

Abstract
A new class of carbamylating agents based on the cyclosulfamide scaffold is reported. These compounds were found to be efficient time-dependent inhibitors of human neutrophil elastase (HNE). Exploitation of the three sites of diversity present in the cyclosulfamide scaffold yielded compounds which inhibited HNE but not proteinase 3 (PR 3) or bovine trypsin. The findings reported herein suggest that the introduction of appropriate recognition elements into the cyclosulfamide scaffold may lead to highly selective agents of potential value in the design of activity-based probes suitable for investigating proteases associated with the pathogenesis of chronic obstructive pulmonary disease.
Introduction
Chronic obstructive pulmonary disease (COPD) is a major health problem that affects 16 million people in the U.S., and is currently the fourth most common cause of death [1–2]. COPD is a complex disorder associated with an influx of neutrophils, macrophages and CD8+ T cells into the lungs. This is followed by the release of a range of pro-inflammatory chemokines and cytokines, adhesion molecules, transcription factors, as well as an array of proteases [3]. The pathogenesis of COPD is currently unknown, consequently there is a need for (a) a rigorous definition of the cellular and molecular mechanisms of the inflammatory and immune processes which play a role in the pathogenesis and progression of COPD and, (b) illuminating the identity and function(s) of the various proteases involved in COPD [4]. The identification and validation of new molecular targets would likely pave the way toward the development of new and improved therapeutic interventions [5].
During the course of exploratory studies related to the utilization of the cyclosulfamide scaffold in the design of reversible competitive inhibitors of COPD-relevant serine proteases [6], it was observed that urea-type cyclosulfamide derivatives inhibited HNE in a time-dependent manner. We report herein a new class of carbamylating agents (I) (Figure 1) of serine proteases having three points of diversity and potentially amenable to the construction of activity-based probes [7].
Figure 1.
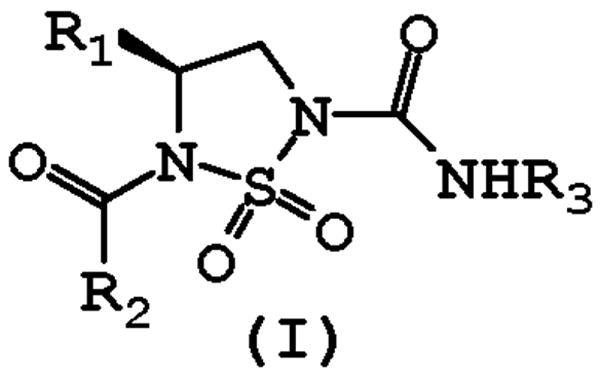
General structure of inhibitor (I).
Materials and methods
General
The 1H and 13C NMR spectra were recorded on a Varian XL-300 or XL-400 NMR spectrometer. A Hewlett-Packard diode array UV/VIS spectrophotometer was used in the in vitro evaluation of the inhibitors. Human neutrophil elastase, proteinase 3, cathepsin G and Boc-Ala-Ala-Nva thiobenzyl ester were purchased from Elastin Products Company, Owensville, MO. Bovine trypsin, methoxysuccinyl Ala-Ala-Pro-Val p-nitroanilide, succinyl Ala-Ala-Pro-Phe p-nitroanilide, 5, 5′-dithio-bis(2-nitrobenzoic acid), and Nα-benzoyl-L-Arg p-nitroanilide were purchased from Sigma Chemicals, St. Louis, MO. Melting points were determined on a Mel-Temp apparatus and are uncorrected. Reagents and solvents were purchased from various chemical suppliers (Aldrich, Acros Organics, TCI America, and Bachem). Silica gel (230–450 mesh) used for flash chromatography was purchased from Sorbent Technologies (Atlanta, GA). Thin layer chromatography was performed using Analtech silica gel plates. The TLC plates were visualized using iodine and/or UV light.
Chemistry
Compounds 7a–g were synthesized using the reaction sequence shown in Scheme 1(a) . Compounds 7a–g and 8–9 are listed in Scheme 1(a) and Scheme 1(b), respectively. The synthetic methodology employed in Scheme 1 is highly versatile and permits the facile introduction of a large number of diverse fragments at the R1, R2, and R3 positions using commercially available natural and unnatural amino acids, carboxylic acids and isocyanates. Intermediate 4 can also be prepared directly from 3 using the Mitsunobu reaction.
Scheme 1.

Synthesis of compounds 7a-g
Representative Syntheses
Compound 1
A solution of N-chlorosulfonyl isocyanate (13.0 mL, 0.15 mol) in methylene chloride (200 mL) was cooled in an ice bath and a solution of t-butyl alcohol (11.12 g, 0.15 mol) in methylene chloride (100 mL) was added dropwise with stirring. After the solution was stirred for 10 minutes, the reaction mixture was transferred to a separatory funnel and added dropwise to a solution of (L) leucine methyl ester hydrochloride (27.25 g, 0.15 mol) and triethylamine (30.4 g, 0.30 mol) in methylene chloride (300 mL) kept in an ice bath. The resulting mixture was then stirred at room temperature overnight. The reaction mixture was washed with 5% HCl (2 × 75 mL) and brine (75 mL). The organic layer was dried over anhydrous Na2SO4 and then concentrated to yield a white solid (45.84 g, 94%): mp 89–91 °C; 1H NMR (CDCl3): δ 0.93 (t, 6H), 1.44 (s, 9H), 1.59 (m, 2H), 1.81 (m, 1H), 3.74 (s, 3H), 4.19 (m, 1H), 5.56 (d, 1H), 7.18 (bs, 1H).
