

Abstract
Propofol (2,6‐diisopropylphenol) is a versatile, short‐acting, intravenous (i.v.) sedative‐hypnotic agent initially marketed as an anesthetic, and now also widely used for the sedation of patients in the intensive care unit (ICU). At the room temperature propofol is an oil and is insoluble in water. It has a remarkable safety profile. Its most common side effects are dose‐dependent hypotension and cardiorespiratory depression. Propofol is a global central nervous system (CNS) depressant. It activates γ‐aminobutyric acid (GABAA) receptors directly, inhibits the N‐methyl‐d‐aspartate (NMDA) receptor and modulates calcium influx through slow calcium‐ion channels. Furthermore, at doses that do not produce sedation, propofol has an anxiolytic effect. It has also immunomodulatory activity, and may, therefore, diminish the systemic inflammatory response believed to be responsible for organ dysfunction. Propofol has been reported to have neuroprotective effects. It reduces cerebral blood flow and intracranial pressure (ICP), is a potent antioxidant, and has antiinflammatory properties. Laboratory investigations revealed that it might also protect brain from ischemic injury. Propofol formulations contain either disodium edetate (EDTA) or sodium metabisulfite, which have antibacterial and antifungal properties. EDTA is also a chelator of divalent ions such as calcium, magnesium, and zinc. Recently, EDTA has been reported to exert a neuroprotective effect itself by chelating surplus intracerebral zinc in an ischemia model. This article reviews the neuroprotective effects of propofol and its mechanism of action.
Keywords: Anesthesia, chelation, disodium edetate (EDTA), middle cerebral artery occlusion (MCAO), neuroprotection, propofol, zinc
Introduction
Propofol is an intravenous (i.v.) agent that is widely used for the induction and maintenance of anesthesia, as well as for sedation in intensive care units (ICUs). Laboratory investigations revealed that propofol might also protect brain from ischemic injury. Propofol has been reported to have many pharmacological effects: (a) it reduces cerebral blood flow, cerebral metabolic rate, and intracranial pressure (ICP) (Murphy et al. 1992), (b) it acts as an antioxidant: it scavenges free radicals, and decreases lipid peroxidation (Sagara et al. 1999; Wilson and Gelb 2002), (c) it activates γ‐aminobutyric acid (GABAA) receptors (Ito et al. 1998), inhibits glutamate receptors (Zhan et al. 2001), and reduces extracellular glutamate levels by either inhibiting Na+ channel‐dependent glutamate release or by enhancing glutamate uptake (Sitar et al. 1999), and (d) it reduces ischemic neuronal injury in animal models of transient global or focal cerebral ischemia (Young et al. 1997).
Disodium edetate (EDTA) or metabisulfite is added to propofol preparations to retard bacterial and fungal growth. As EDTA is a chelator of divalent ions such as calcium, magnesium, and zinc, we recently hypothesized that chelation of excessive neuronal zinc by EDTA might ameliorate zinc‐induced neurotoxicity and reduce subsequent neuronal injury. In this article, we review the literature on the neuroprotective effects of propofol, including the most recent reports, and discuss whether EDTA has indeed a neuroprotective effect.
Chemistry
The empirical formula of propofol (2,6‐diisopropylphenol, Fig. 1) is C12H18O, and its molecular weight is 178.27. The octanol/water partition coefficient for propofol is 6,761:1 at a pH of 6–8.5, and its pKa is 11. As propofol is only very slightly soluble in water, it is formulated in a white, oil‐in‐water emulsion. Current formulations consist of 1% or 2% (w/v) propofol, 10% soya bean oil, 1.2% egg phosphatide, and 2.25% glycerol. To this, 0.005% disodium edetate (EDTA) or metabisulfite is added to retard bacterial and fungal growth.
Figure 1.
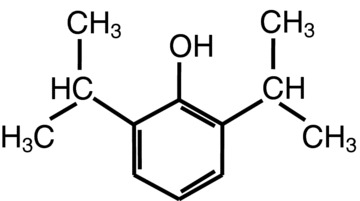
The chemical structure of propofol.
Pharmacology
Propofol is an i.v. sedative‐hypnotic agent with amnestic properties, it causes rapid and reliable loss of consciousness (Godambe et al. 2003). In addition, because of its easy titration, it is widely used as a sedative for intensive care patients. Induction of anesthesia can be achieved by administering propofol at doses of 40 mg every 10 second, until the clinical signs indicate the onset of anesthesia (Langley and Heel 1988). The dose of propofol required to induce anesthesia in adults is normally between 2 and 2.5 mg/kg i.v. (Bryson et al. 1995), although older patients may require a lower dose. Propofol anesthesia can be maintained either with a continuous infusion (approximately 6–12 mg/kg/h) or with intermittent bolus injections (20–50 mg). Among the favorable characteristics of propofol are the lack of accumulation and the short recovery time, both of which are essential for neurological examination after the operative procedure (Mirski et al. 1995). Propofol is lipophilic, it easily crosses the blood–brain barrier, and has been shown to depress electroencephalographic activity (Kochs et al. 1992). It decreases cerebral metabolic rate dose‐dependently (Dam et al. 1990; Ridenour et al. 1992), and reduces cerebral blood flow (Ergun et al. 2002). Propofol may, therefore, be viewed as a global central nervous system (CNS) depressant.
Activation of GABAA or GABAB Receptors
It is thought that propofol directly activates GABAA receptors. Ito et al. (1998) found that pharmacological agents that either act directly on GABAA receptors or modulate GABAA receptor activity (such as propofol, midazolam, and muscimol) are capable of reducing the severity of brain injury following ischemia in gerbils. It has been reported that at clinically relevant concentrations, propofol inhibits glutamate release by blocking current through sodium channels or by activating GABAA receptors (Ratnakumari and Hemmings 1997; Rehberg and Duch 1999; Buggy et al. 2000). Excessive glutamate accumulation in the extracellular space due to ischemia within the CNS is believed to initiate a cascade toward irreversible neuronal damage. Following an ischemic insult, there is a massive release of glutamate, which acts on both ionotropic and metabotropic glutamate receptors. In the pathophysiology of cerebral ischemia, such an uncontrolled release of glutamate during the ischemia and the consequent excessive stimulation of postsynaptic glutamate receptors (excitotoxicity), play a major role in the initiation of neuronal injury (Kawaguchi et al. 2005). Furthermore, glutamate receptor antagonists protect rodents from cerebral injury in both focal (Park et al. 1988; Dezsi et al. 1992; Sarraf‐Yazdi et al. 1998) and transient global ischemia (Gill et al. 1988) models.
Some studies assessed whether propofol has the potential to modify glutamate dynamics during cerebral ischemia. Yano et al. (2000) examined the hypothesis that by intracerebroventricular (i.c.v.) administration propofol, 3 or 10 mg/kg, would reduce the extracellular glutamate levels during global ischemia leading to neuroprotection. They concluded that, although i.c.v. propofol exhibits neuroprotection in transient global forebrain ischemia, the extracellular glutamate level during ischemia is not a major determinant of the neuroprotective activity of propofol. Using rat synaptosomes Bianchi et al. (1991) showed that the propofol (with an approximate IC50 of 3.0 × 10−5 M), slightly inhibited glutamate release, glutamate‐dependent Ca2+ entry, and voltage‐gated calcium channels. According to Feiner et al. (2005), propofol (10–100 μM) lacks the glutamate‐receptor antagonist potency required for neuroprotection by an antiexcitotoxicity mechanism. Amorim et al. (1995) reported that in hippocampal slices during hyperthermal anoxia, propofol (20 μg/mL) attenuated the increase in intracellular Ca2+, but did not depress the release of glutamate. Further examination of these findings is needed.
