

Abstract
Biological pathways are abstract and functional visual representations of existing biological knowledge. By mapping high-throughput data on these representations, changes and patterns in biological systems on the genetic, metabolic and protein level are instantly assessable. Many public domain repositories exist for storing biological pathways, each applying its own conventions and storage format. A pathway-based content review of these repositories reveals that none of them are comprehensive. To address this issue, we apply a general workflow to create curated biological pathways, in which we combine three content sources: public domain databases, literature and experts. In this workflow all content of a particular biological pathway is manually retrieved from biological pathway databases and literature, after which this content is compared, combined and subsequently curated by experts. From the curated content, new biological pathways can be created for a pathway analysis tool of choice and distributed among its user base. We applied this procedure to construct high-quality curated biological pathways involved in human fatty acid metabolism.
Introduction
In recent years, biological pathway analysis has become common in biochemical research. There is a plethora of pathway analysis tools available. In general, such tools map multidimensional experimental data to biological pathways, which are abstract and functional visual representations of existing biological knowledge [1]. Pathways aid the understanding of changes and patterns in biological systems from various types of organisms on the genetic, protein and metabolic level.
A pathway can encompass one or several types of biological processes [2], the key ones being regulatory processes, metabolic processes, protein–protein interactions and signaling processes. A regulatory process could, for instance, involve transcription factors and the genes whose expression they activate or inhibit, an example of which is the regulation of genes involved in fatty acid metabolism by peroxisome proliferator-activated receptor alpha (PPAR-α). A metabolic process describes flows of physiological reactions, involving substrates, products and commonly a catalyst, such as the series of reactions describing fatty acid β-oxidation. An example of a protein–protein interaction is the binding of a ligand to a receptor, which in turn may activate a signaling process like the mitogen-activated protein kinase (MAPK) cascade. A pathway always shows direction, but it can contain more knowledge than a simple network, such as information on the subcellular localization of components, regulatory mechanisms and connections to other pathways.
Over the past decade several online databases were created to store biological pathways [3]. Each database has its own conventions, level of interactivity and storage format, but in the end all of them store information covering biological pathways, from the low-level processes of metabolism to high-level processes like regulation. Some databases store only a static picture of a biological pathway, such as the BioCarta database (http://www.biocarta.com/), while others, such as Reactome [4] and KEGG [5], store extensive annotation for each of the elements in a pathway by using a custom XML format, in addition to a graphical layout. Another growing repository is Science Signaling (http://stke.sciencemag.org/), developed by American Association for the Advancement of Science (AAAS), the publisher of Science. It includes more than 60 curated signaling pathways and additionally provides short lists of key references and evaluations of the strength of existing evidence for associations within the database. WikiPathways (http://www.wikipathways.org/) applies a similar concept. It is a public Wiki platform dedicated to the creation, storing and curation of biological pathways by, and for, the scientific community. WikiPathways contains copies of all GenMAPP pathways plus additional pathways in GenMAPP Pathway Markup Language (GPML) format.
GeneGO Metacore [6], GenMAPP [7] and Metacyc (http://www.metacyc.org/) also offer data visualization and statistical analysis tools to analyze experimental data on a pathway level. This brings us to the most important application of pathways: pathway analysis.
Pathway analysis tools
Pathway analysis of gene or protein expression data applies genomic information to couple the expression data to known biological pathways. Usage of extensive collections of such pathways allows a quickly assessable overview of expression results in relation to biological mechanisms, facilitating the understanding of gene, protein and metabolite interactions at higher physiological levels. Cavalieri and De Filippo [8] reviewed tools that automatically display functional genomics results on biological pathways and tools that test for statistical significance of enrichment of genes belonging to a biological pathway. Among these tools are several commercial applications, such as GeneGO Metacore, Rosetta Resolver and Acuity, which are commonly used for high-throughput data analysis. Open-source programs such as GenMAPP with MAPPFinder [9], Cytoscape [10] and DAVID (http://david.abcc.ncifcrf.gov/) also offer the possibility of interactively visualizing expression datasets on biological pathways. When large amounts of expression data need to be analyzed on a large collection of pathways, a need for automation arises. Several statistical methods were developed to assess the significance of changes in gene expression in a pathway or a collection of pathways. Pathway analysis programs such as Pathway Miner [11], Eu.gene Analyzer [8], MetaCyc (http://www.metacyc.org/) and GenMAPPs MAPPFinder each have their own statistical approaches. An overview of some popular pathway editing and analysis tools is found in Table 1.
TABLE 1.