Compound 2
To a solution of compound 1 (4.86 g, 15 mmol) in dry THF (20 mL) kept in a dry ice-acetone bath was added dropwise 2 M lithium borohydride in THF (7.5 mL, 15 mmol). After the reaction was stirred for 3 h, absolute ethanol (50 mL) was added dropwise. The reaction mixture was stirred overnight at room temperature, then cooled to 0 °C, and neutralized with 5% aqueous HCl. The resulting mixture was concentrated and then diluted with H2O (10 mL). The resulting solution was extracted with ethyl acetate (3 × 25 mL), the organic phase was dried over anhydrous sodium sulfate, and then concentrated. The crude product was purified using flash chromatography (hexane/EtOAc, 75:25) to afford a white solid (1.93 g, 43%): mp 114–116 °C; 1H NMR (CDCl3): δ 0.92 (d, 6H), 1.36 (m, 1H), 1.42 (m, 1H), 1.44 (s, 9H), 1.71 (m, 1H), 3.47 (m, 2H), 3.71 (m, 1H), 5.50 (d, 1H).
Compound 3
To a solution of compound 2 (2.96 g, 10 mmol) in dry THF (25 mL) kept in an ice bath was added methanesulfonyl chloride (1.15 g, 10 mmol). Triethylamine (1.35 g, 13.3 mmol) was then added dropwise. After the reaction mixture was stirred for 4 h at room temperature, the solvent was evaporated and the residue was taken up in ethyl acetate (50 mL) and washed with 5% HCl (2 × 10 mL) and brine (10 mL). The organic phase was dried over anhydrous Na2SO4, then concentrated to yield an oil (3.32 g, 89%): 1H NMR (CDCl3): δ 0.92 (t, 6H), 1.42 (m, 2H), 1.44 (s, 9H), 1.71 (m, 1H), 3.03 (s, 3H), 3.71 (m, 1H), 4.19 (dd, 1H), 4.30 (dd, 1H), 5.40 (dd, 1H).
Compound 4
To a solution of compound 3 (3.74 g, 10 mmol) in dry acetonitrile (45 mL) was added dropwise a solution of DBU (1.83 g, 12 mmol) in dry acetonitrile (25 mL). The mixture was stirred for 5 h and the solvent was then evaporated. The residue was taken up in ethyl acetate (50 mL) and washed with 5% aqueous HCl (2 × 15 mL), 5% NaHCO3 (2 × 15 mL) and brine (15 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the solvent evaporated off. The crude product was purified using flash chromatography (hexane/EtOAc, 85:15) to afford a white solid (1.50 g, 54%): mp 97–99 °C; 1H NMR (CDCl3): δ 0.93 (d, 6H), 1.46 (m, 1H), 1.56 (s, 9H), 1.59 (m, 1H), 1.74 (m, 1H), 3.41 (t, 2H), 3.81 (m, 1H), 4.00 (dd, 1H), 4.27 (d, 1H).
Representative procedure for the synthesis of compounds 5a–g
To a solution of (3-methoxyphenyl) acetic acid (0.25 g, 1.5 mmol) in dry methylene chloride (8 mL) was added HATU (0.68 g, 1.8 mmol), followed by TEA (0.30 g, 3 mmol) and compound 4 (0.42 g, 1.5 mmol) in dry methylene chloride (5 mL). The reaction mixture was stirred at room temperature for 4 h and the solvent was evaporated off. The residue was taken up in ethyl acetate (50 mL) and washed with 5% aqueous HCl (2 × 8 mL), 5% NaHCO3 (2 × 8 mL) and brine (2 × 8 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and the solvent evaporated off to give a crude product which was purified using flash chromatography (hexane/EtOAc, 95:5) to give compound 5a as an oil (0.32 g, 50%): 1H NMR (CDCl3): δ 0.92 (t, 6H), 1.57 (m, 2H), 1.58 (s, 9H), 1.63 (m, 1H), 3.78 (m, 2H), 3.79 (s, 3H), 4.03 (q, 2H), 4.54 (m, 1H), 6.80–7.22 (m, 4H).
Compound 5b
white solid (44% yield), mp 98–100 °C; 1H NMR (CDCl3): δ 0.92 (t, 6H), 1.57 (m, 2H), 1.58 (s, 9H), 1.63 (m, 1H), 3.78 (m, 2H), 3.79 (s, 3H), 4.01 (q, 2H), 4.54 (m, 1H), 6.82–7.23 (m, 4H).
Compound 5c
oil (22% yield). 1H NMR (CDCl3): δ 0.92 (t, 6H), 1.57 (m, 2H), 1.58 (s, 9H), 1.63 (m, 1H), 3.78 (m, 2H), 4.02 (q, 2H), 4.54 (m, 1H), 6.90–7.30 (m, 4H).
Compound 5d
oil (10% yield). 1H NMR (CDCl3): δ 0.96 (dd, 6H), 1.58 (s, 9H), 1.62 (m, 2H), 1.96 (m, 1H), 3.63 (dd, 2H), 4.00 (dd, 2H), 4.99 (m, 1H), 7.45–7.79 (m, 5H).
Compound 5e
oil (20% yield). 1H NMR (CDCl3): δ 0.94 (dd, 6H), 1.57 (m, 2H), 1.58 (s, 9H), 1.61 (m, 1H), 2.98 (m, 3H), 3.10 (m, 1H), 3.73 (m, 2H), 4.53 (m, 1H), 7.20 (m, 5H).