Using a different approach Schwieler et al. (2003) analyzed whether propofol (1–16 mg/kg i.v.) might, by activating somatodendritic GABAB receptors, decrease the firing rate and the burst firing activity of nigral dopamine neurons. They concluded that an activation of central GABAB receptors may, at least partially, contribute to the anesthetic properties of propofol.
Inhibition of N‐Methyl‐d‐Aspartate (NMDA) Receptors
Stimulation of the NMDA receptors leads to Ca2+ and Na2+ influx into the cells. The subsequent excessive accumulation of intracellular calcium activates enzymes such as proteases, lipases, and endonucleases (Kawaguchi et al. 2005). Propofol inhibits the NMDA receptor and reduces calcium influx through slow calcium channels. As calcium influx into the tissues is believed to be responsible for cellular dysfunction and tissue injury, propofol may have organ protective activity. Indeed, antagonists of either NMDA or α‐amino‐3hydroxy‐5‐methylisoxazole‐4‐propionic acid (AMPA) receptors have been shown to be neuroprotective in global and focal cerebral ischemia (Kawasaki‐Yatsugi et al. 1998; Sarraf‐Yazdi et al. 1998). Zhan et al. (2001) used an in vitro cerebral ischemic model to examine the relation between the effects of i.v. anesthetics on neurotransmission and their effects on the NMDA receptors during ischemia. They reported that propofol (100 μM), rather than inhibiting NMDA receptors, slightly augments the NMDA‐mediated effect on the intracellular calcium. In contrast, Hans et al. (1994) showed that in cultured hippocampal neurons, propofol attenuates the neurotoxic effect of glutamate exerted via NMDA receptors. Other reports suggested that the neuroprotective efficacy of propofol might not be sustained. For example, an NMDA antagonist dizocilpine maleate (MK‐801), at 0.5 mg/kg i.v., reduced neuronal injury when the injury was evaluated after a short recovery period (3 days), but not when it was evaluated at 4 weeks after initiation of ischemia (Valtysson et al. 1994). Moreover, Zhu et al. (1997), using the recovery of the population spike amplitude as an indicator of neuronal viability, demonstrated that propofol (112 μM) may enhance NMDA‐induced neuronal damage. Taking all these data together, it appears that propofol may, at best, weakly and incompletely inhibit NMDA receptors.
Antioxidant Activity
Propofol is a lipophilic and phenolic antioxidant, it scavenges free radicals, reduces lipid peroxidation, inhibits cellular oxidative damage, and increases glutathione levels in the tissues. Its chemical structure has some similarity with that of phenol‐based free‐radical scavengers, such as vitamin E (Murphy et al. 1992; Kahraman and Demiryurek 1997). Murphy et al. (1992) demonstrated that propofol (10−6–10−5 M) has an antioxidant capacity equal to that to “Trolox C” (Sigma‐Aldrich, St Louis, MO, USA) a water‐soluble analogue of vitamin E and a known antioxidant. They also reported that Trolox C and propofol are equipotent in reducing the rate of oxygen consumption and lipid peroxidation. Further, Hans et al. (1996) noted that in isolated cell membranes propofol and vitamin E produce qualitatively similar improvements in cell function and that their effects can be correlated with the measurements of lipid peroxidation, so that propofol can replace vitamin E as an antioxidant. Vincenti et al. (1991) examined the antioxidant and antiglutamatergic activities of propofol and found that it inhibits lipid peroxidation in rat liver microsomes and mitochondria, inhibits glutamatergic responses in rat brain synaptosomes, and interfers with the glutamic acid‐mediated calcium entry. Navapurkar et al. (1998) further proposed that propofol (28 μM) may exert a protective action against the oxidative stress caused by free radicals in the liver. Scavenging of free radicals by propofol may, in presence of Ca2+, contribute to the stabilization of the mitochondrial membrane during oxidative stress (Eriksson 1991). Taken together, these results indicate that propofol is a free radical scavenger with a significant antioxidant activity.
Immunomodulatory Activity
Propofol possesses immunomodulatory activity, and may thus diminish the systemic inflammatory response believed to be responsible for organ dysfunction (Crozier et al. 1994; Matsushita et al. 1996; Helmy et al. 1999). The lipid component of propofol EDTA is based on soybean oil, and contains a variety of triglycerides, phospholipids, glycerol, vitamins, and minerals. The primary lipid in soybean oil is linoleic acid, an omega‐6 long‐chain polyunsaturated fatty acid, and there are smaller amounts of omega‐3 long‐chain fatty acids. Long‐chain fatty acids are bioactive and affect the synthesis and secretion of cytokines, free radicals, and other inflammatory mediators (Herr et al. 2000). These lipids integrate into cellular membranes altering membrane structure and function, as well as ion‐channel flow, second‐messenger generation, and production of eicosanoids.
Anxiolytic Effects
Propofol, like benzodiazepines, has anxiolytic effect at doses that do not induce sedation. Smith et al. (1994) found that by i.v. infusion at 0.2, 0.4, 0.5, or 0.7 mg/kg propofol decreases anxiety scores in patients undergoing urologic surgery under regional anesthesia. The mechanism of its anxiolytic action is likely to involve a positive modulation of the inhibitory function of GABA through GABAA receptors (Ito et al. 1999).
Analgesic Effects
Anwar and Abdel‐Rahman (1998) reported that at 25 or 50 mg/kg i.p. propofol may control pain via an opioid system, with the onset of action of 5 up to 20 min. The analgesic effect of propofol was also reported by other investigators (Briggs et al. 1982; Anker‐Moller et al. 1991; Jewett et al. 1992).
In contrast, Godambe et al. (2003) found that at 1 mg/kg i.v. propofol is a poor analgesic, and that it usually requires an adjunctive analgesic agent, and Ewen et al. (1995) found that propofol (340 or 680 μg/kg/min, 20 min, i.v.) may produce a hyperalgesic effect. Overall, these results suggest that pain control by propofol may be weak and incomplete.
Anticonvulsant Activity
The anticonvulsant properties of propofol were examined some years ago in two experimental models of status epilepticus in rabbits (de Riu et al. 1992). In that study, it was found that propofol (12 mg/kg) suppressed electroencephalographic and pharmacological seizures in a pentylenetetrazole‐induced generalized epileptic status. It also reduced focal epilepsy induced by cortically applied penicillin G, although its efficacy was low and the effect was short‐lasting. In a later in vitro study, Rasmussen et al. (1996) showed that in rat hippocampal slices propofol markedly reduces epileptiform activity induced by picrotoxin, bicuculline, pilocarpine, K+, or by omission of Mg2+ from the medium (1.7, 50, 16.9–56.2, 168, 337 μM propofol, respectively). Furthermore, in humans propofol is well known to be effective against status epilepticus that was refractory to standard anticonvulsants (Marik 2004).
Neuroprotective Properties
The reports that propofol depresses electroencephalographic activity (at 0.8–1.2 mg/kg/min i.v., Kochs et al. 1992), decreases cerebral metabolic rate (at 20 mg/kg i.v., Dam et al. 1990), and reduces cerebral blood flow (at 50 mg/kg i.p., Ergun et al. 2002) suggested that it may have a neuroprotective effect against brain ischemia. Actually, it has certain properties that might well be neuroprotective, including free‐radical scavenging (Grasshoff and Gillessen 2002), augmentation of aminobutyric acid (GABA)‐receptor currents, and inhibition of NMDA‐type glutamate receptor currents (Orser et al. 1995; Yamakura et al. 1995; Wakasugi et al. 1999). Indeed, the neuroprotective effect of propofol has been attributed to its antioxidant properties, potentiation of GABAA‐mediated inhibition of synaptic transmission, and inhibition of glutamate release in cerebral ventricles (at 3 or 10 mg/kg, Yano et al. 2000).
