

Abstract
The purpose of this study was to prepare lipospheres containing aceclofenac intended for topical skin delivery with the aim of exploiting the favorable properties of this carrier system and developing a sustained release formula to overcome the side effects resulting from aceclofenac oral administration. Lipospheres were prepared using different lipid cores and phospholipid coats adopting melt and solvent techniques. Characterization was carried out through photomicroscopy, scanning electron microscopy, particle size analysis, DSC, In vitro drug release and storage study. The anti-inflammatory effect of liposphere systems was assessed by the rat paw edema technique and compared to the marketed product. Results revealed that liposphere systems were able to entrap aceclofenac at very high levels (93.1%). The particle size of liposphere systems was well suited for topical drug delivery. DSC revealed the molecular dispersion of aceclofenac when incorporated in lipospheres. Both entrapment efficiency and release were affected by the technique of preparation, core and coat types, core to coat ratio and drug loading. Lipospheres were very stable after 3 months storage at 2–8°C manifested by low leakage rate (less than 7%) and no major changes in particle size. Finally, liposphere systems were found to possess superior anti-inflammatory activity compared to the marketed product in both lotion and paste consistencies. Liposphere systems proved to be a promising topical system for the delivery of aceclofenac as they possessed the ability to entrap the drug at very high levels and high stability, and to sustain the anti-inflammatory effect of the drug.
Key words: aceclofenac, animal experiment, entrapment, formulation, lipospheres, stability, sustained release
INTRODUCTION
In recent years, it has become more and more evident that the development of new drugs alone is not sufficient to ensure progress in drug therapy. A promising strategy involves the development of suitable drug carrier systems. Lipid particles based on triglycerides, waxes or fatty acids as matrix lipids are being intensively investigated as potential carrier systems, in particular for lipophilic substances (1).
The lipospheres system is a newly introduced lipid-based carrier system developed for parenteral and topical drug delivery of bioactive compounds. Lipospheres consist of water-dispersible solid microparticles of particle size between 0.2–100 µm in diameter and composed of a solid hydrophobic fat core stabilized by one monolayer of phospholipid molecules embedded in their surface which are a potential group of penetration enhancers. Both egg and soybean phosphatidylcholine contain unsaturated fatty acids which may be responsible for the penetration enhancement (2). The packing nature of unsaturated fatty acids disrupts the stratum corneum lipid structure and enhances the percutaneous penetration of drugs; also they strongly raise the fluidity of the stratum corneum. In addition, lecithin has a high affinity for epidermal tissue and even seem to improve skin hydration (2). Being biodegradable and composed of natural body constituents, topically administered phospholipids can be generally considered as safe (2).
Lipospheres like solid lipid nanoparticles are one of the carriers of choice for topically applied drugs because their lipid components have an approved status or are excipients used in commercially available topical cosmetic or pharmaceutical preparations. The small size of the lipid particles ensures close contact to the stratum corneum and can increase the amount of the drug penetrating into the mucosa or skin. Due to their solid lipid matrix, controlled release from these carriers is possible which is important to supply the drug over a prolonged period of time and to reduce systemic absorption, increased drug stability can be achieved and finally lipospheres possess a film forming ability leading to occlusive properties (3).
The lipospheres carrier system has several advantages over other delivery systems, including emulsions, liposomes and microspheres, such as: better physical stability, low cost of ingredients, ease of preparation and scale-up, high dispersability in an aqueous medium, high entrapment of hydrophobic drugs, controlled particle size and extended release of entrapped drug (4).
Aceclofenac is a potent analgesic, antipyretic and anti-inflammatory agent with side effects affecting the gastrointestinal tract, liver, kidney and platelet functions (5). The aim of this work was to formulate aceclofenac in lipospheres to be applied topically on the skin for the purpose of sustaining its release to avoid the side effects resulting from the oral administration of this drug. The prepared lipospheres were characterized and the anti-inflammatory activity of the selected formulation was determined and compared to that of the marketed product.
MATERIALS AND METHODS
Materials
Acelofenac was kindly provided by Bristol-Myers Squibb Company, Cairo, Egypt (99.9% purity). l-α-Phosphatidylcholine, type X-E: from dried egg yolk, stearic acid, stearyl alcohol and carrageenan were purchased from Sigma Chemical Co., St. Louis, USA. Tristearin was purchased from Fluka Chemical Co., Buchs, Switzerland. Soybean lecithin (Phospholipon® 90G) was purchased from Nattermann, Cologne, Germany (PC content 94–102%). Chloroform, absolute ethyl alcohol, potassium dihydrogen phosphate, disodium hydrogen phosphate and sodium chloride were purchased from Adwic, El-Nasr chemical Co., Cairo, Egypt according to the methods of Prolabo, Paris, France. Spectra/Por dialysis membrane, 12.000–14.000 molecular weight cut off was purchased from Spectrum Laboratories Inc., Rancho Dominguez, Canada.
Preparation of Aceclofenac Lipospheres
Melt Method
In this method, 400, 600 and 800 mg of different lipid materials (glyceryl stearate, stearic acid and stearyl alcohol) were used as the core material, an amount of 200 mg of egg phosphatidylcholine or soybean phosphatidylcholine was used as the coat material corresponding to core to coat ratio (Cr/Ct) of 2:1, 3:1 and 4:1 respectively (4). Aceclofenac was used in different amounts viz. 25, 50 and 100 mg.
The lipid core material was melted on a water bath then the required amount of aceclofenac was dispersed in this molten lipid. A volume of 10 ml of phosphate buffered saline (PBS) at 75°C was added at once with 200 mg of phospholipid. The mixture was homogenized for 15 min using a bath type sonicator (Crest Ultrasonics Carp, Trenton, USA), after which a uniform emulsion was obtained. The milky formulation was then rapidly cooled down to 20°C with continued shaking for another 5 min for formation of uniform dispersion of lipospheres.
