J, John M, Tregear G, Corvol P (1988) Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc Natl Acad Sci U S A 85:9386–9390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernstein KE, Martin BM, Edwards AS, Bernstein EA (1989) Mouse angiotensin-converting enzyme is a protein composed of two homologous domains. J Biol Chem 264:11945–11951 [PubMed] [Google Scholar]
- 4.Ehlers MR, Fox EA, Strydom DJ, Riordan JF (1989) Molecular cloning of human testicular angiotensin-converting enzyme: the testis isozyme is identical to the C-terminal half of endothelial angiotensin-converting enzyme. Proc Natl Acad Sci U S A 86:7741–7745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Howard TE, Shai SY, Langford KG, Martin BM, Bernstein KE (1990) Transcription of testicular angiotensin-converting enzyme (ACE) is initiated within the 12th intron of the somatic ACE gene. Mol Cell Biol 10:4294–4302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langford KG, Shai SY, Howard TE, Kovac HJ, Overbeek PA, Bernstein KE (1991) Transgenic mice demonstrate a testisspecific promoter for angiotensin-converting enzyme. J Biol Chem 266:15559–15562 [PubMed] [Google Scholar]
- 7.Ehlers MR, Chen YN, Riordan JF (1992) The unique N-terminal sequence of testis angiotensin-converting enzyme is heavily O-glycosylated and unessential for activity or stability. Biochem Biophys Res Commun 183:199–205 [DOI] [PubMed] [Google Scholar]
- 8.Wei L, Alhenc-Gelas F, Soubrier F, Michaud A, Corvol P, Clauser E (1991) Expression and characterization of recombinant human angiotensin I-converting enzyme. Evidence for a C-terminal transmembrane anchor and for a proteolytic processing of the secreted recombinant and plasma enzymes. J Biol Chem 266:5540–5546 [PubMed] [Google Scholar]
- 9.Williams TA, Michaud A, Houard X, Chauvet MT, Soubrier F, Corvol P (1996) Drosophila melanogaster angiotensin I-converting enzyme expressed in Pichia pastoris resembles the C domain of the mammalian homologue and does not require glycosylation for secretion and enzymic activity. Biochem J 318(Pt 1):125–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riviere G, Michaud A, Corradi HR, Sturrock ED, Ravi Acharya K, Cogez V, Bohin JP, Vieau D, Corvol P (2007) Characterization of the first angiotensin-converting like enzyme in bacteria: ancestor ACE is already active. Gene 399:81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coates D (2003) The angiotensin converting enzyme (ACE). Int J Biochem Cell Biol 35:769–773 [DOI] [PubMed] [Google Scholar]
- 12.Harmer D, Gilbert M, Borman R, Clark KL (2002) Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett 532:107–110 [DOI] [PubMed] [Google Scholar]
- 13.Ryan US, Ryan JW, Whitaker C, Chiu A (1976) Localization of angiotensin converting enzyme (kininase II). II. Immunocytochemistry and immunofluorescence. Tissue Cell 8:125–145 [DOI] [PubMed] [Google Scholar]
- 14.Cushman DW, Cheung HS (1971) Concentrations of angiotensin-converting enzyme in tissues of the rat. Biochim Biophys Acta 250:261–265 [DOI] [PubMed] [Google Scholar]
- 15.Danilov SM, Sadovnikova E, Scharenborg N, Balyasnikova IV, Svinareva DA, Semikina EL, Parovichnikova EN, Savchenko VG, Adema GJ (2003) Angiotensin-converting enzyme (CD143) is abundantly expressed by dendritic cells and discriminates human monocyte-derived dendritic cells from acute myeloid leukemia-derived dendritic cells. Exp Hematol 31:1301–1309 [DOI] [PubMed] [Google Scholar]
- 16.Saijonmaa O, Nyman T, Fyhrquist F (2007) Atorvastatin inhibits angiotensin-converting enzyme induction in differentiating human macrophages. Am J Physiol Heart Circ Physiol 292:H1917–H1921 [DOI] [PubMed] [Google Scholar]
- 17.Erdos EG, Schulz WW, Gafford JT, Defendini R (1985) Neutral metalloendopeptidase in human male genital tract. Comparison to angiotensin I-converting enzyme. Lab Invest 52:437–447 [PubMed] [Google Scholar]
- 18.van Sande ME, Scharpe SL, Neels HM, Van Camp KO (1985) Distribution of angiotensin converting enzyme in human tissues. Clin Chim Acta 147:255–260 [DOI] [PubMed] [Google Scholar]