In contrast, propofol failed to protect cells in vivo (at 10 mg/kg i.v. bolus or 16 mg/kg/h continuous i.v. infusion for 4 h, Tsai et al. 1994) or in in vitro models of cerebral ischemia (at 20 μg/mL, Amorim et al. 1995; or at 100 μM, Zhan et al. 2001). Kawaguchi et al. (2005) suggested that the propofol may be neuroprotective in mild ischemic insults during a long postischemic recovery period, but that its neuroprotective effect is not adequate for sustained protection from moderate to severe insults. They concluded that sustained anesthetic neuroprotection might be limited to brief ischemia. On the other hand, Engelhard et al. (2004) compared the effect of propofol (by i.v. infusion at 0.8–1.2 mg/kg/min) with that of nitrous oxide (FiO2= 0.33) and fentanyl (i.v. bolus: 10 μg/kg, i.v. infusion: 25 μg/kg/min) and demonstrated that propofol reduces neuronal damage and favorably modulates apoptosis‐regulating proteins for at least 28 days, suggesting long‐term neuroprotection by this agent. In a permanent middle cerebral arteryocclusion (MCAO) model, we recently demonstrated (Kotani et al. 2008) that propofol (10 mg/kg, i.v.) reduces infarct volume and neuronal damage for 7 days, suggesting a long‐term neuroprotection by propofol.
In Vitro Studies
Several researchers reported that propofol reduces cell injury in cellular preparations (1–8 μM, Daskalopoulos et al. 2001; 1,10, or 100 μM, Grasshoff and Gillessen 2002; 20–100 μM, Sagara et al. 1999). Velly et al. (2003) studied the relationship between propofol‐induced neuroprotection, glutamate extracellular concentrations, and glutamate transporter activity in a cell‐culture model of cortical ischemia. They found that propofol (0.05–10 μM) displayed a neuroprotective effect against oxygen‐glucose deprivation (OGD). Adembri et al. (2006) reported that propofol (10–100 μM) attenuated CA1 injury in hippocampal slices in vitro when it was present in the incubation medium during both OGD and the subsequent 24 h recovery period. However, Feiner et al. (2005) were unable to demonstrate neuroprotection with 10–100 μM propofol in a similar organotypic hippocampal slice model. We recently examined the in vitro effects of propofol and EDTA against OGD‐induced cell damage in cultures of PC12 cells (Fig. 2). The results showed that either propofol (20 μM) or EDTA (0.05 μM) protected PC12 cells from OGD‐induced damage, and that this effect of propofol was enhanced by EDTA.
Figure 2.
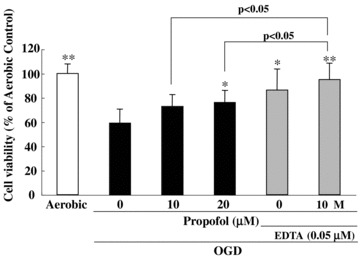
Both propofol and propofol EDTA reduced the cell damage induced by OGD in PC12 culture. Cell viability was assessed following immersion in 10% resazurin solution for 3 h at 37 °C, and fluorescence was recorded at 560/590 nm. OGD induced cell death, and propofol and EDTA each inhibited this OGD‐induced cell death. Propofol plus EDTA reduced cell death by more than propofol alone. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01 versus OGD‐treatment alone (Dunnett's test or Student's t‐test); n= 4. Data from Kotani et al. (2008).
In Vivo Studies
Permanent middle cerebral artery occlusion
Adembri et al. (2006) reported that at twenty‐four hours after permanent MCAO in rats, infarct size was reduced by approximately 30% when propofol (100 mg/kg, i.p.) was administered immediately after or up to 30 min after the occlusion. On the other hand, Tsai et al. (1994) failed to demonstrate a protective action of propofol (10 mg/kg i.v. bolus or 16 mg/kg/h continuous i.v. infusion for 4 h) after permanent MCAO in rats. Recently, we evaluated the neuroprotective properties of propofol by determining whether or not it alters the volume of the infarct resulting from permanent focal cerebral ischemia in mice. By i.v. administration at 5 or 10 mg/kg propofol EDTA or propofol (but not at 1 mg/kg) significantly reduced infarct area, infarct volume, and brain swelling (Fig. 3A, B). Propofol EDTA reduced infarct area and brain swelling significantly better than propofol alone. Likewise, propofol EDTA or propofol, at 5 or 10, but not at 1 mg/kg i.v., 10 min before ischemia, appeared to reduce neurological deficits, although only propofol EDTA reduced them significantly (Fig. 3C). In our study, TUNEL‐positive cells were predominantly located in the ischemic core region rather than in the ischemic penumbra, although propofol EDTA significantly reduced the number of TUNEL‐positive cells only in the ischemic penumbra (Fig. 4C, D). Thus, our findings suggest that propofol EDTA inhibits apoptosis mainly within ischemic penumbra.
Figure 3.
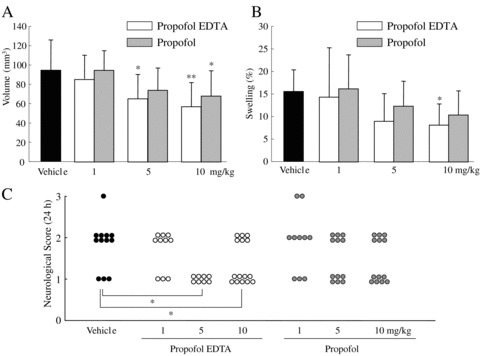
(A) Effects of propofol EDTA and propofol on infarct volume at 24 h after permanent MCA occlusion (each treatment being given intravenously [1, 5, or 10 mg/kg, given over 90 second, at 0.1 mL/10 g] 10 min before MCA occlusion). *P < 0.05, **P < 0.01 versus vehicle (Dunnett's test); n= 10–14. (B) The effects of propofol EDTA and propofol on brain swelling 24 h after permanent MCA occlusion in mice. *P < 0.05 versus vehicle (Dunnett's test); n= 10–14. (C) Effects of propofol EDTA and propofol (details as in B) on neurological deficits at 24 h after permanent MCA occlusion. *P < 0.05 versus vehicle (Mann‐Whitney U‐test); n= 10–14. Values are mean ± SD. Data from Kotani et al. (2008).
Figure 4.
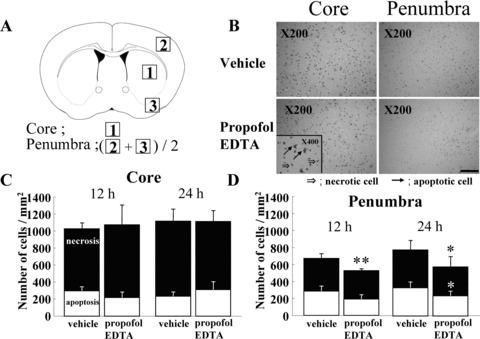
Effect of propofol EDTA on TUNEL staining after MCA occlusion. Propofol EDTA (10 mg/kg i.v.) was administered 10 min before MCA occlusion, and mice were sacrificed at 12 or 24 h after the occlusion. (A) Schematic drawing showing the brain regions at a level 0.4–1.0 mm anterior to bregma (through the anterior commissure); 1, ischemic core; 2 and 3, ischemic penumbra. The number of TUNEL‐positive cells was counted in each of these areas, the average for areas 2 and 3 being taken as the number for the ischemic penumbra. (B) Propofol EDTA appeared to reduce the number of TUNEL‐positive cells (versus vehicle treatment) in the ischemic penumbra, but not in the ischemic core. (C and D) Quantitative representation of TUNEL‐positive cells in ischemic brains treated with propofol EDTA or vehicle. White part of bar shows number of apoptotic cells among all positive cells. Note that at 24 h in the ischemic penumbra, a considerably smaller number of TUNEL‐positive cells was observed in mice treated with propofol EDTA than in mice treated with vehicle. However, in the ischemic core no such difference was detected. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01 versus vehicle (Student's t‐test); n= 4–6. Scale bar = 100 μm. Data from Kotani et al. (2008).