Solvent Evaporation Method
Accurately weighed amounts of the drug, lipid core and phospholipids were dissolved in chloroform (4). The organic solvent was slowly evaporated under reduced pressure at 50–60°C using a rotary evaporator (Janke and Kunkel, model RVO5-ST, IKA Laboratories, Staufen, Germany). The resulting solid was mixed with 10 ml PBS at 50–60°C with continuous mixing till the formation of a homogenous dispersion. The temperature was then reduced to 10°C with continuous rotation at 150 rpm for 5 min (6). The system was sonicated for 15 min in a bath type sonicator then cooled down to 20°C with continued shaking for another 5 min. Table I summarizes the different formulations of aceclofenac lipospheres.
Table I.
The Composition of Different Lipospheres Preparations, entrapment Efficiency Percentages, T 8hr a and Particle Size of Different Lipospheres Preparations
| Code | Method of Preparation | Core Material | Coat Material | Cr/Cte | Amount of Drug (mgs) | Entrapment Efficiency Percent ± S.D | T 8hr ± S.D | Particle Size (µm) |
|---|---|---|---|---|---|---|---|---|
| F1 | Melt | Tristearin | E.P.C.c | 2:1 | 50 | 93.10 ± 0.29 | 58.33 ± 0.31 | 0.69 |
| F2 | Melt | Tristearin | S.P.C.d | 2:1 | 50 | 83.96 ± 1.42 | 66.02 ± 0.93 | 0.86 |
| F3 | Solvent | Tristearin | E.P.C. | 2:1 | 50 | 86.40 ± 0.47 | 73.06 ± 2.63 | 0.66 |
| F4 | Solvent | Tristearin | S.P.C. | 2:1 | 50 | 80.67 ± 0.91 | 74.84 ± 2.39 | 0.63 |
| F5 | Melt | Tristearin | E.P.C. | 3:1 | 50 | 94.31 ± 0.89 | 44.87 ± 1.48 | 0.72 |
| F6 | Solvent | Tristearin | E.P.C. | 3:1 | 50 | 92.00 ± 0.48 | 41.60 ± 1.06 | 0.59,0.84 |
| F7 | Melt | Tristearin | E.P.C. | 4:1 | 50 | 94.60 ± 0.12 | 41.66 ± 0.25 | 0.66,1.30 |
| F8 | Solvent | Tristearin | E.P.C. | 4:1 | 50 | 92.10 ± 0.26 | 41.18 ± 1.93 | 0.61,0.85 |
| F9 | Melt | Tristearin | E.P.C. | 2:1 | 25 | 93.76 ± 0.30 | 58.25 ± 3.40 | 0.67 |
| F10 | Melt | Tristearin | E.P.C. | 2:1 | 100 | 51.75 ± 0.72 | 62.85 ± 1.28 | 0.84 |
| F11 | Melt | Stearic acid | E.P.C. | 2:1 | 50 | 53.88 ± 0.65 | 23.67 ± 0.48 | 1.19 |
| F12 | Melt | Stearic acid | S.P.C. | 2:1 | 50 | 49.76 ± 0.49 | 33.10 ± 1.89 | 0.84 |
| F13 | Solvent | Stearic acid | E.P.C. | 2:1 | 50 | 44.37 ± 1.20 | 38.34 ± 1.32 | 0.84 |
| F14 | Solvent | Stearic acid | S.P.C. | 2:1 | 50 | 37.60 ± 0.81 | 38.12 ± 1.57 | 0.56 |
| F15 | Melt | S.A.b | E.P.C. | 2:1 | 50 | 47.00 ± 1.90 | 31.74 ± 0.66 | 0.69 |
| F16 | Melt | S.A. | S.P.C. | 2:1 | 50 | 34.00 ± 2.90 | 32.73 ± 1.27 | 0.58 |
| F17 | Solvent | S.A. | E.P.C. | 2:1 | 50 | 40.24 ± 1.47 | 71.24 ± 1.65 | 0.62 |
| F18 | Solvent | S.A. | S.P.C. | 2:1 | 50 | 22.90 ± 0.28 | 81.02 ± 0.18 | 0.61 |
| F19 | Melt | S.A. | S.P.C. | 3:1 | 50 | 41.83 ± 0.62 | 30.86 ± 0.23 | 0.60,0.83 |
| F20 | Solvent | S.A. | S.P.C. | 3:1 | 50 | 29.44 ± 0.46 | 46.49 ± 2.49 | 0.66 |
| F21 | Melt | S.A. | S.P.C. | 4:1 | 50 | 45.82 ± 0.43 | 29.69 ± 1.15 | 0.85 |
| F22 | Solvent | S.A. | S.P.C. | 4:1 | 50 | 31.57 ± 0.90 | 40.79 ± 0.29 | 0.85 |
| F23 | Solvent | S.A. | S.P.C. | 2:1 | 25 | 45.43 ± 1.70 | 79.68 ± 1.76 | 0.56 |
| F24 | Solvent | S.A. | S.P.C. | 2:1 | 100 | 11.93 ± 0.45 | 85.52 ± 1.84 | 0.67 |
aCumulative percent of aceclofenac released after 8 h
bStearyl alcohol
cEgg phosphatidylcholine
dSoybean phosphatidylcholine
eLipid core: phospholipid coat ratio
Separation of Unentrapped Aceclofenac from the Prepared Lipospheres
Aceclofenac lipospheres were separated from free unentrapped aceclofenac by centrifugation at 20,000 g for 30 min at 4°C (7–9). The pellets formed were washed twice each with 10 ml phosphate buffered saline and recentrifuged again for 30 min. The lipospheres were decanted and kept in the refrigerator for further investigations.
Determination of Entrapment Efficiency
The entrapped drug concentration was determined by lysis of the lipospheres with absolute alcohol and sonication (8,9). Accurately weighed amount of loaded lipospheres (50 mg) was dissolved in 10 ml absolute alcohol and covered well with parafilm to prevent evaporation.