Compound 5f
oil (13% yield). 1H NMR (CDCl3): δ 0.96 (dd, 6H), 1.58 (s, 9H), 1.60 (m, 2H), 1.96 (m, 1H), 3.63 (dd, 2H), 4.00 (dd, 2H), 4.99 (m, 1H), 6.99–7.58 (m, 9H).
Compound 5g
oil (11% yield). 1H NMR (CDCl3): δ 0.92 (dd, 6H), 1.59 (m, 1H), 1.60 (s, 9H), 1.70 (m, 2H), 3.82 (m, 2H), 4.56 (q, 2H), 4.58 (m, 1H), 7.40–7.84 (m, 7H).
Representative procedure for the synthesis of compounds 6a–g
Compound 5a (0.58 g, 1.36 mmol) was treated with TFA/methylene chloride (1:1 v/v, 15 mL) and stirred for 3 h at room temperature. The solvent and excess TFA were removed and the residue was taken up in ethyl acetate (35 mL) and washed with saturated NaHCO3 (2 × 10 mL) and brine (10 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and the solvent evaporated off to yield compound 6a as an oil (0.38 g, 87%). 1H NMR (CDCl3): δ 0.92 (dd, 6H), 1.55 (m, 2H), 1.72 (m, 1H), 3.23 (dd, 1H), 3.64 (dd, 1H), 3.79 (s, 3H), 4.03 (q, 2H), 4.54 (m, 1H), 6.78–7.22 (m, 4H).
Compound 6b
white solid (100% yield), mp 135–137 °C. 1H NMR (CDCl3): δ 0.92 (dd, 6H), 1.57 (m, 2H), 1.72 (m, 1H), 3.23 (dd, 1H), 3.64 (dd, 1H), 3.79 (s, 3H), 4.01 (q, 2H), 4.54 (m, 1H), 6.83–7.21 (m, 4H).
Compound 6c
white solid (91% yield), mp 141–144 °C. 1H NMR (CDCl3): δ 0.92 (t, 6H), 1.57 (m, 2H), 1.72 (m, 1H), 3.23 (d, 1H), 3.64 (dd, 1H), 4.01 (q, 2H), 4.54 (m, 1H), 6.82–7.32 (m, 4H).
Compound 6d
oil (100% yield). 1H NMR (CDCl3): δ 0.96 (d, 6H), 1.60 (m, 2H), 1.84 (m, 1H), 3.21 (dd, 1H), 3.64 (dd, 2H), 4.20 (bs, 1H), 4.83 (m, 1H), 7.42–7.80 (m, 5H).
Compound 6e
oil (98% yield). 1H NMR (CDCl3): δ 0.94 (dd, 6H), 1.57 (m, 2H), 1.68 (m, 1H), 2.98 (m, 4H), 3.24 (dd, 1H), 3.64 (dd, 1H), 4.54 (m, 1H), 7.26 (m, 5H).
Compound 6f
oil (89% yield). 1H NMR (CDCl3): δ 0.96 (dd, 6H), 1.61 (m, 2H), 1.82 (m, 1H), 3.20 (m, 1H), 3.62 (m, 1H), 4.71 (m, 1H), 6.90–7.60 (m, 9H).
Compound 6g
oil (84% yield). 1H NMR (CDCl3): δ 0.90 (dd, 6H), 1.50 (m, 2H), 1.70 (m, 1H), 3.22 (d, 1H), 3.64 (dd, 1H), 3.80 (bs, 1H), 4.51 (m, 2H), 4.52 (m, 1H), 7.40–7.84 (m, 7H).
Representative procedure for the synthesis of compounds 7a–g
To a solution of compound 6a (0.37 g, 1.13 mmol) in dry methylene chloride (10 mL) was added TEA (0.12 g, 1.13 mmol) and 2-phenethyl isocyanate (0.24 g, 1.58 mmol) successively. The reaction mixture was gently refluxed for 1 h. The solvent was evaporated off and the residue was taken up in ethyl acetate (50 mL) and washed with 5% aqueous HCl (2 × 10 mL) and brine (10 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and the solvent evaporated off. The crude product was purified using flash chromatography (hexanes/EtOAc, 85:15) to afford 7a (oil, 0.41 g, 77%). 1H NMR (CDCl3): δ 0.92 (dd,6H), 1.59 (m, 3H), 2.86 (t, 2H), 3.57 (m, 2H), 3.80 (s, 3H), 3.86 (m, 2H), 4.00 (q, 2H), 4.50 (m, 1H), 6.05 (t, 1H), 6.81–7.38 (m, 9H).
Compound 7b
oil (95% yield). 1H NMR (CDCl3): δ 0.92 (dd, 6H), 1.59 (m, 3H), 2.86 (t, 21 2H), 3.57 (m, 2H), 3.80 (s, 3H), 3.86 (m, 2H), 4.00 (q, 2H), 4.50 (m, 1H), 6.05 (t, 1H), 6.86–7.38 (m, 9H).
Compound 7c
oil (72% yield). 1H NMR (CDCl3): δ 0.96 (dd, 6H), 1.59 (m, 3H), 2.86 (t, 2H), 3.59 (m, 2H), 3.86 (m, 2H), 4.02 (q, 2H), 4.50 (m, 1H), 6.05 (t, 1H), 6.95–7.38 (m, 9H).