Transient middle cerebral artery occlusion
Lee et al. (2000) reported that pretreatment with propofol (administered at 96 mg/kg/h for 20 min and maintained at 72 mg/kg/h until the initiation of carotid occlusion) could markedly reduce the extent of the 2,3,5‐triphenyltetrazolium chloride (TTC)‐infarcted area following incomplete global cerebral ischemia and reperfusion. Arcadi et al. (1996) investigated the hippocampal cell death in the gerbil that occurred as a result of transient cerebral ischemia. They concluded that propofol (50 or 100 mg/kg, i.p.) has protective activity against transient forebrain ischemia‐induced delayed hippocampal neuronal death without improving overall survival rate. A few years later, Wang et al. (2002) used microdialysis technique to evaluate the effects of propofol infusion on infarct size and the striatal dopamine levels following temporary MCA occlusion in rats. They showed that when propofol (36 mg/kg/h) was infused during ischemia and reperfusion, it reduced the cerebral infarct size and significantly decreased dopamine accumulation in the striatum. They concluded that the neuroprotective effect of propofol might be partially due to its ability to inhibit dopamine accumulation. Engelhard et al. (2004) used rats to investigate the long‐term effects of propofol against neuronal damage and apoptosis‐related proteins after cerebral ischemia and reperfusion. They analyzed the amounts of the apoptosis‐related proteins Bax, p53, Bcl‐2, and Mdm‐2, and the number of neurons positive for activated caspase‐3, and found: (1) that propofol (at 1 mg/kg i.v. bolus, immediately followed by an infusion at 10 mg/kg/h for 10 min, at 8 mg/kg/h for the next 10 min, and thereafter at 6 mg/kg/h) had a sustained neuroprotective effect that was associated with reduced eosinophilic and apoptotic injury, and (2) that activated caspase‐3‐dependent apoptotic pathways were not affected by propofol, suggesting the presence of activated caspase‐3‐independent apoptotic pathways.
Some studies failed, however, to detect the neuroprotective effect of propofol. In rat hippocampal slices propofol, at 100 μM, had no protective effect in terms of the recovery of population spikes in the CA‐1 pyramidal layer following transient ischemia (Zhan et al. 2001). Thus, opinions are divided as to whether propofol has a neuroprotective effect in transient cerebral ischemia, although most investigators seem to believe that propofol is neuroprotective.
Zinc Chelation
In our recent studies, we focused on zinc because it is one of the most abundant transition metals in the brain, and is essential for development, growth, DNA synthesis, immunity, and a wide array of cellular processes. The physiological significance of neuronal zinc release within the CNS is not clear, and its role in ischemic brain injury is controversial. After brain ischemia, there is a depletion of presynaptic bouton zinc and a concurrent accumulation of zinc in the cell bodies of vulnerable neurons (Koh et al. 1996). In fact, it has been proposed that synaptic zinc or extracellular zinc acts as “the cell‐death ion” during neuronal damage, both in vitro and in vivo (Choi et al. 1998; Canzoniero et al. 1999; Shabanzadeh et al. 2004).
In contrast, some authors believe that zinc may have a protective function (Matsushita et al. 1996; Bancila et al. 2004). Nakatani et al. (2000) reported that Zn2+ ions play an important role in free radical metabolism and apoptosis, while Calderone et al. (2004) reported that when the influx of extracellular Zn2+ into postsynaptic neurons is blocked by intraventricular injection of a Zn2+‐chelating agent, neurodegeneration is prevented.
Disodium edetate (EDTA) or metabisulfite is added to conventional pharmaceutical preparations of propofol to retard bacterial and fungal growth. EDTA is a potent chelator of heavy metals, including zinc, iron, copper, manganese, chromium, cobalt, and lead (Guldager et al. 1996; Powell et al. 1999). In our recent study, we asked whether EDTA could modulate the neuroprotective effect of propofol, and studies the effects of EDTA on intracerebral zinc levels during cerebral ischemia. When propofol EDTA at 10 mg/kg i.v. was injected at 10 min before the onset of cerebral ischemia (permanent MCAO), the zinc levels decreased significantly (vs. vehicle) in the cortical area, but not in the subcortex (Table 1) (Kotani et al. 2008). In contrast, in the propofol EDTA‐treated group that did not undergo MCAO, the intracerebral zinc levels in the cortex were not significantly altered (Kotani et al. 2008). This finding indicates that chelation of zinc by EDTA occurs in the cortex during ischemia, possibly leading to neuroprotection. Therefore, the dynamics of microelements (in particular, zinc) at the time of cerebral ischemia should be considered in the future studies, and chelation therapy using EDTA may prove to be a new treatment for cerebral ischemia.
Table 1.
Zinc levels in mouse brains
| Treatments | Ischemia | Cortex (μg/g) | Subcortex (μg/g) |
|---|---|---|---|
| Vehicle | − | 139.9 ± 42.6 | 110.3 ± 30.1 |
| Propofol EDTA | − | 137.0 ± 32.0 | 108.0 ± 36.5 |
| Vehicle | + | 150.2 ± 36.7 | 107.3 ± 30.4 |
| Propofol EDTA | + | 120.8 ± 22.4* | 98.6 ± 19.2 |
Propofol EDTA (10 mg/kg i.v.) or vehicle (Intralipid) was administered 10 min before the onset of ischemia. Mice were anesthetized using 2.0–3.0% isoflurane and maintained using 1.0–1.5% isoflurane in 70% N2O and 30% O2. After a midline skin incision, the left external carotid artery was exposed, and its branches were occluded. An 8–0 nylon monofilament (Ethicon, Somerville, NJ) coated with a silicone resin mixture (Xantopren; Bayer Dental, Osaka, Japan) was introduced into the left internal carotid artery through the external carotid artery stump so as to occlude the origin of the middle cerebral artery (MCA). Afterwards, the left common carotid artery was occluded. Mice were killed 1 h after the onset of ischemia. Nonischemic groups were killed 1 h after drug (or vehicle) administration. Values are mean ± SD (n= 10). *P < 0.05 versus vehicle + ischemia group (Bonferroni correction). Data from Kotani et al. (2008).
Pharmacokinetics
The pharmacokinetics of propofol can be well described by a three‐compartment linear model, with the compartments representing plasma, rapidly and slowly equilibrating tissues (AstraZeneca 2001). Following an i.v. bolus dose, there is rapid equilibration of propofol levels between plasma and highly perfused tissue of the brain, accounting for the rapid onset of anesthesia. Propofol has been shown to be extensively (97–98%) bound to plasma proteins (Altmayer et al. 1995), although the plasma level initially shows a steep decline as a result of both rapid distribution and high metabolic clearance (the latter being from 23 to 50 mL/kg/min [1.6 to 3.4 L/min in a 70 kg adult]). A difference in pharmacokinetics due to gender has not been observed (AstraZeneca 2001). Propofol is mainly eliminated by hepatic conjugation to inactive glucuronide metabolites (Raoof et al. 1996), which are excreted by the kidney, and glucuronide conjugates account for about 50% of the administered dose.