The solution was sonicated for 15 min to obtain a clear solution. An aliquot of 1 ml of this solution was added to 9 ml of absolute alcohol. The solution was sonicated for another 15 min. The concentration of aceclofenac in absolute alcohol was determined spectrophotometrically (Shimadzu, model UV-1601 PC, Kyoto, Japan) at 276 nm after appropriate dilution. Unloaded lipospheres produced insignificant absorbance values at the same wavelength. Each sample was analyzed in triplicate.
The entrapment efficiency was calculated through the following relationship:
 |
1 |
Characterization of Aceclofenac Lipospheres
Photomicroscopic Analysis
A drop of lipospheres preparation was placed on a slide for morphological examination under binocular optical microscope (Carl Zeiss, Model Axiolab A, Berlin, Germany) and photographed at a magnification of ×400 by means of a fitted camera (Panasonic, Japan).
Electron Microscopy
The detailed surface characteristics of the selected aceclofenac lipospheres formulation were observed using a scanning electron microscope (Model JEM-100S, Jeol, Tokyo, Japan). The lipospheres sample was attached to the specimen holder using a double coated adhesive tape and gold coated (~20 nm thickness) under vacuum using a sputter coater (Model JFC-1100, Jeol, Japan) for 5–10 min at 40 mA and then investigated at 30 kV (10).
Particle Size Analysis
The size of the lipospheres was determined by light scattering based on laser diffraction using the laser diffraction particle size analyzer, Malvern Master Sizer (Malvern Instruments Ltd., WorcesterShire, UK) (11) and the distribution modal size was estimated. Measurements were performed using a 45 mm focus objective, a beam length of 2.4 mm and obscuration levels from 5 to 10%.
Differential Scanning Calorimetry (DSC)
Samples of aceclofenac, plain and drug loaded lipospheres of the selected formulation were submitted to DSC analysis using differential scanning calorimeter (Schimadzu, model TA-50 WSI, Kyoto, Japan) calibrated with indium. The analysis was carried out on 1 mg samples sealed in standard aluminum pans. Thermograms were obtained at a scanning rate of 10°C/min using dry nitrogen flow of (25 ml/min). Each sample was scanned between zero and 400°C.
In Vitro Release of Aceclofenac from Lipospheres
The release of aceclofenac from the prepared lipospheres was determined applying the membrane diffusion technique (6) using molecular porous membrane (Spectra/Por dialysis membrane 12–14.000 M.wt cut off). An accurately measured amount of aceclofenac lipospheres, equivalent to 2 mg aceclofenac, was suspended in 1 ml phosphate buffered saline (pH 7.4) and transferred to a glass cylinder having a length of 7 cm and diameter of 2.5 cm. This cylinder was fitted, before addition of lipospheres suspension, with the presoaked dialysis membrane and was suspended in the dissolution flask of the USP dissolution tester (Pharma Test, Hainburg, Germany) containing 100 ml phosphate buffered saline (pH 7.4). The apparatus was adjusted to a constant speed (50 rpm) and a temperature of 32°C. Samples were collected after 15 min, 30 min, 1 h and every hour over a period of 8 h and assayed spectrophotometrically for drug content at 274 nm. Results were the mean of three runs. The release data was subjected to kinetic treatment to determine the order of release.
Storage Study for Lipospheres
Aceclofenac lipospheres were incubated at refrigeration temperature (2–8°C) in sealed containers. Samples were taken at definite time intervals and the percent of aceclofenac retained at all intervals as well as the particle size after the end of the storage study were determined.
In Vivo Anti-inflammatory Study of Lipospheres
The anti-inflammatory activity of the selected lipospheres formulation (F1) applied both in solid and in dispersion form was estimated and compared to the marketed product using the rat paw edema test. The protocol of the present work was approved by Experiments and Advanced Pharmaceutical Research Unit (EAPRU), Faculty of Pharmacy, Ain Shams University, Cairo, Egypt (12).
Adult male albino rats of 6–8 weeks of age, each weighing 130–150 g were divided into five groups, of five animals each. Group I received topical saline application, group II received plain lipospheres, group III received lipospheres (F1) in solid pellet form as a paste, group IV received the same formula of group III but in dispersion form as a lotion, group V received the marketed Bristaflam cream®. The volume of paw edema (milliliter) was measured in each animal using a plethysmometer (UGO-Basile, 7140, Comerio, Italy) to a precision of two decimal places. The rats were marked on the left hind paw just beyond the tibiotarsal junction, so that every time the paw was dipped in the electrolyte fluid column up to a fixed mark to ensure constant paw volume.
The tested preparations were applied to the left hind paws of rats using an amount equivalent to 1 mg of aceclofenac. The area of application was occluded with a parafilm® and was left in place for 2 h. The parafilm was then removed and the residual formulation on the surface was wiped off with cotton wool.
After 2 h of topical application, initial paw volume of the rats was measured by dipping the rat paw into the electrolyte column just before carrageenan injection and the increase in volume due to fluid displacement was noted from a digital display, followed by the injection of 0.1 ml of 1% (w/v) carrageenan solution in saline in the subplantar region of left hind paw of the rats. Measurement of paw volume was done after 1, 2, 3, 4, 5, 6, 7 and 8 h. The edema rate and inhibition rate of each group was calculated as follows:
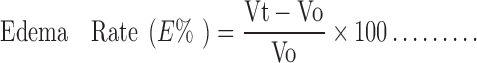 |
2 |
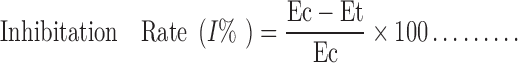 |
3 |
Where: Vo is the mean paw volume before carrageenan injection (ml), Vt is the mean paw volume after carrageenan injection (ml), Ec is the edema rate of control group, and Et is the edema rate of the treated group (13).