Overview of some popular pathway editing and analysis tools
| Pathway editing | Pathway analysis | License | Textmininga | Pathway databases | |
|---|---|---|---|---|---|
| Biocarta | Yes | No | Free | No | Proprietary |
| EU.gene | Yes | Yes | Free | No | Proprietary, WikiPathwaysb |
| GenMAPP | Yes | Yes | Free | No | Proprietary, KEGG, WikiPathwaysb |
| Genomatix | No | Yes | Commercial | Yes | Proprietary |
| Ingenuity | Yes | Yes | Commercial | Yes | Proprietary, KEGG |
| MetaCore | Yes | Yes | Commercial | Yes | Proprietary |
| Pathway Studio | Yes | Yes | Commercial | Yes | ResNet mammalian database, ResNet plant, ResNet targeted databases, KEGG, Science signaling, Prolexys HyNet yeast two-hybrid database |
| Reactome | Yes | No | Free | No | Proprietary |
| WikiPathways | Yes | No | Free | No | Proprietary, GenMAPP |
For pathway expansion.
Through online converter on WikiPathways.
GenMAPP is a popular, freely available biological pathway analysis tool developed at the Conklin Lab at the J. David Gladstone Institutes of the University of California. Several gene properties can be displayed simultaneously on pathways by creating a lookup table, linking colors and descriptions to user-specified criteria that, for instance, indicate changes in the gene expression level. GenMAPPs built-in statistical tool MAPPFinder enables a pathway-based enrichment analysis. MAPPFinder calculates a statistical p-value for each pathway entity using the hypergeometrical distribution, after which pathways are ranked by significance.
Another popular tools-suite is Ingenuity Pathways Analysis (IPA, http://www.ingenuity.com/), an all-in-one commercial software application that enables modeling, analysis and understanding of the complex biological and chemical systems at the core of life-science research. It consists of several tools enabling one to mine the scientific literature easily, build dynamic pathway models and analyze high-throughput experimental data quickly to identify key insights. IPAs dynamic pathway modeling tool applies textmining approaches for the construction of novel relations between pathway entities. As an alternative, the textmining tool Bibliosphere by Genomatix (http://www.genomatix.de) offers similar functionality, combining textmining results from literature with sequence data to identify relations more robustly. Bibliosphere is available from the Genomatix website. Likewise, Pathway Studio, a commercial application suite from Ariadne Genomics, implements textmining approaches to find relationships among pathway entities. In addition, Pathway Studio can perform Gene Set Enrichment analysis on sets of genes that share a functional, biological or other relation.
Pathway repositories
Every pathway analysis tool uses pathways that are created locally or downloaded from a central repository. To assess the quality and completeness of such repositories, we extracted and compared the pathway content from processes involved in fatty acid metabolism from several free pathway databases. We focused on fatty acid oxidation, fatty acid synthesis and the regulation of these processes. These pathways are well described in literature. Since most pathway repositories curate their pathways with literature, one would expect excellent entries. A full overview is given in Table 2.
TABLE 2.
Fatty acid metabolism pathway contenta of selected pathway repositories
|
Biosynthesis
|
Fatty acid degradationb
|
Transport | Regulation
|
Fatty acid elongation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fatty acids | Triacyl glyceride | β-ox M | β-ox U | β-ox P | ω-ox | Carnitine shuttle | Synthesis | Degradation | Elongation | |
| Reactome | i | c | c | c | m | m | m | i | m | m |
|
| ||||||||||
| KEGG | c | m | c | m | m | m | m | m | m | c |
|
| ||||||||||
| GenMAPP | c | m | i | i | m | m | i | m | m | m |
|
| ||||||||||
| BioCarta | m | m | c | c | i | c | c | i | m | m |
c, complete pathway in database; m, missing in database; i, present but incomplete in database.
β-ox M, mitochondrial β-oxidation; β-ox U, mitochondrial β-oxidation of unsaturated fatty acids; β-ox P, peroxisomal β-oxidation.
KEGG stores 33,679 pathways for over 100 species generated from 269 reference pathways concerning metabolism, genetic information processing, environmental response, cellular processes, human diseases and drug response. Most pathway tools make extensive use of the KEGG database. Although generally considered a robust database, some of the fatty acid metabolism pathways in KEGG have not been updated for years. The database contains the pathways for fatty acid biosynthesis, fatty acid elongation, fatty acid desaturation and fatty acid mitochondrial β-oxidation, but no entries related to the regulation of these processes.