The terminal half‐life of propofol after a 10‐day infusion is 1–3 days (AstraZeneca 2001). If there are higher than necessary infusion levels of plasma, this return of propofol from the peripheral tissues will delay recovery. In other words, failure to reduce the infusion rate in patients receiving propofol for extended periods may result in excessively high blood concentrations of the drug. Therefore, titration to the clinical response and daily evaluation of sedation levels are important during the use of propofol infusion for ICU sedation, especially if it is of long duration.
Clinical Efficacy
Propofol causes hypotension (particularly in volume‐depleted patients), decreases cerebral oxygen consumption, reduces ICP, and has potent anticonvulsant properties. Therefore, propofol is being increasingly used in the management of traumatic head injury, status epilepticus, delirium tremens, status asthmaticus, and in septic patients (Cosmo et al. 2005). In patients with severe head injury, the goal of therapy is to avoid secondary brain damage, with i.v. sedation being an integral part of the therapy. Herregods et al. (1988) investigated the effects of a bolus injection of propofol (2 mg/kg) on mean ICP in six adult, comatose patients who had severe head injuries. They demonstrated that the mean ICP was decreased significantly (from 25 to 11 mmHg; p < 0.05) at 30 seconds and at 1 and 2 min, and that the cerebral perfusion pressure was decreased significantly from 92 mmHg at all measurement‐point (p < 0.05) (Herregods et al. 1988). Merlo et al. (1991) investigated the decrease in ICP in 11 patients who had an ICP above 20 mmHg despite hyperventilation and neurosedation. They observed statistically significant (p < 0.05) decreases in ICP and systolic arterial pressure after a bolus of propofol (1.5 mg/kg i.v.). They concluded that propofol could be used to treat intracranial hypertension, but that the hemodynamic effects in hypovolemic patients need to be taken into consideration (Merlo et al. 1991). Thus, the available data permit the conclusion that propofol can be used effectively in patients with an elevated ICP.
Mizuno et al. (2002) reported a case in which propofol (a bolus of 60 mg i.v., followed by continuous i.v. infusion at 2 mg/kg/h) was effective in controlling myoclonus during rewarming of a hypothermic patient. In addition, propofol is frequently used in cardiac surgery. CNS dysfunction is a common consequence of otherwise uncomplicated cardiac surgery. This led Ederberg et al. (1998) to investigate the effects of burst‐suppression doses of propofol (a bolus dose of 1 mg/kg immediately followed by an infusion of 10 mg/kg/h for 10 min, 8 mg/kg/h for the next 10 min, and thereafter at 6 mg/kg/h) on cerebral blood flow velocity (CBFV), cerebral oxygen extraction (COE), and dynamic autoregulation in 20 patients undergoing cardiac surgery. They reported that propofol induced 35% and 10% decreases in CBFV and COE, respectively, and concluded that propofol decreases CBFV and improves dynamic autoregulation during moderate hypothermic nonpulsatile cardiopulmonary bypass (Ederberg et al. 1998). Consequently, Souter et al. (1998) prospectively investigated the effects of propofol (at a subanesthetic dose of 30 mg/min, causing a burst‐suppression rate of 80%, throughout cardiopulmonary bypass) on cerebral venous oxyhemoglobin saturation (SjO2) (SjO2 < 50%). They concluded that, when administered at doses sufficient to produce electroencephalographic burst suppression, propofol did not attenuate either the frequency or extent of the reductions in cerebral venous oxyhemoglobin saturation (Souter et al. 1998). Thus, propofol appears to have a neuroprotective effect only when given at anesthetic doses.
Toxicology
Acute Toxicity
Propofol has a remarkable safety profile. However, the effects of acute hepatic or renal failure on the pharmacokinetics of propofol have not been studied. The i.v. LD50 values for propofol, administered as the emulsion formulation, average 53 and 42 mg/kg in mice and rats, respectively, while the oral LD50 values for propofol, administered as a solution in soybean oil, are 1,230 and 600 mg/kg in mice and rats, respectively (Stuart Pharmaceuticals 1989). Overdosage with propofol would be expected to produce manifestations that principally represent extensions of the drug's pharmacologic and adverse effects and are associated with cardiorespiratory depression. It is recommended that in the case of an overdose, propofol should be discontinued immediately, and appropriate symptomatic therapy initiated (AstraZeneca 2001).
Adverse Effects
The reported adverse effects of propofol are: pain on injection (Boysen et al. 1989), bradycardia (Tramer et al. 1997), arterial hypotension (Nimmo et al. 1994), bloodstream infection (Bennett et al. 1995), airway obstruction, changes in serum lipids (Barrientos‐Vega et al. 1997), and excitation of the CNS (Stark et al. 1985), including seizures in susceptible patients (Bredahl 1990). Pain on injection is the most frequently observed adverse effect, its incidence varies from 28.5% (for small veins) to 6% (for larger veins) (Mackenzie and Grant 1987), but the incidence of thrombophlebitis is very low (about 0.5%) (Stark et al. 1985). Other uncommon complications include hypertriglyceridemia and pancreatitis.
High‐dose propofol infusions may be associated with the “propofol syndrome,” a potentially fatal complication characterized by severe metabolic acidosis and circulatory collapse. This is a rare complication first reported in pediatric patients, and believed to be due to a decreased transmembrane electrical potential and alteration in electron transport across the inner mitochondrial membrane (Marik 2004).
Herr et al. (2000) compared the safety of propofol with that of propofol EDTA. Each drug was given initially for sedation of critically ill postsurgical or trauma patients in ICU by continuous infusion at a rate of 5 μg/min, with the rate adjusted, if necessary. The infusion continued till the patients achieved the Modified Ramsay Sedation Scale score. They investigators reported that: (1) the addition of EDTA to propofol appeared to have no effect on calcium or magnesium homeostasis, and (2) because propofol has little effect on renal function, adding EDTA, at a low concentration, to propofol produced no untoward effects on renal function in critically ill patients. In addition, Zaloga and Teres (2000) observed that the most notable abnormality in ICU patients given propofol containing EDTA was a low blood zinc level, although no adverse events indicative of zinc deficiency occurred.
Conclusions
Propofol is a versatile i.v. sedative‐hypnotic agent. Many preliminary studies suggest that it may have a neuroprotective effect against brain ischemia. Indeed, it seems to afford neuroprotection against both in vivo and in vitro ischemic damage. Such effects may be enhanced when EDTA is added, and the addition of EDTA appears to have no detrimental effect on the safety or efficacy of propofol when it is used for sedation in critically ill surgical ICU patients. However, the evidence that propofol may reduce ischemic cerebral damage is inconclusive, and further research is needed.
Conflict of Interest
The authors have no conflict of interest.