Compound 7d
white solid (78% yield), mp 77–78 °C. 1H NMR (CDCl3): δ 0.96 (dd, 6H), 1.64 (m, 2H), 1.86 (m, 1H), 2.78 (t, 2H), 3.45 (m, 2H), 3.62 (dd, 1H), 4.23 (dd, 1H), 4.84 (m, 1H), 5.95 (t, 1H), 7.12–7.80 (m, 10H).
Compound 7e
white solid (87% yield), mp 88–90 °C. 1H NMR (CDCl3): δ 0.96 (d, 6H), 1.59 (m, 3H), 2.84 (t, 2H), 3.01 (m, 4H), 3.53 (m, 2H), 3.83 (m, 2H), 4.44 (m, 1H), 5.98 (t, 1H), 7.20 (m, 10H).
Compound 7f
oil (70% yield). 1H NMR (CDCl3): δ 0.96 (dd, 6H), 1.60 (m, 2H), 1.81 (m, 1H), 2.80 (t, 2H), 3.45 (m, 2H), 3.62 (dd, 1H), 4.21 (dd, 1H), 4.80 (m, 1H), 5.95 (t, 1H), 7.01–7.50 (m, 14H).
Compound 7g
oil (69% yield). 1H NMR (CDCl3): δ 0.90 (dd, 6H), 1.60 (m, 3H), 2.90 (t, 2H), 3.60 (m, 2H), 3.94 (d, 2H), 4.51 (m, 2H), 4.52 (m, 1H), 6.08 (t, 1H), 7.22–7.84 (m, 12H).
Compound 8
To a solution of compound 6b (0.55 g, 1.68 mmol) in dry methylene chloride (10 mL) was added TEA (0.17 g, 1.68 mmol) and 2-phenethyl isothiocyanate (0.38 g, 2.35 mmol) successively. The reaction mixture was gently refluxed for one hour. The solvent was then evaporated and the residue was taken up in ethyl acetate (50 mL) and washed with 5% aqueous HCl (2 × 10 mL) and brine (10 mL). The organic phase was dried over anhydrous Na2SO4, filtered and the solvent was evaporated off, leaving a crude product which was purified using flash chromatography (hexane/EtOAc, 95:5) to afford 8 as an oily product (0.03 g, 4%): 1H NMR (CDCl3): δ 0.92 (dd, 6H), 1.60 (m, 3H), 2.96 (t, 2H), 3.79 (s, 3H), 3.90 (m, 2H), 3.95 (t, 2H), 4.10 (m, 1H), 4.24 (d, 1H), 4.44 (m, 1H), 6.82–7.58 (m, 9H).
Compound 9
colorless oil (29 % yield). 1H NMR (CDCl3): δ 0.93 (m, 9H), 1.40 (m, 2H), 1.58 (m, 5H), 3.33 (m, 2H), 3.80 (s, 3H), 3.89 (d, 2H), 4.02 (dd, 2H), 4.50 (m, 1H), 6.02 (t, 1H), 6.83 (m, 3H), 7.25 (m, 1H).
Biochemical studies: enzyme assays and inhibition studies
Human neutrophil elastase
HNE was assayed by mixing 10 μL of a 70 μM enzyme solution in 0.05 M sodium acetate/0.5 M NaCl buffer, pH 5.5, 10 μL dimethyl sulfoxide and 980 μL of 0.1 M HEPES buffer containing 0.5 M NaCl, pH 7.25, in a thermostated cuvette. A 100 μL aliquot was transferred to a thermostated cuvette containing 880 μL 0.1 M HEPES/0.5 M NaCl buffer, pH 7.25, and 20 μL of a 70 μM solution of MeOSuc-Ala-Ala-Pro-Val p-nitroanilide, and the change in absorbance was monitored at 410 nm for 60 seconds. In a typical inhibition run, 10 μL of inhibitor (3.5 mM) in dimethyl sulfoxide was mixed with 10 μL of 70 μM enzyme solution and 980 μL 0.1 M HEPES/0.5 M NaCl buffer, pH 7.25, and placed in a constant temperature bath. Aliquots (100 μL) were withdrawn at different time intervals and transferred to a cuvette containing 20 μL of MeOSuc-Ala-Ala-Pro-Val p-nitroanilide (7 mM) and 880 μL 0.1 M HEPES/0.5 M NaCl buffer. The absorbance was monitored at 410 nm for 60 seconds. Bovine trypsin, proteinase 3 and cathepsin G were assayed as described previously [8].
Progress curve method
The inhibitory activity of compounds 7a–g and 9 toward HNE was determined by the progress curve method [9, 8(b)]. Thus, in a typical run 5 μL of a 2.0 μM HNE solution in 0.05 M sodium acetate buffer containing 0.5 M NaCl, pH 5.5 was added to 10 μL of inhibitor (0.2 mM solution in DMSO), 15 μL of substrate (MeOSuc-Ala-Ala-Pro-Val pNA, 7 mM in DMSO) and 970 μL 0.1 M HEPES buffer/0.5 M NaCl buffer, pH 7.25, and the absorbance was monitored at 410 nm for ten minutes. Typical progress curves for the hydrolysis of MeOSuc-AAPV-pNA by HNE in the presence of inhibitor 7g are shown in Figure 2. Control curves in the absence of inhibitor were linear. The release of p-nitroaniline was continuously monitored at 410 nm. The pseudo first-order rate constants (kobs) for the inhibition of HNE by derivatives of (I) as a function of time were determined according to eq 1 below, where A is the absorbance at 410 nm, vo is the reaction velocity at t = 0, vs is the final steady-state velocity, kobs is the observed first-order rate constant, and Ao is the absorbance at t = 0. The kobs values were obtained by fitting the A versus t data to eq 1 using nonlinear regression analysis (SigmaPlot, Jander Scientific). The second order rate constants (kinact/KI M−1 s−1) were then determined by calculating kobs/[I] and then correcting for the substrate concentration using eq 2. The apparent second-order rate constants (kinact/KI M−1 s−1) were determined in duplicate and are listed in Table 1.