The BioCarta pathway database offers a free collection of pathways and is hosted by a company that supplies antibodies for entities on several pathways. It contains key information for over 120,000 genes from multiple species through a user-friendly interface. The database contains 296 regularly updated pathways. Pathways describing mitochondrial and peroxisomal β-oxidation of fatty acids, oxidation of odd-numbered chain fatty acids, oxidation of polyunsaturated fatty acids, disease-related ω-oxidation and a separate pathway of the carnitine-mediated transport system are all found in BioCarta.
GenMAPP stores more than 200 contributed pathways as well as hundreds of pathways derived from the KEGG database. The contributed pathways related to human fatty acid metabolism are fatty acid synthesis, mitochondrial fatty acid β-oxidation and fatty acid degradation, but these have not been updated or checked for years.
Reactome is an online bioinformatics database of biology described in molecular terms, storing pathways as a series of separate biochemical reactions. Most pathways involved in human fatty acid metabolism are present. Some pathways, like the oxidation of unsaturated fatty acids, are not found in other databases. Several reactions, like the one converting palmitate to long-chain fatty acids, are only mentioned, whereas other databases give a complete set of reactions.
A pathway curation workflow
Biological pathway content varies greatly in quality and completeness among the tools and databases described above, even at first glance. Additionally, pathway repositories in general apply different storage formats and conventions. Ideally, one would want to integrate all knowledge of a particular pathway from the various tools and databases available to create one ‘complete’ pathway. This is especially attractive when performing pathway analysis. Yet, researchers that possess the knowledge needed to create and curate such an integrated pathway are often discouraged to do so. A possible reason is the lack of a general guideline for pathway creation and curation, in addition to the false assumption that it is a time-consuming process.
The authors of this manuscript apply a general biological pathway curation workflow (Fig. 1) comprising four phases and three main data sources: online public domain databases, literature and experts. The databases used to collect content are KEGG, Reactome, GenMAPP and BioCarta, using the content from Reactome as a basis, because its content is the most complete, heavily curated and, therefore, of the highest quality. PubMed (http://www.pubmed.org/) is used to find relevant literature.
FIGURE 1.
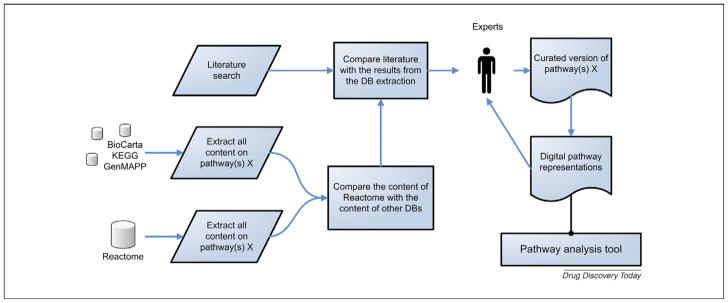
Pathway curation workflow. This figure shows the outline of the biological pathway curation workflow. The workflow applies content from online databases, literature and experts. First, all biological information associated with the biological pathways in question is retrieved from Reactome and compared to content from KEGG, GenMAPP and BioCarta. Scientific literature is used to fill in the gaps and, finally, the results are curated with the help of experts to remove ambiguities and inconsistencies. The curated content is used to make new digital pathway representations, suitable for analysis in a pathway analysis tool of choice. These digital representations are distributed among the user base of the pathway analysis tool of choice, resulting in additional curation feedback.
The first step in this manual workflow is to collect all biological information associated with the biological pathways in question by (i) retrieving biological pathway content from several curated and highly regarded biological pathway databases; (ii) comparing and structuring content from different databases; (iii) searching for scientific literature referring to the processes described in these biological pathways and finally (iv) curating the results with the help of experts to remove ambiguities and inconsistencies. The resulting curated content is subsequently used to create new pathways in a format suitable for a pathway analysis tool of choice, resulting in additional curation feedback.
Creating high-quality curated fatty acid metabolism pathways
The manual curation process was used to create high-quality curated pathways of the key processes involved in human fatty acid metabolism: fatty acid oxidation, fatty acid synthesis and regulation of these processes. Pathways involved in fatty acid metabolism are essential pathways in nutritional research. Curated up-to-date versions of these pathways are therefore in high demand.
Biological pathways in Reactome are presented as a collection of biochemical reactions and lack a visual overview showing connections between these reactions. Hence, comparing pathway content from Reactome to the content found in other databases is a difficult task. To facilitate this process, graphical overviews were created manually by starting with the first listed reaction, connecting its product as the substrate of the second reaction and so on, thus creating a series of representations of all the reactions involved in human fatty acid metabolism. Once these representations were created, differences with pathway content found in the biological pathway repositories described above were annotated and literature was searched simultaneously to check everything found up to that point. Some of the pathways involved in human fatty acid metabolism were found to be incomplete or completely missing in the various biological pathway repositories. The results are best discussed on a pathway-to-pathway basis, highlighting the differences in completeness and accuracy between databases (summarized in Table 2).