References
- Adembri C, Venturi L, Tani A, Chiarugi A, Gramigni E, Cozzi A, Pancani T, De Gaudio RA, Pellegrini‐Giampietro DE (2006) Neuroprotective effects of propofol in models of cerebral ischemia: inhibition of mitochondrial swelling as a possible mechanism. Anesthesiology 104:80–89 [DOI] [PubMed] [Google Scholar]
- Altmayer P, Buch U, Buch HP (1995) Propofol binding to human blood proteins. Arzneimittelforschung 45:1053–1056. [PubMed] [Google Scholar]
- Amorim P, Chambers G, Cottrell J, Kass IS (1995) Propofol reduces neuronal transmission damage and attenuates the changes in calcium, potassium, and sodium during hyperthermic anoxia in the rat hippocampal slice. Anesthesiology 83:1254–1265. [DOI] [PubMed] [Google Scholar]
- Anker‐Moller E, Spangsberg N, Arendt‐Nielsen L, Schultz P, Kristensen MS, Bjerring P (1991) Subhypnotic doses of thiopentone and propofol cause analgesia to experimentally induced acute pain. Br J Anaesth 66:185–188. [DOI] [PubMed] [Google Scholar]
- Anwar MM, Abdel‐Rahman MS (1998) Effect of propofol on perception of pain in mice: mechanisms of action. Comp Biochem Physiol Part A 120:249–253. [DOI] [PubMed] [Google Scholar]
- Arcadi FA, Rapisarda A, De Luca R, Trimarchi GR, Costa G (1996) Effect of 2,6‐diisopropylphenol on the delayed hippocampal cell loss following transient forebrain ischemia in the gerbil. Life Sci 58:961–970. [DOI] [PubMed] [Google Scholar]
- AstraZeneca : Diprivan® (propofol) injectable emulsion for IV administration prescribing information. Wilmington , DE, 2001. [Google Scholar]
- Bancila V, Nikonenko I, Dunant Y, Bloc A (2004) Zinc inhibits glutamate release via activation of pre‐synaptic KATP channels and reduces ischaemic damage in rat hippocampus. J Neurochem 90:1243–1250. [DOI] [PubMed] [Google Scholar]
- Barrientos‐Vega R, Mar Sanchez‐Soria M, Morales‐Garcia C, Robas‐Gomez A, Cuena‐Boy R, Ayensa‐Rincon A (1997) Prolonged sedation of critically ill patients with midazolam or propofol: impact on weaning and costs. Crit Care Med 25:33–40. [DOI] [PubMed] [Google Scholar]
- Bennett SN, McNeil MM, Bland LA, Arduino MJ, Villarino ME, Perrotta DM, Burwen DR, Welbel SF, Pegues DA, Stroud L, et al. (1995) Postoperative infections traced to contamination of an intravenous anesthetic, propofol. N Engl J Med 333:147–154. [DOI] [PubMed] [Google Scholar]
- Bianchi M, Battistin T, Galzigna L (1991) 2,6‐diisopropylphenol, a general anesthetic, inhibits glutamate action on rat synaptosomes. Neurochem Res 16:443–446. [DOI] [PubMed] [Google Scholar]
- Boysen K, Sanchez R, Krintel JJ, Hansen M, Haar PM, Dyrberg V (1989) Induction and recovery characteristics of propofol, thiopental and etomidate. Acta Anaesthesiol Scand 33:689–692. [DOI] [PubMed] [Google Scholar]
- Bredahl C (1990) Seizures and opistotonus after propofol anesthesia. A possible connection. Ugeskr Laeger 152:748–749. [PubMed] [Google Scholar]
- Briggs LP, Dundee JW, Bahar M, Clarke RS (1982) Comparison of the effect of diisopropyl phenol (ICI 35, 868) and thiopentone on response to somatic pain. Br J Anaesth 54:307–311. [DOI] [PubMed] [Google Scholar]
- Bryson HM, Fulton BR, Faulds D (1995) Propofol: an update of its use in anaesthesia and conscious sedation. Drugs 50:513–559. Review. [DOI] [PubMed] [Google Scholar]
- Buggy DJ, Nicol B, Rowbotham DJ, Lambert DG (2000) Effects of intravenous anesthetic agents on glutamate release: a role for GABAA receptor‐mediated inhibition. Anesthesiology 92:1067–1073. [DOI] [PubMed] [Google Scholar]
- Calderone A, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, Zukin RS (2004) Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia‐induced death. J Neurosci 24:9903–9913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzoniero LM, Turetsky DM, Choi DW (1999) Measurement of intracellular free zinc concentrations accompanying zinc‐induced neuronal death. J Neurosci 19:RC31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Yokoyama M, Koh J (1998) Zinc neurotoxicity in cortical cell culture. Neurosci 24:67–79. [DOI] [PubMed] [Google Scholar]
- Crozier TA, Muller JE, Quittkat D, Sydow M, Wuttke W, Kettler D (1994) Effect of anesthesia on the cytokine responses to abdominal surgery. Br J Anaesth 72:2280–2285. [DOI] [PubMed] [Google Scholar]
- Dam M, Ori C, Pizzolato G, Ricchieri GL, Pellegrini A, Giron GP, Battistin L (1990) The effects of propofol anesthesia on local cerebral glucose utilization in the rat. Anesthesiology 73:499–505. [DOI] [PubMed] [Google Scholar]
- Daskalopoulos R, Korcok J, Farhangkhgoee P, Karmazyn M, Gelb AW, Wilson JX (2001) Propofol protection of sodium‐hydrogen exchange activity sustains glutamate uptake during oxidative stress. Anesth Analg 93:1199–1204. [DOI] [PubMed] [Google Scholar]
- De Cosmo G, Congedo E, Clemente A, Aceto P (2005) Sedation in PACU: the role of propofol. Curr Drug Targets 6:741–744. Review. [DOI] [PubMed] [Google Scholar]
- De Riu PL, Petruzzi V, Testa C, Mulas M, Melis F, Caria MA, Mameli O (1992) Propofol anticonvulsant activity in experimental epileptic status. Br J Anaesth 69:177–181. [DOI] [PubMed] [Google Scholar]
- Dezsi L, Greenberg JH, Hamar J, Sladky J, Karp A, Reivich M (1992) Acute improvement in histological outcome by MK‐801 following focal cerebral ischemia and reperfusion in the cat independent of blood flow changes. J Cereb Blood Flow Metab 12:390–399. [DOI] [PubMed] [Google Scholar]
- Ederberg S, Westerlind A, Houltz E, Svensson SE, Elam M, Ricksten SE (1998) The effects of propofol on cerebral blood flow velocity and cerebral oxygen extraction during cardiopulmonary bypass. Anesth Analg 86:1201–1206. [DOI] [PubMed] [Google Scholar]
- Engelhard K, Werner C, Eberspacher E, Pape M, Stegemann U, Kellermann K, Hollweck R, Hutzler P, Kochs E (2004) Influence of propofol on neuronal damage and apoptotic factors after incomplete cerebral ischemia and reperfusion in rats: a long‐term observation. Anesthesiology 101:912–917. [DOI] [PubMed] [Google Scholar]
- Ergun R, Akdemir G, Sen S, Tasci A, Ergungor F (2002) Neuroprotective effects of propofol following global cerebral ischemia in rats. Neurosurg Rev 25:95–98. [DOI] [PubMed] [Google Scholar]
- Eriksson O (1991) Effects of the general anaesthetic propofol on the Ca2+‐induced permeabilization of rat liver mitochondria. FEBS Lett 279:45–48. [DOI] [PubMed] [Google Scholar]
- Ewen A, Archer DP, Samanani N, Roth SH (1995) Hyperalgesia during sedation: effects of barbiturates and propofol in the rat. Can J Anaesth 42:532–540. [DOI] [PubMed] [Google Scholar]
- Feiner JR, Bickler PE, Estrada S, Donohoe PH, Fahlman CS, Schuyler JA (2005) Mild hypothermia, but not propofol, is neuroprotective in organotypic hippocampal cultures. Anesth Analg 100:215–225. [DOI] [PubMed] [Google Scholar]