Figure 2.
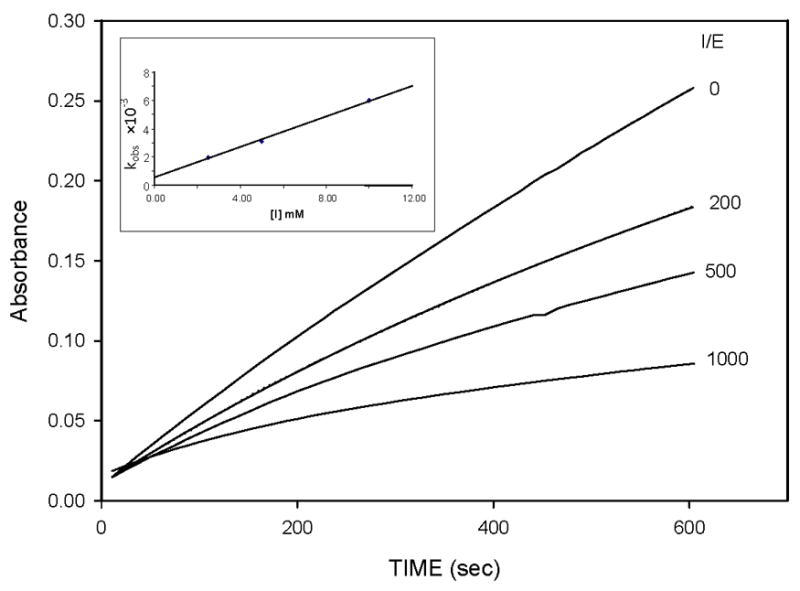
Progress curves for the inhibition of human neutrophil elastase (HNE) by inhibitor 7g. Absorbance was monitored at 410 nm for reaction solutions containing 10 nM HNE, 105 μM MeOSuc-AAPV p-nitroanilide, and the inhibitor at the indicated inhibitor to enzyme ratios in 0.1 M HEPES buffer containing 0.5 M NaCl, pH 7.25, and 2.5% DMSO. The temperature was maintained at 25°C, and reactions were initiated by the addition of enzyme. Inset shows the linear dependence of kobs with increasing [I].
Table 1.
Inhibition of Human Neutrophil Elastase by Derivatives of Compound (I).
| Compound | kinact/KI M−1 s−1 |
|---|---|
| 7a | 350 |
| 7b | 350 |
| 7c | 750 |
| 7d | 890 |
| 7e | 1260 |
| 7f | 980 |
| 7g | 1050 |
| 8 | Inactive |
| 9 | 860 |
| eq 1 |
| eq 2 |
Hydroxylamine Reactivation
Human neutrophil elastase (10 μL, 70 μM) was incubated with a 50-fold excess of inhibitor 7g (10 μL of a 3.5 mM solution in dimethyl sulfoxide) and 970 μL of 0.1 M HEPES buffer containing 0.5 M NaCl, pH 7.25 for 15 min. The enzyme was totally inactivated (as shown by withdrawing an aliquot and assaying for remaining enzyme activity). Excess hydroxylamine hydrochloride (10 μL of a 70 mM solution) was then added to the fully inactivated enzyme. Aliquots (100 μL) were removed at various time intervals (from 15 min to 24 h) and assayed for enzyme activity by mixing with MeOSuc-Ala-Ala-Pro-Val-p-NA (20 μL, 7 mM) and 0.1 M HEPES buffer containing 0.5 M NaCl (880 μL), pH 7.25, and monitoring the absorbance at 410 nm. Enzyme activity was determined by comparing the activity of an enzyme solution containing no inhibitor (control) with the activity of an enzyme solution containing inhibitor at the same time point.
Results and Discussion
The structural motif, robustness, ease of assembly, and three points of diversity embodied in the cyclosulfamide scaffold make it particularly attractive with respect to the generation of lead discovery prospecting libraries. Consequently, we recently utilized this versatile scaffold in the design of reversible inhibitors of (chymo)trypsin-like serine proteases, specifically human neutrophil elastase (HNE) [6]. Those studies revealed that cyclosulfamide-based derivatives inhibited the enzyme by an apparent partial mixed-type inhibition and, furthermore, that the mode of binding to the active site was greatly influenced by the nature of recognition elements R1, R2 and R3.
In an attempt to probe further the nature of the interaction of (I) with HNE, as well as its mode of binding, it was observed that the presence of a urea-like functionality at R3 led to time-dependent inactivation of HNE. This observation suggested that compounds represented by structure (I) may constitute a new class of carbamylating agents capable of exhibiting optimal enzyme selectivity through the judicious selection of the three recognition elements attached to the scaffold. Indeed, incubation of compound 7e with HNE led to rapid, time-dependent, irreversible inactivation of the enzyme (Figure 3).
Figure 3.