Fatty acid biosynthesis has three major functions: the storage of excess energy intake, synthesis of fat from carbohydrates or proteins if the quantity of fat in the diet is low and synthesis of fat for lactation [12].
Reactome gives a good overview of the transport of citric acid from the mitochondria to the cytosol, where the citrate ligase catalyzes the production of acetyl-coenzyme A (acetyl-CoA) from citrate to the creation of long-chain fatty acids. This pathway is followed by the triacylglycerol biosynthesis pathway.
Several essential parts of the fatty acid metabolism were found to be missing from the Reactome database. No details are given regarding the reaction that transforms palmitate into a long-chain fatty acid and there is only a schematic overview of the reaction that transfers butyryl-acyl carrier protein (butyryl-ACP) into palmitoyl-ACP. Pathways involved in fatty acid elongation and desaturation are missing entirely.
The elongation of fatty acids proceeds through a repeated cycle of reactions. This cycle starts by the conversion of acyl-CoA to 3-ketoacyl-CoA, which is catalyzed by acetyl-CoA C-acyltransferase. The 3-ketoacyl-CoA intermediate undergoes the same three reactions that form the basis of β-oxidation, only in reverse order. Reduction of the keto group is followed by dehydration to form a double bond. Reduction of the double bond results in an acyl-CoA that is two carbons longer than the acyl-CoA at the beginning of the cycle [13]. The desaturation takes place after elongation in the endoplasmic reticulum and is catalyzed by acyl-CoA desaturase.
KEGG describes the elongation cycles in detail and, in addition, gives all the enzyme codes of the different fatty acid synthases. GenMAPP has MAPPs describing fatty acid elongation and desaturation, but the reactions are not visualized in a cyclic manner.
There are several processes involved in the degradation of fatty acids, with β-oxidation being the most important [14]. β-Oxidation is the process by which fatty acids are degraded in the mitochondria. The carnitine shuttle [15] is essentially the first step of mitochondrial fatty acid β-oxidation. It is involved in the transport of long-chain fatty acids through both mitochondrial membranes, from the cytosol to the mitochondrial matrix where β-oxidation takes place [16].
Collecting and comparing the information on the several fatty acid degradation processes showed that Reactome does not contain any information on reactions related to the fatty acid carnitine transport system (the transport of fatty acid into mitochondria) or the fatty acid cell transport system (the transport of fatty acid into the cell). The KEGG database refers to the carnitine O-palmitotransferase enzyme in the β-oxidation pathway, but fails to clarify the uptake into mitochondria. GenMAPP and BioCarta give a full overview of the carnitine shuttle, but disagree on some minor details concerning the subcellular location of each step in the process. These ambiguities were clarified using literature [15,16].
Mitochondrial fatty acid β-oxidation of saturated and unsaturated fatty acids is well described in Reactome. In KEGG, β-oxidation of unsaturated fatty acids is missing. GenMAPP gives an abbreviated description and BioCarta gives a small overview. Neither BioCarta nor GenMAPP details the seven different cycles of the β-oxidation process, falsely assuming a reiteration of one general cycle.
Peroxisomal β-oxidation [17] applies the same mechanism as mitochondrial β-oxidation, but the peroxisomal fatty acid β-oxidation system is able to shorten only fatty acid chains and cannot degrade fatty acids completely. The shorter chains are transported as carnitine-ester from the peroxisomes to the mitochondria, where the degradation is completed. Specific information on peroxisomal β-oxidation is missing from most databases. Reactions describing the process are present, but incomplete in the Reactome database. BioCarta gives a short, but otherwise complete, overview.
Another form of fatty acid oxidation is ω-oxidation [17]. ω-Oxidation is a minor process that takes place in the endoplasmic reticulum, but only occurs when the β-oxidation processes are somehow impaired by disease or fasting. Information on ω-oxidation is found only in BioCarta. ω-Oxidation was not mentioned in any other database. Additions were found in literature [17].
There are five main regulatory proteins involved in the regulation of fatty acid synthesis: liver X receptor/retinoid X receptor (LXR-α/RXR-α) heterodimer, nuclear transcription factor Y (NF-Y), sterol regulatory element binding protein 1 and 2 (SREBP1, SREBP2) and carbohydrate regulatory element binding protein (ChREBP) [18–23]. The main regulator in the regulation of β-oxidation is PPAR-α [24–26].