Data were expressed as mean ± S.D and statistically assessed by one way analysis of variance (ANOVA). Values for edema rate percentage for lipospheres were compared to the saline control and the differences were determined statistically using Dunnett’s t test. P < 0.05 was considered significant.
RESULTS AND DISCUSSION
Lipospheres represent a promising lipid based topical carrier system consisting of a lipid core stabilized by a layer of phospholipid coat embedded in their surface. Different formulation variables have been found to affect the drug entrapment inside the lipospheres as well as its release profile.
Entrapment Efficiency for Lipospheres
Effect of Method of Preparation
From the results in Table I, it is obvious that under any condition, the melt technique produced lipospheres with significantly higher entrapment efficiency than the solvent technique (P < 0.05). This came in accordance with other research workers (7,14). The melt method’s superiority over the solvent evaporation method could be attributed to the fact that the former permits better drug incorporation into the solid lipid core in contrary to the latter which permits better drug entrapment in the coat. Also, this may be explained on the basis of the higher stability of the primary emulsion formed in the case of melt method due to the higher viscosity of the lipid phase acting as an efficient barrier and thus preventing the diffusion of the drug to the outer phase. The stability of this emulsion is claimed to be a key factor in the encapsulation process (14) suggesting that the drug loading is highly dependant on the method of preparation (7,15). Furthermore, the use of the melt technique in the preparation is an advantage since it is a one step technique which doesn’t involve the use of solvents.
Effect of Lipid Core Type
Aceclofenac showed significant differences in entrapment efficiency for various lipid cores as shown in Table I (P < 0.001). The observed high entrapment efficiency of the drug in the hydrophobic cores is believed to be caused by the poor aqueous solubility of the drug (16). On the basis of aceclofenac entrapment efficiency into lipospheres, the utilized lipid core categories can be arranged in the following decreasing order: triglyceride > fatty acid > fatty alcohol due to the fact that tristearin is a non polar triglyceride while stearic acid and stearyl alcohol are polar hydrophilic lipids (17).
Effect of Phospholipid Type
Egg phosphatidylcholine gave significantly higher entrapment for aceclofenac than soybean phosphatidylcholine (P < 0.05) as shown in Table I due to the difference in fatty acid distribution of both phosphatidylcholine types. Egg phosphatidylcholine contains (palmitic acid 33%, stearic acid 13%, oleic acid 31% and linoleic acid 15% with minor contributions from other fatty acids; Sigma certificate of analysis) while for soybean phosphatidylcholine, the fatty acid content is (palmitic acid 12.9%, stearic acid 4.4%, oleic acid 10.5%, linoleic acid 66.5%, linolenic acid 5.7%; Nattermann phospholipid GmbH certificate of analysis). In the case of soybean phosphatidylcholine, the predominant fatty acid is the unsaturated linoleic acid, representing 67% of the total composition while egg phosphatidylcholine is less unsaturated and the linoleic acid represents 15% only. The increased membrane fatty acid unsaturation causes an increase in the membrane fluidity which is one of the major factors affecting the encapsulation efficiency of the drug. The more unsaturated the phospholipid the more leakage of trapped drug occurs. Also, unsaturated fatty acid phospholipid chains are able to pack less efficiently than saturated ones and are more loosely spaced than the later (18,19). Similar results were obtained in liposomes prepared by both types of phosphatidylcholine in which the difference between the phospholipid fatty acid composition (including chain length and degree of unsaturation) influenced liposomal membrane properties (18).
Effect of Core to Coat Ratio
Tristearin lipospheres F1 and F3 and stearyl alcohol lipospheres F16 and F18 were selected for this study as they represented formulations with the highest and lowest entrapment efficiency respectively among those prepared by the melt and solvent techniques.
Despite being statistically significant, no remarkable increase in entrapment efficiency upon increasing Cr/Ct from 2:1 to 3:1 and 4:1 was observed for tristearin lipospheres but was more obvious for stearyl alcohol lipospheres as shown in Table I. This came in accordance with other researchers claiming that the percentage of drug incorporated into the solid hydrophobic core increases as the amount of triglyceride was increased at fixed phospholipid ratio (11,15) which may be attributed to the increased amount of the dissolved drug in the melted lipid.
Effect of Amount of Drug Used in Lipospheres Preparation
Aceclofenac lipospheres formulations F1 and F18 were selected for this study as they represented the highest and the lowest entrapment efficiency percent respectively among all other formulations. By inspection of Table I it is obvious that by increasing the amount of drug used in the preparation of lipospheres from 25 to 50 and 100 mg, the entrapment efficiency significantly decreased (P < 0.05). This may be explained by the fact that as the ratio of drug to the lipid matrix increases, a reduced space becomes available for the drug to be entrapped. Similar results were obtained with verapamil solid lipid nanoparticles (20).
Characterization of Aceclofenac Lipospheres
Photomicroscopic Analysis
The photomicrograph of aceclofenac lipospheres of formula F1 is represented by Fig. 1. It reveals the uniform spherical shape of lipospheres (4) showing the solid lipid core and the phospholipid coat.
Fig. 1.

Photomicrograph of aceclofenac lipospheres prepared by melt method at a magnification of ×400
Electron Microscopy
Figure 2 illustrates the SEM of freshly prepared aceclofenac lipospheres. The particles are spherical in shape with irregular surface as usually obtained when phosphatidylcholine is used as the coat (9).
Fig. 2.

Scanning electron micrographs of aceclofenac lipospheres prepared by melt method
Particle Size Analysis
Results shown in Table I reveal that the mean particle size for lipospheres ranged from 0.56 µm with F14 and F23 to 1.19 µm with F11 which makes them suitable candidates for topical route. It was reported that particles less than 3 µm are randomly distributed in the stratum corneum and penetrate the skin (21).