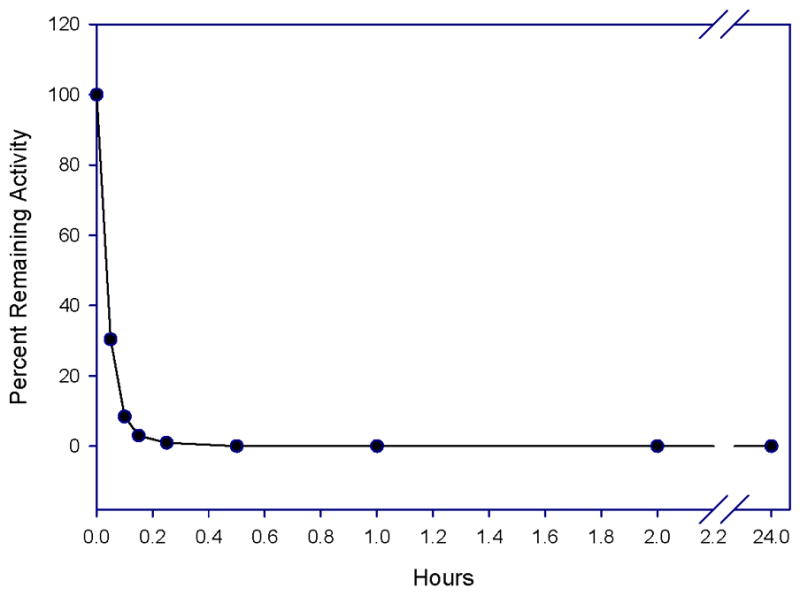
Time dependent loss of enzymatic activity. Percent remaining activity versus time plot obtained by incubating inhibitor 7e (37 μM) with human neutrophil elastase (700 nM) in 0.1 M HEPES buffer containing 0.5 M NaCl, pH 7.25, and 1% DMSO. Aliquots were withdrawn at different time intervals and assayed for enzymatic activity using MeOSuc-AAPV p-NA by monitoring the absorbance at 410 nm.
The urea-like group NCONHR3 was found to be essential for inhibitory activity. All compounds lacking the urea-like functionality (compounds 6a–g, Scheme 1(b) ) were devoid of inhibitory activity. The less reactive thiourea derivative 8 ( Scheme 1(b) ) was likewise found to be inactive, further reinforcing the requirement for the presence of a urea-like functionality for the manifestation of inhibitory activity. These observations suggest that the mode of binding likely involves accommodation of the R3 moiety in the primary specificity (S1) subsite of HNE [11].
The tentative inference that (I) binds to the active site with the R3 group accommodated at S1 is further supported by the following observations: since HNE shows a strong preference for small hydrophobic (n-propyl/isopropyl, n-butyl/isobutyl) P1 residues, the 2-phenethyl group in compound 7a was replaced by an n-butyl group and the resulting compound 9 ( Scheme 1(b) ) was found to exhibit significant inhibitory activity (kinact/KI 860 M−1 s−1 ) toward HNE but was devoid of any inhibitory activity toward cathepsin G and bovine trypsin. This is in accord with the known substrate specificity of Cat G and bovine trypsin which show a strong preference for a Phe or Lys residue at P1, respectively. This compound was also inactive toward proteinase 3 (PR 3), a serine protease that prefers small hydrophobic (ethyl, n-propyl) residues at S1 [12–13]. This is probably due to the more constricted S1 subsite of PR 3. Lastly, compounds 7a–g (listed in Scheme 1(a) ) were all found to be inactive toward bovine trypsin but inhibited human neutrophil cathepsin G weakly (40–65% inhibition following a 30 minute incubation at an [I]/[E] ratio of 250).
In order to determine how the nature of the acyl group (R2CO) influences potency and selectivity, a series of compounds was synthesized and their inhibitory activity toward HNE was determined using the progress curve method. It is evident from Table 1 that varying R2 leads to a 2-3-fold improvement in potency. This may simply reflect the increased hydrophobicity of the more potent compounds (compound 9, for example) rather than an effect of a specific hydrophobic interaction [14]. Importantly, replacement of the R2CO group with a benzyl group yielded a compound devoid of any inhibitory activity (30 minute incubation period at an [I]/[E] ratio of 500). Replacement of the isobutyl group at R1 with an n-propyl group increased potency more than 2-fold (compare compounds 7a and 7d, Table 1).
Mechanism
The mechanism of inactivation of HNE by (I) was probed by:
assessing the stability of the enzyme-inhibitor adduct. Thus, HNE was inactivated with excess inhibitor 7g (0% remaining activity) and the inactive enzyme was then treated with excess buffered hydroxylamine and its activity was monitored over a 24 h period. There was no regain in enzymatic activity (Figure 4), attesting to the robustness of the enzyme-inhibitor adduct (carbamylated enzyme as opposed to acyl enzyme) and suggesting that a covalent, irreversible mechanism is involved;
as mentioned earlier, the presence of urea-type and R2CO functionalities at N5 and N3 respectively is necessary for inhibitory activity, however, the lack of inhibitory activity by compounds 6a–g suggests that carbamylation of the enzyme takes place at the urea-type functionality. Taken together, the binding and inhibition of HNE by (I) can be tentatively proposed to occur as illustrated in Figure 5 where binding of the inhibitor to the active site places the R3 at the S1 subsite and orients the rest of the molecule toward the S′ subsites. With respect to the kinetics of inhibition, it appears that the interaction of (I) with HNE likely involves the rapid establishment of an initial equilibrium leading to the formation of an EI complex which is followed by a slower chemical step (carbamylation). This is reminiscent of the similar behavior of ONO-4056 [15], a known HNE inhibitor that rapidly forms a complex with the enzyme, followed by slow acylation of the active site serine.