- 19.Hooper NM (1991) Angiotensin converting enzyme: implications from molecular biology for its physiological functions. Int J Biochem 23:641–647 [DOI] [PubMed] [Google Scholar]
- 20.Natesh R, Schwager SL, Sturrock ED, Acharya KR (2003) Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature 421:551–554 [DOI] [PubMed] [Google Scholar]
- 21.Krege JH, John SW, Langenbach LL, Hodgin JB, Hagaman JR, Bachman ES, Jennette JC, O’Brien DA, Smithies O (1995) Male–female differences in fertility and blood pressure in ACE-deficient mice. Nature 375:146–148 [DOI] [PubMed] [Google Scholar]
- 22.Esther CR Jr, Howard TE, Marino EM, Goddard JM, Capecchi MR, Bernstein KE (1996) Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology, and reduced male fertility. Lab Invest 74:953–965 [PubMed] [Google Scholar]
- 23.Fuchs S, Xiao HD, Cole JM, Adams JW, Frenzel K, Michaud A, Zhao H, Keshelava G, Capecchi MR, Corvol P, Bernstein KE (2004) Role of the N-terminal catalytic domain of angiotensin-converting enzyme investigated by targeted inactivation in mice. J Biol Chem 279:15946–15953 [DOI] [PubMed] [Google Scholar]
- 24.Fuchs S, Xiao HD, Hubert C, Michaud A, Campbell DJ, Adams JW, Capecchi MR, Corvol P, Bernstein KE (2008) Angiotensin-converting enzyme C-terminal catalytic domain is the main site of angiotensin i cleavage in vivo. Hypertension 51(2):267–274 [DOI] [PubMed] [Google Scholar]
- 25.Lenfant M, Wdzieczak-Bakala J, Guittet E, Prome JC, Sotty D, Frindel E (1989) Inhibitor of hematopoietic pluripotent stem cell proliferation: purification and determination of its structure. Proc Natl Acad Sci U S A 86:779–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole J, Quach DL, Sundaram K, Corvol P, Capecchi MR, Bernstein KE (2002) Mice lacking endothelial angiotensin-converting enzyme have a normal blood pressure. Circ Res 90:87–92 [DOI] [PubMed] [Google Scholar]
- 27.Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC Jr, Kasi VS, Hoit BD, Keshelava G, Zhao H, Capecchi MR, Bernstein KE (2004) Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am J Pathol 165:1019–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dahlof B (1995) Effect of angiotensin II blockade on cardiac hypertrophy and remodelling: a review. J Hum Hypertens 5(9 Suppl):S37–S44 [PubMed] [Google Scholar]
- 29.Pillai JB, Gupta M, Rajamohan SB, Lang R, Raman J, Gupta MP (2006) Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol 291:H1545–H1553 [DOI] [PubMed] [Google Scholar]
- 30.Kasi VS, Xiao HD, Shang LL, Iravanian S, Langberg J, Witham EA, Jiao Z, Gallego CJ, Bernstein KE, Dudley SC Jr (2007) Cardiac-restricted angiotensin-converting enzyme overexpression causes conduction defects and connexin dysregulation. Am J Physiol Heart Circ Physiol 293:H182–H192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao HD, Fuchs S, Bernstein EA, Li P, Campbell DJ, Bernstein KE (2008) Mice expressing ACE only in the heart show that increased cardiac angiotensin II is not associated with cardiac hypertrophy. Am J Physiol Heart Circ Physiol 294:H659–H667 [DOI] [PubMed] [Google Scholar]
- 32.Reudelhuber TL, Bernstein KE, Delafontaine P (2007) Is angiotensin II a direct mediator of left ventricular hypertrophy? Time for another look. Hypertension 49:1196–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen XZ, Li P, Weiss D, Fuchs S, Xiao HD, Adams JA, Williams IR, Capecchi MR, Taylor WR, Bernstein KE (2007) Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am J Pathol 170:2122–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uemura H, Ishiguro H, Kubota Y (2006) Angiotensin II receptor blocker: possibility of antitumor agent for prostate cancer. Mini Rev Med Chem 6:835–844 [DOI] [PubMed] [Google Scholar]
- 35.Ino K, Shibata K, Kajiyama H, Nawa A, Nomura S, Kikkawa F (2006) Manipulating the angiotensin system—new approaches to the treatment of solid tumours. Expert Opin Biol Ther 6:243–255 [DOI] [PubMed] [Google Scholar]
- 36.Egami K, Murohara T, Shimada T, Sasaki K, Shintani S, Sugaya T, Ishii M, Akagi M, Ikeda H, Matsuishi T, Imaizumi T (2003) Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J Clin Invest 112:67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]