All lipospheres prepared using Cr/Ct ratio 2:1 had a unimodal size. However, by increasing Cr/Ct from 2:1 to 3:1 and 4:1 a slight increase in the mean particle size occurred with the appearance of bimodal frequency distribution curves (not shown) as exhibited in lipospheres F6, F7, F8 and F19. Similar results were obtained by Domb et al. who stated that the average size of particles increases with increasing fat to phospholipid molar ratio (4) which may be attributed to the increased viscosity of the emulsion formed as a result of higher amount of lipid used in the preparation (22). By further inspection of Table I, data reveal that increasing the amount of drug used for the preparation of lipospheres, from 25 to 50 to 100 mg, was accompanied with a slight increase in particle size. Similar results were also obtained with cyclosporine lipospheres (23).
Differential Scanning Calorimetry
DSC is considered as a tool to investigate the melting behavior of crystalline materials like solid lipid nanoparticles (24). Formula F1 was chosen as a representative of aceclofenac lipospheres as it possessed the highest entrapment efficiency. DSC thermogram of aceclofenac showed endothermic peak at 150.4°C as evident in Fig. 3 which is the reported melting point of the drug (25). Plain tristearin lipospheres prepared by melt method showed endothermic peak at 65.74°C. Drug loaded lipospheres prepared by the same method showed a large endothermic peak at 66.15°C and disappearance of the drug peak suggesting a molecular dispersion of aceclofenac into the loaded lipospheres (9) and that aceclofenac exists in amorphous state rather than in crystalline state (26). Also, the melting point decreased from 67.2°C for the bulk tristearin (Fluka certificate of analysis) to 65.74 and 66.15°C for the plain and drug loaded lipospheres respectively which could be attributed to the small size of the prepared lipospheres. Furthermore, the inclusion of drug molecules in the lipid is normally accompanied by a depression in the lipid’s melting point (24).
Fig. 3.

DSC thermogram of aceclofenac, plain and drug loaded lipospheres
In Vitro Release of Aceclofenac from Lipospheres
By inspection of Table I, it is obvious that with all lipospheres prepared using egg phosphatidylcholine, the percentages of drug released after 8 h (T8hr) were overall significantly lower than with those prepared with soybean phosphatidylcholine under the same conditions (same core type, method of preparation, Cr/Ct and amount of drug incorporated; P < 0.05) since the efflux of drugs increases by membrane fatty acid unsaturation (27). In addition, soybean phosphatidylcholine membranes are known to be more fluid than egg phosphatidylcholine membranes resulting in faster drug release (28).
Moreover, T8hr of lipospheres prepared using melt method was overall significantly less than those prepared by solvent evaporation method under the same conditions (P < 0.05). This may be attributed to the fact that the melt method produced lipospheres with higher amount of the drug in the lipid core, while the solvent method resulted in higher amount of the drug in the phospholipid bilayers (7,15). Drug enriched solid cores are known to achieve sustained release of the drug (29).
The significantly different T8hr values among the various core types can be attributed to the physical and chemical properties of the respective cores (10). The highest T8hr value was obtained with stearyl alcohol formulation F24 (85.52%) which may indicate inefficient coating of the drug or may be attributed to the fact that stearyl alcohol has hydroxyl group making the matrix more susceptible to hydration in the dissolution medium thus providing a hydrophilic pathway for water molecules to access the drug and increase the rate of dissolution (10). The lowest T8hr value was obtained with stearic acid, formulation F11 (23.67%). This was explained by other research workers who suggested that fatty acids may potentially undergo a range of interactions with an alkaline medium. The release mechanism is associated with an interaction between the stearic acid and the medium that involves the presence of sodium ions leading to the formation of acid soaps which are crystals that contain the fatty acid and the metal carboxylate (soap) ion pairs. The proportion of the two components is stochiometrically discrete and the influence of sodium is complex with evidence presented for there being an optimal level for maximum release. The increased sodium molarity of the buffer can decrease the overall level of drug release (30).
By increasing the ratio of Cr/Ct from 2:1 to 3:1 to 4:1, T8hr values were significantly decreased for all selected formulations (P < 0.05) which may be attributed to the increased viscosity by an increase in the content of the insoluble ingredients (lipids) (4).
Finally, it was obvious that the release of aceclofenac was independent to some extent on the amount of the loaded drug. These results came in accordance with other research workers who found that the drug dose within the inner triglyceride core can change the amount of drug released without appreciably altering the percentages of released drug under ideal sink conditions (7,20).
The release data was kinetically treated as observed in Table II. Results revealed that aceclofenac was released from lipospheres by a diffusion controlled mechanism similar to results obtained with carbamazepine lipospheres in which the Higuchi profile was the mode of drug release from fatty materials (16). This may be due to the formation of a core enriched with active compound leading to a membrane release governed by Fick’s law of diffusion (31).
Table II.
Kinetic Parameters of the Release Data of Aceclofenac from Lipospheres
| Code | r 2 | Order of Release | ||
|---|---|---|---|---|
| Zero Order | First Order | Diffusion Model | ||
| F1 | 0.935 | 0.975 | 0.992 | Diffusion |
| F2 | 0.950 | 0.989 | 0.992 | Diffusion |
| F3 | 0.861 | 0.936 | 0.955 | Diffusion |
| F4 | 0.899 | 0.966 | 0.974 | Diffusion |
| F5 | 0.964 | 0.979 | 0.985 | Diffusion |
| F6 | 0.943 | 0.953 | 0.967 | Diffusion |
| F7 | 0.935 | 0.959 | 0.991 | Diffusion |
| F8 | 0.935 | 0.963 | 0.985 | Diffusion |
| F9 | 0.954 | 0.986 | 0.997 | Diffusion |
| F10 | 0.951 | 0.985 | 0.988 | Diffusion |
| F11 | 0.974 | 0.983 | 0.996 | Diffusion |
| F12 | 0.973 | 0.984 | 0.988 | Diffusion |
| F13 | 0.963 | 0.976 | 0.982 | Diffusion |
| F14 | 0.962 | 0.979 | 0.988 | Diffusion |
| F15 | 0.953 | 0.966 | 0.991 | Diffusion |
| F16 | 0.954 | 0.970 | 0.991 | Diffusion |
| F17 | 0.931 | 0.977 | 0.986 | Diffusion |
| F18 | 0.920 | 0.974 | 0.976 | Diffusion |
| F19 | 0.927 | 0.943 | 0.971 | Diffusion |
| F20 | 0.950 | 0.976 | 0.993 | Diffusion |
| F21 | 0.975 | 0.983 | 0.989 | Diffusion |
| F22 | 0.977 | 0.987 | 0.989 | Diffusion |
| F23 | 0.952 | 0.993 | 0.996 | Diffusion |
| F24 | 0.958 | 0.982 | 0.990 | Diffusion |
Storage Study
Lipospheres formulation F1 were stored at 2–8°C because it was reported that the gelation and rate of crystal growth of drug was found to be faster at elevated temperature like room temperature due to the increase in fluidity and permeability of the phospholipid membrane surrounding the liposphere particles at a high temperature (11).