Figure 4.
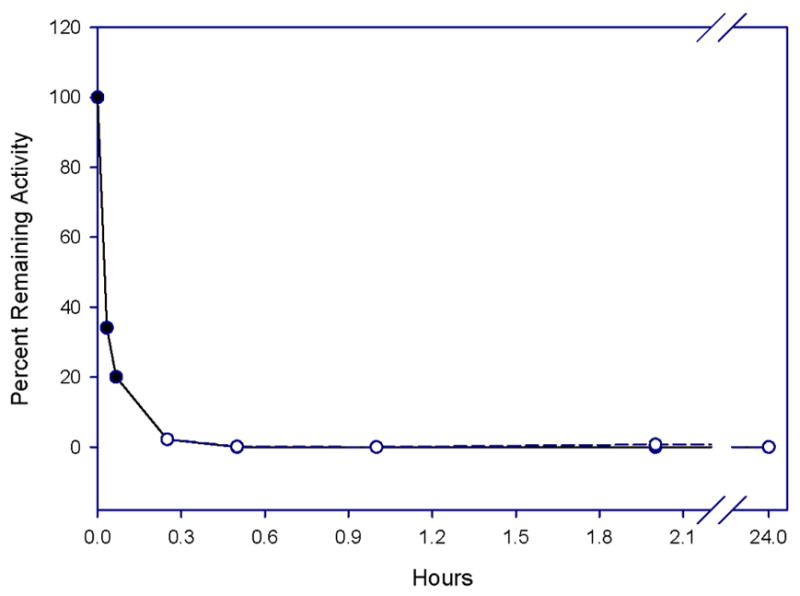
Effect of hydroxylamine on enzyme reactivation. Human neutrophil elastase (700 nM) was totally inactivated by incubating the enzyme with a 20-fold excess of inhibitor 7e (35 μM) for 15 minutes in 0.1 M HEPES buffer/0.5 M NaCl, pH 7.25. Excess hydroxylamine (0.7 mM) was then added, and aliquots were removed at different time intervals and assayed for enzyme activity using MeOSuc-Ala-Ala-Pro-Val pNA (o). Enzyme activity was determined by comparison with a control (absence of hydroxylamine) at the same time point (•).
Figure 5.
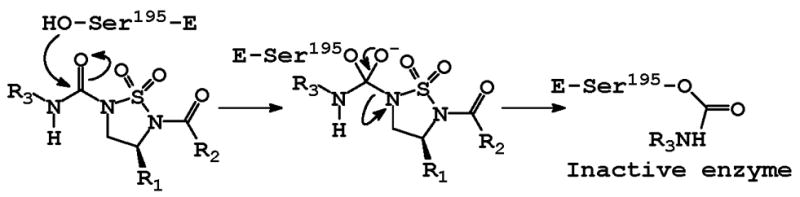
Postulated mechanism of action of (I).
The moderate chemical reactivity of inhibitor (I) and its structural versatility suggest that it may be of value as an activity-based probe. Activity-based probes (ABPs) have been successfully used to profile enzyme activities in vivo, including the role of proteases in disease states [16]. Tracking protease activities in complex proteomes typically entails the utilization of ABPs that exploit differences in the substrate specificity and/or mechanism of action of the target proteases. The use of ABPs that exhibit high labeling specificity is particularly advantageous because of the complexity of proteomes. This is especially true for COPD-relevant proteases which include serine (neutrophil elastase, proteinase 3, cathepsin G), cysteine (cathepsin S), and matrix metalloproteases (MMP-9, MMP-12). In the case of human neutrophil elastase (HNE) and proteinase 3 (PR 3), an additional challenge is the preference of both enzymes for a small hydrophobic residue at P1 which necessitates the exploitation of differences in the S2 and S′ subsites of the two enzymes [12–13].
In summary, a versatile class of carbamylating agents (I) that exhibit excellent selectivity toward HNE has been reported. Further studies with (I) via the use of amino acid-derived isocyanates with attached alkyne and/or azide groups are currently in progress.
Acknowledgments
This work was supported in part by the National Institutes of Health (HL 57788).