- Gill R, Foster AC, Woodruff GN (1988) MK‐801 is neuroprotective in gerbils when administered during the post‐ischaemic period. Neuroscience 25:847–855. [DOI] [PubMed] [Google Scholar]
- Godambe SA, Elliot V, Matheny D, Pershad J (2003) Comparison of propofol/fentanyl versus ketamine/midazolam for brief orthopedic procedural sedation in a pediatric emergency department. Pediatrics 112:116–123. [DOI] [PubMed] [Google Scholar]
- Grasshoff C, Gillessen T (2002) The effect of propofol on increased superoxide concentration in cultured rat cerebrocortical neurons after stimulation of N‐methyl‐d‐aspartate receptors. Anesth Analg 95:920–922. [DOI] [PubMed] [Google Scholar]
- Guldager B, Jorgensen PJ, Grandjean P (1996) Metal excretion and magnesium retention in patients with intermittent claudication treated with intravenous disodium EDTA. Clim Chem 42:1938–1942. [PubMed] [Google Scholar]
- Hans P, Bonhomme V, Collette J, Albert A, Moonen G (1994) Propofol protects cultured rat hippocampal neurons against N‐methyl‐D‐aspartate receptor‐mediated glutamate toxicity. J Neurosurg Anesthesiol 6:249–253. [DOI] [PubMed] [Google Scholar]
- Hans P, Deby C, Deby‐Dupont G, Vrijens B, Albert A, Lamy M (1996) Effect of propofol on in vitro lipid peroxidation induced by different free radical generating systems: a comparison with vitamin E. J Neurosurg Anesthesiol 8:154–158. [DOI] [PubMed] [Google Scholar]
- Helmy SAK, Wahby MAM, El‐Na‐waway M (1999) The effect of anaesthesia and surgery on plasma cytokine production. Anaesthesia 54:733–738. [DOI] [PubMed] [Google Scholar]
- Herr DL, Kelly K, Hall JB, Ulatowski J, Fulda GJ, Cason B, Hickey R, Nejman AM, Zaloga GP, Teres D (2000) Safety and efficacy of propofol with EDTA when used for sedation of surgical intensive care unit patients. Intensive Care Med 26:S452–S462. [DOI] [PubMed] [Google Scholar]
- Herregods L, Verbeke J, Rolly G, Colardyn F (1988) Effect of propofol on elevated intracranial pressure. Preliminary results. Anaesthesia 43:107–109. [DOI] [PubMed] [Google Scholar]
- Ito H, Watanabe Y, Isshiki A, Uchino H (1998) Suppression of parasympathetic reflex vasodilatation in the lower lip of the cat by isoflurane, propofol, ketamine and pentobarbital: implications for mechanisms underlying the production of anaesthesia. Br J Anaesth 81:563–568. [DOI] [PubMed] [Google Scholar]
- Ito H, Watanabe Y, Isshiki A, Uchino H (1999) Neuroprotective properties of propofol and midazolam, but not pentobarbital, on neuronal damage induced by forebrain ischemia, based on the GABAA receptors. Acta Anaesthesiol Scand 43:153–162. [DOI] [PubMed] [Google Scholar]
- Jewett BA, Gibbs LM, Tarasiuk A, Kendig JJ (1992) Propofol and barbiturate depression of spinal nociceptive neurotransmission. Anesthesiology 77:1148–1154. [DOI] [PubMed] [Google Scholar]
- Kahraman S, Demiryurek AT (1997) Propofol is a peroxynitrite scavenger. Anesth Analg 84:1127–1129. [DOI] [PubMed] [Google Scholar]
- Kawaguchi M, Furuya H, Patel PM (2005) Neuroprotective effects of anesthetic agents. J Anesth 19:150–156. [DOI] [PubMed] [Google Scholar]
- Kawasaki‐Yatsugi S, Yatsugi S, Takahashi M, Toya T, Ichiki C, Shimizu‐Sasamata M, Yamaguchi T, Minematsu K (1998) A novel AMPA receptor antagonist, YM872, reduces infarct size after middle cerebral artery occlusion in rats. Brain Res 793:39–46. [DOI] [PubMed] [Google Scholar]
- Kochs E, Hoffman WE, Werner C, Thomas C, Albrecht RF, Schulte am Esch J (1992) The effects of propofol on brain electrical activity, neurologic outcome, and neuronal damage following incomplete ischemia in rats. Anesthesiology 76:245–252. [DOI] [PubMed] [Google Scholar]
- Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW (1996) The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 272:1013–1016. [DOI] [PubMed] [Google Scholar]
- Kotani Y, Nakajima Y, Hasegawa T, Masahiko S, Nagase H, Shimazawa M, Yoshimura S, Iwama T, Hara H (2008) Propofol exerts greater neuroprotection with disodium edetate (EDTA) than without it. J Cereb Blood Flow Metab 28:354–366. [DOI] [PubMed] [Google Scholar]
- Langley MS, Heel RC (1988) Propofol: a review of its pharmacodynamic and pharmacokinetic properties and use as an intravenous anaesthetic. Drugs 35:334–372. [DOI] [PubMed] [Google Scholar]
- Lee Y, Chung C, Oh YS (2000) Effectiveness of propofol pretreatment on the extent of deranged cerebral mitochondrial oxidative enzyme system after incomplete forebrain ischemia/reperfusion in rats. J Korean Med Sci 15:627–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie N, Grant IS (1987) Propofol for intravenous sedation. Anaesthesia 42:3–6. [DOI] [PubMed] [Google Scholar]
- Marik PE. (2004) Propofol: therapeutic indications and side‐effects. Curr Pharm 10:3639–3649. [DOI] [PubMed] [Google Scholar]
- Matsushita K, Kitagawa K, Matsuyama T, Ohtsuki T, Taguchi A, Mandai K, Mabuchi T, Yagita Y, Yanagihara T, Matsumoto M (1996) Effect of systemic zinc administration on delayed neuronal death in the gerbil hippocampus. Brain Res 743:362–365. [DOI] [PubMed] [Google Scholar]
- Merlo F, Demo P, Lacquaniti L, Tricarico L, Faccin G, Irone M (1991) Propofol in single bolus for treatment of elevated intracranial hypertension. Minerva Anestesiol 57:359–363 (In Italian). [PubMed] [Google Scholar]
- Mirski MA, Muffelman B, Ulatowski JA, Hanley DF (1995) Sedation for the critically ill neurologic patient. Crit Care Med 23:2038–20. Review. [DOI] [PubMed] [Google Scholar]
- Mizuno J, Sugimoto S, Tsutsui T, Machi‐da K, Sakai K (2002) Efficacy of propofol in controlling myoclonus during rewarming in a brain hypothermia patient. Masui 51:186–189 (In Japanese). [PubMed] [Google Scholar]
- Murphy PG, Myers DS, Davies MJ, Webster NR, Jones JG (1992) The antioxidant potential of propofol (2,6‐diisopropylphenol). Br J Anaesth 68:613–618. [DOI] [PubMed] [Google Scholar]
- Nakatani T, Tawaramoto M, Opare Kennedy D, Kojima A, Matsui‐Yuasa I (2000) Apoptosis induced by chelation of intracellular zinc is associated with depletion of cellular reduced glutathione level in rat hepatocytes. Chem Biol Interact 125:151–163. [DOI] [PubMed] [Google Scholar]
- Navapurkar VU, Skepper JN, Jones JG, Menon DK (1998) Propofol preserves the viability of isolated rat hepatocyte suspensions under an oxidant stress. Anesth Analg 87: 1152–1157. [DOI] [PubMed] [Google Scholar]
- Nimmo GR, Mackenzie SJ, Grant IS (1994) Haemodynamic and oxygen transport effects of propofol infusion in critically ill adults. Anaesthesia 49:485–489. [DOI] [PubMed] [Google Scholar]