After storage for 90 days, 93.7% of aceclofenac was retained in lipospheres. The leakage of aceclofenac from lipospheres could have resulted from the crystallization of lipids directly after production in higher energy modifications (α, ß′) with more imperfections in the crystal lattice followed by the polymorphic transition to the stable ß form during storage causing the expulsion of the drug from the lipid matrix (32). However, the low percent of drug leakage could be ascribed to the role of egg lecithin in the retardation of polymorphic transition from α to ß form in tristearin dispersions. This retardation effect persists during storage of the tristearin dispersions due to the high similarity (saturation) between the acyl chains in egg lecithin and the dispersed tristearin as well as spatial restrictions imposed by the latter (1).
The particle size distribution was recorded after 3 months of storage at 2–8°C. It was found that the particle size slightly increased as size growth for glyceride particles is common. The distribution modal size increased from 0.69 to 0.84. However, this increase was minimal and preserved the unimodal shape as well as lacked the presence of micrometer aggregates as shown in Table III.
Table III.
The Effect of 3 Months Storage on the Particle Size Distribution of the Selected Liposphere Formula F1
| Cumulative Distribution Percentile | Mean Particle Diameter (µm) of Lipospheres Formulations | |
|---|---|---|
| Freshly Prepared | Stored Lipospheres | |
| 10% | 0.45 | 0.51 |
| 20% | 0.50 | 0.56 |
| 50% | 0.63 | 0.70 |
| 80% | 0.71 | 0.84 |
| 90% | 0.75 | 0.89 |
| Distribution modal sizes | 0.69 | 0.84 |
In Vivo Anti-Inflammatory Study
Lipospheres formulation F1 was administered in two forms, paste and lotion consistencies as it was demonstrated that the existence of a solid particle matrix with a size in the nanometer range forms a semisolid dispersion having the appropriate consistency for topical application which represents an alternative drug carrier system to emulsions and polymeric nanoparticles (33). Phosphate buffered saline was used as the aqueous phase in lipospheres preparation. For topical applications, lotion and paste consistencies are desired, which are achieved by adding sodium chloride to the water phase (4).
As evident in Table IV, differences between anti-inflammatory activity of the saline control and plain lipospheres were statistically insignificant at P < 0.05 indicating that plain lipospheres components possess no in vivo anti-inflammatory activity. Depending on the paste and lotion based formulations, the behavior of the lipospheres could change enormously as shown in Table IV and Fig. 4.
Table IV.
The Anti-inflammatory Activity of Plain and Aceclofenac Loaded Lipospheres Compared to the Marketed Bristaflam Cream
| Tested Formulation (Group) | Edema Rate (%) ±S.D. | |||||||
|---|---|---|---|---|---|---|---|---|
| [Inhibition Rate (%)] | ||||||||
| 1 h | 2 h | 3 h | 4 h | 5 h | 6 h | 7 h | 8 h | |
| Control saline (Group I) | 50.13 ± 8.50 | 53.33 ± 3.20 | 63.47 ± 5.66 | 66.07 ± 3.84 | 59.33 ± 6.41 | 52.27 ± 1.86 | 48.69 ± 3.81 | 46.40 ± 3.59 |
| Plain lipospheres (Group II) | 50.71 ± 1.27 | 53.26 ± 1.80 | 56.37 ± 5.90 | 58.92 ± 5.96 | 55.81 ± 1.39 | 52.13 ± 3.38 | 50.11 ± 1.09 | 41.93 ± 2.80 |
| F1 lipospheres pellet form (Group III) | 37.70 ± 1.59** | 38.48 ± 0.18** | 40.31 ± 2.60** | 41.99 ± 2.96** | 38.51 ± 3.70** | 35.10 ± 1.11** | 30.89 ± 4.62** | 22.36 ± 2.40** |
| [24.80] | [27.85] | [36.49] | [36.45] | [35.09] | [32.85] | [36.56] | [51.81] | |
| F1 lipospheres dispersion form (Group IV) | 29.67 ± 1.56** | 35.55 ± 2.21** | 37.60 ± 6.40** | 42.20 ± 4.34** | 36.06 ± 4.33** | 37.08 ± 1.99** | 33.76 ± 3.20** | 23.53 ± 0.51** |
| [40.81] | [33.34] | [40.76] | [36.13] | [39.22] | [29.06] | [30.66] | [49.29] | |
| Marketed product (Group V) | 26.83 ± 1.92** | 31.52 ± 2.92** | 38.04 ± 1.39** | 51.09 ± 1.07** | 40.22 ± 2.31** | 40.43 ± 1.56** | 41.30 ± 2.92* | 41.96 ± 5.22 |
| [46.48] | [40.90] | [40.07] | [22.67] | [32.21] | [22.65] | [15.18] | [9.57] | |
Edema rate % values were statistically significant from saline control using Dunnett’s t test at P < 0.01 (**) or P < 0.05 (*)
PC Phosphatidylcholine
Fig. 4.