Footnotes
Nonstandard abbreviations: TFA, trifluoroacetic acid; TEA, triethylamine; DBU, 1,8-diazabicyclo[5.4.0]-7-undecene; HATU, o-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-te-tramethyluronium hexafluorophosphate
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mannino DM, Braman S. Proc Am Thorac Soc. 2007;4:502–506. doi: 10.1513/pats.200701-001FM. [DOI] [PubMed] [Google Scholar]
- 2.Halpin DMG, Miravitlles M. Proc Am Thorac Soc. 2006;3:619–623. doi: 10.1513/pats.200603-093SS. [DOI] [PubMed] [Google Scholar]
- 3.(a) Barnes PJ. PLoS Medicine. 2007;4:779–780. doi: 10.1371/journal.pmed.0040112. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) MacNee W. Clin Chest Med. 2007;28:479–513. doi: 10.1016/j.ccm.2007.06.008. [DOI] [PubMed] [Google Scholar]; (c) Yoshida T, Tuder RM. Physiol Rev. 2007;87:1047–1082. doi: 10.1152/physrev.00048.2006. [DOI] [PubMed] [Google Scholar]
- 4.(a) MacNee W. Proc Am Thorac Soc. 2005;2:258–266. doi: 10.1513/pats.200504-045SR. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Croxton TL, Weinmann GG, Senior RM, Wise RA, Crapo JD, Buist SA. Am J Respir Crit Care Med. 2003;167:1142–1149. doi: 10.1164/rccm.200207-756WS. [DOI] [PubMed] [Google Scholar]; (c) Barnes PJ, Shapiro SD, Pauwels RA. Eur Respir J. 2003;22:672–688. doi: 10.1183/09031936.03.00040703. [DOI] [PubMed] [Google Scholar]
- 5.(a) Barnes PJ. J Allergy Clin Immunol. 2007;119:1055–1062. doi: 10.1016/j.jaci.2007.01.015. [DOI] [PubMed] [Google Scholar]; (b) Barnes PJ, Stockley RA. Eur Respir J. 2005;25:1084–1106. doi: 10.1183/09031936.05.00139104. [DOI] [PubMed] [Google Scholar]
- 6.Zhong J, Gan X, Alliston KR, Lai Z, Yu H, Groutas CS, Wong T, Groutas WC. J Comb Chem. 2004;6:556–563. doi: 10.1021/cc030047r. [DOI] [PubMed] [Google Scholar]
- 7.Evans MJ, Cravatt BF. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 8.(a) Lai Z, Gan X, Wei L, Alliston KR, Yu H, Li YH, Groutas WC. Arch Biochem Biophys. 2004;429:191–197. doi: 10.1016/j.abb.2004.06.014. [DOI] [PubMed] [Google Scholar]; (b) Groutas WC, Kuang R, Venkataraman R, Epp JB, Ruan S, Prakash O. Biochemistry. 1997;36:4739–4750. doi: 10.1021/bi9628937. [DOI] [PubMed] [Google Scholar]
- 9.Morrison JF, Walsh CT. Adv Enzymol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 10.(a) Dixon M. Biochem J. 1953;55:170–171. doi: 10.1042/bj0550170. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wei L, Gan X, Zhong J, Alliston KR, Groutas WC. Bioorg Med Chem. 2003;11:5149–5153. doi: 10.1016/j.bmc.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 11.Nomenclature used is that of Schechter I, Berger A. Biochem Biophys Res Comm. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. where S1, S2, S3,…Sn and S1′, S2′, S3′,…Sn′ correspond to the enzyme subsites on either side of the scissile bond. Each subsite accommodates a corresponding amino acid residue side chain designated P1, P2, P3, ….Pn and P1′, P2′, P3′, ….Pn′ of the substrate or (inhibitor). S1 is the primary substrate specificity subsite, and P1-P1′ is the scissile bond
- 12.(a) Brubaker MJ, Groutas WC, Hoidal JR, Rao NV. Biochem Biophys Res Comm. 1992;188:1318–1324. doi: 10.1016/0006-291x(92)91375-z. [DOI] [PubMed] [Google Scholar]; (b) Kam CM, Kerrigan JE, Dolman KM, Goldschmeding R, Von dem Borne AE, Powers JC. FEBS Lett. 1992;297:119–123. doi: 10.1016/0014-5793(92)80340-m. [DOI] [PubMed] [Google Scholar]
- 13.(a) Koehl C, Knight CG, Bieth JG. J Biol Chem. 2003;278:12609–12612. doi: 10.1074/jbc.M210074200. [DOI] [PubMed] [Google Scholar]; (b) Kormaz B, Attucci S, Hazouard E, Ferrandiere M, Jourdan ML, Brillard-Bourdet M, Juliano L, Gauthier F. J Biol Chem. 2002;277:39074–39081. doi: 10.1074/jbc.M202918200. [DOI] [PubMed] [Google Scholar]; (c) Kormaz B, Attucci S, Moreau T, Godat E, Juliano L, Gauthier F. Am J Respir Cell Mol Biol. 2004;30:801–807. doi: 10.1165/rcmb.2003-0139OC. [DOI] [PubMed] [Google Scholar]; (d) Hajjar E, Korkmaz B, Gauthier F, Brandsdal BO, Witko-Sarsat V, Reuter N. J Med Chem. 2006;49:1248–1260. doi: 10.1021/jm051018t. [DOI] [PubMed] [Google Scholar]; (e) Kormaz B, Hajjar E, Kalupov T, Reuter N, Brillard-Bourdet M, Moreau T, Juliano L, Gauthier F. J Biol Chem. 2007;282:1989–1997. doi: 10.1074/jbc.M608700200. [DOI] [PubMed] [Google Scholar]
- 14.Davis AM, Teague SJ. Angew Chem Intl Ed. 1999;38:736–749. doi: 10.1002/(SICI)1521-3773(19990315)38:6<736::AID-ANIE736>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 15.Imaki K, Okada T, Nakayama Y, Nagao Y, Kobayashi K, Sakai Y, Mohri T, Amino T, Nakai H, Kawamura M. Bioorg Med Chem. 1996;4:2115. doi: 10.1016/s0968-0896(96)00216-7. [DOI] [PubMed] [Google Scholar]
- 16).a) Barglow KT, Cravatt BF. Nature Meth. 2007;4:822–827. doi: 10.1038/nmeth1092. [DOI] [PubMed] [Google Scholar]; (b) Li W, Blankman JL, Cravatt BF. J Am Chem Soc. 2007;129:9594–9595. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]; (c) Pan Z, Jeffery DA, Chehade K, Beltman J, Clark JM, Grothaus P, Bogyo M, Baruch A. Bioorg Med Chem. 2006;16:2882–2885. doi: 10.1016/j.bmcl.2006.03.012. [DOI] [PubMed] [Google Scholar]; (d) Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]