- Orser BA, Bertlik M, Wang LY, MacDonald JF (1995) Inhibition by propofol (2,6 di‐isopropylphenol) of the N‐methyl‐D‐aspartate subtype of glutamate receptor in cultured hippocampal neurones. Br J Pharmacol 116:1761–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CK, Nehls DG, Graham DI, Teasdale GM, McCulloch J (1988) Focal cerebral ischaemia in the cat: treatment with the glutamate antagonist MK‐801 after induction of ischaemia. J Cereb Blood Flow Metab 8:757–762. [DOI] [PubMed] [Google Scholar]
- Powell JJ, Burden TJ, Greenfield SM, Taylor PD, Thompsson RPH (1999) Urinary excretion of essential metals following intravenous calcium disodium edetate: an estimate of free zinc and zinc status in man. J Inorganic Biochem 75:159–165. [DOI] [PubMed] [Google Scholar]
- Raoof AA, Obbergh LJ, Ville de Goyet J, Verbeeck R (1996) Extrahepatic glucuronidation of propofol in man: possible contribution of gut wall and kidney. Eur J Clin Pharmacol 50:91–96. [DOI] [PubMed] [Google Scholar]
- Rasmussen PA, Yang Y, Rutecki PA (1996) Propofol inhibits epileptiform activity in rat hippocampal slices. Epilepsy Res 25:169–175. [DOI] [PubMed] [Google Scholar]
- Ratnakumari L, Hemmings HC Jr (1997) Effects of propofol on sodium channel‐dependent sodium influx and glutamate release in rat cerebrocortical synaptosomes. Anesthesiology 86:428–439. [DOI] [PubMed] [Google Scholar]
- Rehberg B, Duch DS (1999) Suppression of central nervous system sodium channels by propofol. Anesthesiology 91:512–520. [DOI] [PubMed] [Google Scholar]
- Ridenour TR, Warner DS, Todd MM, Gionet TX (1992) Comparative effects of propofol and halothane on outcome from temporary middle cerebral artery occlusion in the rat. Anesthesiology 76:807–812. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Hendler S, Khoh‐Reiter S, Gillenwater G, Carlo D, Schubert D, Chang J (1999) Propofol hemisuccinate protects neuronal cells from oxidative injury. J Neurochem 73:2524–2530. [DOI] [PubMed] [Google Scholar]
- Sarraf‐Yazdi S, Sheng H, Miura Y, McFarlane C, Dexter F, Pearlstein R, Warner DS (1998) Relative neuroprotective effects of dizocilpine and isoflurane during focal cerebral ischemia in the rat. Anesth Analg 87:72–78. [DOI] [PubMed] [Google Scholar]
- Schwieler L, Delbro DS, Engberg G, Erhardt S (2003) The anaesthetic agent propofol interacts with GABAB‐receptors: an electrophysiological study in rat. Life Sci 72:2793–2801. [DOI] [PubMed] [Google Scholar]
- Shabanzadeh AP, Shuaib A, Yang T, Salam A, Wang CX (2004) Effect of zinc in ischemic brain injury in an embolic model of stroke in rats. Neurosci Lett 356:69–71. [DOI] [PubMed] [Google Scholar]
- Sitar SM, Hanifi‐Moghaddam P, Gelb A, Cechetto DF, Siushansian R, Wilson JX (1999) Propofol prevents peroxide‐induced inhibition of glutamate transport in cultured astrocytes. Anesthesiology 90:1446–1453. [DOI] [PubMed] [Google Scholar]
- Smith I, Monk TG, White PF, Ding Y (1994) Propofol infusion during regional anesthesia: sedative, amnestic, and anxiolytic properties. Anesth Analg 79:313–319. [DOI] [PubMed] [Google Scholar]
- Souter MJ, Andrews PJ, Alston RP (1998) Propofol does not ameliorate cerebral venous oxyhemoglobin desaturation during hypothermic cardiopulmonary bypass. Anesth Analg 86:926–931. [DOI] [PubMed] [Google Scholar]
- Stark RD, Binks SM, Dutka VN, O'Connor KM, Arnstein MJ, Glen JB (1985) A review of the safety and tolerance of propofol (‘Diprivan’). Postgrad Med J 61:152–156. [PubMed] [Google Scholar]
- Stuart Pharmaceutical : Technical brochure on Diprivan® Propofol. Wilmington , DE, 1989. [Google Scholar]
- Tramer MR, Moore RA, McQuay HJ (1997) Propofol and bradycardia: causation, frequency and severity. Br J Anaesth 78:642–651. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Huang SJ, Lai YY, Chang CL, Cheng JT (1994) Propofol does not reduce infarct volume in rats undergoing permanent middle cerebral artery occlusion. Acta Anaesthesiol Sin 32:99–104. [PubMed] [Google Scholar]
- Valtysson J, Hillered L, Andine P, Hagberg H, Persson L (1994) Neuropathological endpoints in experimental stroke pharmacotherapy: the importance of both early and late evaluation. Acta Neurochir (Wien) 129:58–63. [DOI] [PubMed] [Google Scholar]
- Velly LJ, Guillet BA, Masmejean FM, Nieoullon AL, Bruder NJ, Gouin FM, Pisano PM (2003) Neuroprotective effects of propofol in a model of ischemic cortical cell cultures: role of glutamate and its transporters. Anesthesiology 99:368–375. [DOI] [PubMed] [Google Scholar]
- Vincenti E, Michielan F, Feltracco P, Volpin SM (1991) Pharmacological properties of propofol: therapeutic implications In: Focus on infusion: intravenous anesthesia Prys‐Roberts C, editor. London : Medical Literature Ltd, 177–178. [Google Scholar]
- Wakasugi M, Hirota K, Roth SH, Ito Y (1999) The effects of general anesthetics on excitatory and inhibitory synaptic transmission in area CA1 of the rat hippocampus in vitro. Anesth Analg 88:676–680. [DOI] [PubMed] [Google Scholar]
- Wang J, Yang X, Camporesi CV, Yang Z, Bosco G, Chen C, Camporesi EM (2002) Propofol reduces infarct size and striatal dopamine accumulation following transient middle cerebral artery occlusion: a microdialysis study. Eur J Pharmacol 452:303–308. [DOI] [PubMed] [Google Scholar]
- Wilson JX, Gelb AW (2002) Free radicals, antioxidants, and neurologic injury: possible relationship to cerebral protection by anesthetics. J Neurosurg Anesthesiol 14:66–79. [DOI] [PubMed] [Google Scholar]
- Yamakura T, Sakimura K, Shimoji K, Mishina M (1995) Effects of propofol on various AMPA‐, kainate‐ and NMDA‐selective glutamate receptor channels expressed in Xenopus oocytes. Neurosci Lett 188:187–190. [DOI] [PubMed] [Google Scholar]
- Yano T, Nakayama R, Ushijima K (2000) Intracerebroventricular propofol is neuroprotective against transient global ischemia in rats: extracellular glutamate level is not a major determinant. Brain Res 883:69–76. [DOI] [PubMed] [Google Scholar]
- Young Y, Menon DK, Tisavipat N, Matta BF, Jones JG (1997) Propofol neuroprotection in a rat model of ischaemia reperfusion injury. Eur J Anaesthesiol 14:320–326. [DOI] [PubMed] [Google Scholar]
- Zaloga GP, Teres D (2000) The safety and efficacy of propofol containing EDTA: a randomised clinical trial programme focusing on cation and trace metal homeostasis in critically ill patients. Intensive Care Med 26:S398–S399. [DOI] [PubMed] [Google Scholar]
- Zhan RZ, Qi S, Wu C, Fujihara H, Taga K, Shimoji K (2001) Intravenous anesthetics differentially reduce neurotransmission damage caused by oxygen‐glucose deprivation in rat hippocampal slices in correlation with N‐methyl‐d‐aspartate receptor inhibition. Crit Care Med 29:808–813. [DOI] [PubMed] [Google Scholar]
- Zhu H, Cottrell JE, Kass IS (1997) The effect of thiopental and propofol on NMDA‐ and AMPA‐mediated glutamate excitotoxicity. Anesthesiology 87:944–951. [DOI] [PubMed] [Google Scholar]