Inhibition percentage of aceclofenac lipospheres compared to the marketed product
Regarding lipospheres in dispersion form, rapid onset of action was observed. In the first hour the percent edema for lipospheres dispersion was 29.67% (close to that of the marketed product) and the percent edema inhibition was 40.81%, burst release is useful in topical application to improve penetration of drugs (13). The lipospheres dispersion remained superior to the marketed product in its ability to suppress edema starting from the third hour and throughout the whole study time suggesting the sustained anti-inflammatory activity of aceclofenac lipospheres.
In case of lipospheres in solid pellet form, the % edema inhibition was superior to the marketed product starting from the fourth hour (36.45 versus 22.67%) and throughout the whole study time and exhibited the highest percent edema inhibition. This may be attributed to the film forming ability of the lipospheres pellet which reduces transepidermal water loss similar to solid lipid nanoparticles (13). Occlusive effect of the lipospheres pellet can similarly promote the penetration of actives into the skin (34).
Sustained release of the drug is important to supply the skin with the drug over a prolonged period of time (13). Generally the high specific surface area of the submicron sized lipospheres facilitates contact of encapsulated drug with the stratum corneum resulting in sustained release of the drug for a long period of time. It has been previously reported that the in vivo distribution of lipospheres demonstrate a high affinity to inflamed tissue (17) in which they are well suited for being based on non-irritative and non-toxic lipids (13). Integration of phospholipid molecules with the skin lipids can be held responsible for modifying the properties of the keratinized layer and might have served further to help retain the drug molecules within the skin, thus leading to prolonged presence of drug molecules (35). The drug in its lipo-solubilized state might have found facilitated entry into the tough barrier consisting of stratum corneum.
CONCLUSION
Findings of this investigation suggest that lipospheres can be considered a promising delivery system for topical aceclofenac delivery. Lipospheres were able to entrap the drug at very high levels and sustain its release over a prolonged time. Lipospheres possessed a suitable size for topical route and being based on non irritative and non toxic lipids, lipospheres seemed to be well suited for use on damaged or inflamed skin. Furthermore, lipospheres possessed a very high stability as well as superior anti-inflammatory activity compared to the marketed product.
References
- 1.Bunjes H., Koch M.H.J. Saturated phospholipids promote crystallization but slow down polymorphic transitions in triglyceride nanoparticles. J. Control. Release. 2005;107:229–243. doi: 10.1016/j.jconrel.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Valjakka-Koskela R., Kirjavainen M., Monkkonen J., Urtti A., Kiesvaara J. Enhancement of percutaneous absorption of naproxen by phospholipids. Int. J. Pharm. 1998;175:225–230. doi: 10.1016/S0378-5173(98)00285-3. [DOI] [Google Scholar]
- 3.Souto E.B., Wissing S.A., Barbosa C.M., Muller R.H. Development of a controlled release formulation based on SLN and NLC for topical clotrimazole delivery. Int. J. Pharm. 2004;278:71–77. doi: 10.1016/j.ijpharm.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 4.Domb A.J., Bergelson L., Amselem S., Benita S. Microencapsulation, methods and industrial applications. New York, NY: Marcel Dekker; 1996. Lipospheres for controlled delivery of substances; pp. 377–410. [Google Scholar]
- 5.Insel P.A. Analgesic-antipyretics and anti-inflammatory agents: drugs employed in the treatment of rheumatoid arthritis and gout. In: Goodman L.S., Gilman A., Rall T.W., Nies A.S., Taylor P., editors. The pharmacological basis of therapeutics. New York, NY: McGraw-Hill International editions; 1992. pp. 638–681. [Google Scholar]
- 6.A. Domb and M. Maniar, Inventors. Liposphere delivery systems for local anesthetics. US patent 5 227 165. July 13, 1993.
- 7.Masters D.B., Domb A.J. Liposphere local anesthetic timed-release for perineural site application. Pharm. Res. 1998;15:1038–1045. doi: 10.1023/A:1011978010724. [DOI] [PubMed] [Google Scholar]
- 8.Iannuccelli V., Sala N., Tursilli R., Coppi G., Scalia S. Influence of liposphere preparation on butyl-methoxydibenzoyl methane photostability. Eur. J. Pharm. Biopharm. 2006;63:140–145. doi: 10.1016/j.ejpb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Tursilli R., Casolari A., Iannuccelli V., Scalia S. Enhancement of melatonin photostability by encapsulation in lipospheres. J. Pharm. Biomed. Anal. 2006;40:910–914. doi: 10.1016/j.jpba.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 10.El Gibaly I., Abdel-Ghaffar S.K. Effect of hexacosanol on the characteristics of novel sustained release allopurinol solid lipospheres (SLS): factorial design application and product evaluation. Int. J. Pharm. 2005;294:33–51. doi: 10.1016/j.ijpharm.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 11.Toongsuwan S., Li L., Erickson B.K., Chang H. Formulation and characterization of bupivacaine lipospheres. Int. J. Pharm. 2004;280:57–65. doi: 10.1016/j.ijpharm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 12.Habashy R.R., Abdel-Naim A.B., Khalifa A.E., Al-Azizi M.M. Anti-inflammatory effects of jojoba liquid wax in experimental models. Pharmacol. Res. 2005;51:95–105. doi: 10.1016/j.phrs.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 13.Mei Z., Chen H., Weng T., Yang Y., Yang X. Solid lipid nanoparticles and microemulsion for topical delivery of triptolide. Eur. J. Pharm. Biopharm. 2003;56:189–196. doi: 10.1016/S0939-6411(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 14.Reithmeier H., Herrmann J., Gopferich A. Development and characterization of lipid microparticles as a drug carrier for somatostatin. Int. J. Pharm. 2001;218:133–143. doi: 10.1016/S0378-5173(01)00620-2. [DOI] [PubMed] [Google Scholar]
- 15.Maniar M., Hannibal D., Amselem S., Xie X., Burch R., Domb A. Characterization of Lipospheres ™-1: Effect of carrier and phospholipid on the loading of drug into the lipospheres. Pharm. Res. 1991;8 suppl.:S–185. [Google Scholar]
- 16.Barakat N.S., Yassin A.E.B. In vitro characterization of carbamazepine-loaded precifac lipospheres. Drug Delivery. 2006;13:95–104. doi: 10.1080/10717540500313661. [DOI] [PubMed] [Google Scholar]
- 17.Cortesi R., Esposito E., Luca G., Nastruzzi C. Production of lipospheres as carriers for bioactive compounds. Biomaterials. 2002;23:2283–2294. doi: 10.1016/S0142-9612(01)00362-3. [DOI] [PubMed] [Google Scholar]
- 18.Abra R.M., Mihalko P.J., Schreier H. The effect of lipid composition upon the encapsulation and in vitro leakage of metaproterenol sulfate from 0.2 µm diameter, extruded, multilamellar liposomes. J. Control. Release. 1990;14:71–78. doi: 10.1016/0168-3659(90)90062-X. [DOI] [Google Scholar]
- 19.Nii T., Ishii F. Encapsulation efficiency of water-soluble and insoluble drugs in liposomes prepared by the microencapsulation vesicle method. Int. J. Pharm. 2005;298:198–205. doi: 10.1016/j.ijpharm.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 20.Kim B.D., Na K., Choi H.K. Preparation and characterization of solid lipid nanoparticles (SLN) made of cacao butter and curdlan. Eur. J. Pharm. Sci. 2005;24:199–205. doi: 10.1016/j.ejps.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Escribano E., Calpena A.C., Queralt J., Obach R., Domenech J. Assessment of diclofenac permeation with different formulations: anti-inflammatory study of a selected formula. Eur. J. Pharm Sci. 2003;19:203–210. doi: 10.1016/S0928-0987(03)00103-9. [DOI] [PubMed] [Google Scholar]
- 22.Iscan Y., Hekimoglu S., Sargon M.F., Hincal A.A. Deet-loaded solid lipid particles for skin delivery: In vitro release and skin permeation characteristics in different vehicles. J. Microencapsul. 2006;23:315–327. doi: 10.1080/02652040500444198. [DOI] [PubMed] [Google Scholar]
- 23.Bekerman T., Golenser J., Domb A. Cyclosporin nanoparticulate lipospheres for oral administration. J. Pharm. Sci. 2004;93:1264–1270. doi: 10.1002/jps.20057. [DOI] [PubMed] [Google Scholar]
- 24.Hou D., Xie C., Huang K., Zhu C. The production and characteristics of solid lipid nanoparticles (SLNs) Biomaterials. 2003;24:1781–1785. doi: 10.1016/S0142-9612(02)00578-1. [DOI] [PubMed] [Google Scholar]
- 25.S. Budavari, M. J. O’Neil, A. Smith, P. E. Heckelman, J. F. Kinneary (eds.) The Merck Index, an Encyclopedia of Chemicals, Drugs and Biologicals. Whitehouse station, NJ, Merck and Co., 1996.
- 26.Scalia S., Tursilli R., Sala N., Iannuccelli V. Encapsulation in lipospheres of the complex between butyl methoxydibenzoylmethane and hydroxypropyl-ß-cyclodextrin. Int. J. Pharm. 2006;320:79–85. doi: 10.1016/j.ijpharm.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 27.Vora B., Khopade A.J., Jain N.K. Proniosome based transdermal delivery of levonorgesterol for effective contraception. J. Control. Release. 1998;54:149–165. doi: 10.1016/S0168-3659(97)00100-4. [DOI] [PubMed] [Google Scholar]
- 28.Schreier H., Levy M., Mihalko P. Sustained release of liposome-encapsulated gentamycin and fate of phospholipid following intramuscular injection in mice. J. Control. Release. 1987;5:187–192. doi: 10.1016/0168-3659(87)90010-1. [DOI] [Google Scholar]
- 29.Lippacher A., Muller R.H., Mader K. Preparation of semisolid drug carriers for topical application based on solid lipid nanoparticles. Int. J. Pharm. 2001;214:9–12. doi: 10.1016/S0378-5173(00)00623-2. [DOI] [PubMed] [Google Scholar]
- 30.Robson H., Craig D.Q.M., Deutsch D. An investigation into the release of cefuroxime axetil from taste-masked stearic acid microspheres II. The effects of buffer composition on drug release. Int. J. Pharm. 2000;195:137–145. doi: 10.1016/S0378-5173(99)00391-9. [DOI] [PubMed] [Google Scholar]
- 31.Muller R.H., Radtke M., Wissing S.A. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv. Drug Deliv. Rev. 2002;54:S131–S155. doi: 10.1016/S0169-409X(02)00118-7. [DOI] [PubMed] [Google Scholar]
- 32.Wissing S.A., Kayser O., Muller R.H. Solid lipid nanoparticles for parentral drug delivery. Adv. Drug Deliv. Rev. 2004;56:1257–1272. doi: 10.1016/j.addr.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Lippacher A., Muller R.H., Mader K. Semisolid SLN™ dispersions for topical application influence of formulation and production parameters on viscoelastic properties. Eur. J. Pharm. Biopharm. 2002;53:155–160. doi: 10.1016/S0939-6411(01)00233-8. [DOI] [PubMed] [Google Scholar]
- 34.Souto E.B., Muller R.H., Gohla S. A novel approach based on lipid nanoparticles (SLN®) for topical delivery of a-lipoic acid. J. Microencapsul. 2005;22:581–592. doi: 10.1080/02652040500162378. [DOI] [PubMed] [Google Scholar]
- 35.Bhatia A., Kumar R., Katare O.P. Tamoxifen in topical liposomes: development, characterization and in-vitro evaluation. J Pharm Pharmaceut Sci. 2004;7:252–259. [PubMed] [Google Scholar]