Regulation of fatty acid synthesis by ChREBP [18–23] has an entry in the Reactome database, but transcriptional activation of the synthesis via SREBP1 and LXR-α/RXR-α heterodimer and NF-Y is absent. The regulation of fatty acid degradation is not mentioned in Reactome.
BioCarta contains pathways describing regulation of fatty acid synthesis by SREBP1 and SREBP2 and the regulation of genes involved in fatty acid β-oxidation by PPAR-α. LXR is only mentioned in conjunction with farnesoid X receptor (FXR) in a pathway for the regulation of cholesterol metabolism. Likewise, RXR degradation is mentioned, but there is no entry describing the relation to the regulation of fatty acid synthesis.
GenMAPP and KEGG do not contain any pathways describing regulation of processes involved in human fatty acid metabolism. Content from Reactome and BioCarta was expanded with the help of literature [21–26].
After all content was collected it was passed on to experts in the field of fatty acid metabolism. New pathways describing fatty acid synthesis, triacylglyceride synthesis, fatty acid β-oxidation (saturated and unsaturated) and fatty acid ω-oxidation were created from this curated content. Pathways are available from WikiPathways (http://www.wikipathways.org/). Downloading the pathways from this location ensures that you have the most up-to-date version. WikiPathways has the option to export to formats suitable for analysis in Cytoscape, EU.gene Analyzer and Gen- MAPP. This resulted in constructive feedback from the large user base of these tools.
Concluding remarks
Biological pathway databases are far from comprehensive. We have used a rapid pathway curation workflow to collect all content of biological pathways involved in human fatty acid metabolism. At the time of writing, these improved fatty acid metabolism pathways are among the most widely used biological pathways in GenMAPP and, as such, have become part of the standard MAPP archive. The suggested workflow can be seen as a general guideline for anyone looking to create novel high-quality curated biological pathways. The choice for a particular pathway-editing tool, however, is up to the user. Additionally, we use only public domain databases. Although content that comes with commercial tools is often derived from public databases and literature, those tools are, in many cases, well developed and have convenient user interfaces, which makes their usage beneficial for pathway developers that have access to such tools.
A downside of the presented manual curation workflow is that it will not generally yield novel pathways when using database content as the starting point. This can be addressed by consulting experts first, which in most cases will result in a ‘raw pathway’. Literature and database content can then be used to polish this raw pathway. Finding relevant literature is an arduous task as querying articles for single reactions yields excessive amounts of hits. A possible solution for this issue is textmining, which can be used to generate an exhaustive list of relevant literature and networks of possible connections between queried components, for instance based on cocitation in PubMed abstracts. These networks of inferred relations are then integrated into one or several pathways. The content should then still be curated by experts, because textmining will, in general, yield a large amount of false positives. In a similar fashion to manually extracted content from databases and literature, this content may then be transcribed into novel pathways. Textmining is already used in several pathway analysis tools (Table 1) as an alternative to pathway analysis using known curated pathways.
Using the presented workflow, it is possible to create a small collection of curated pathways within 20 ‘man-hours’, of course depending on expert availability. This is not fast enough to fill a repository with all known pathways. But this is not the purpose of the presented workflow. Anyone can make a small pathway in half-an-hour, but this does not imply that the information contained within the pathway is of high quality or even correct. Curation is the key. Pathway curation can normally take months, leading to out-of-date and incomplete content. The presented pathway curation workflow, although possibly less thorough, is faster and is founded on the principle of public demand. Hence, it is a suitable workflow for creating key pathways. A community effort is needed to create a more robust set of pathways in a similar manner, as demonstrated by the Science Signaling and WikiPathways initiatives.
Still, to improve speed, partial automation of the curation process could be implemented, starting with content extraction from Reactome and ending at novel pathways in a suitable exchange format. There are two data flows at the moment, an overview of which can be seen in Fig. 2. Once the content is in GPML format the pathways are easily converted to Cytoscape, EU.gene Analyzer and GenMAPP formats through the WikiPathways interface.
FIGURE 2.
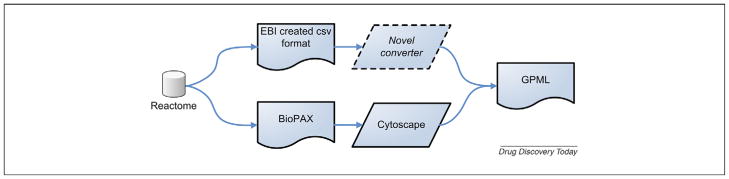
Reactome to GPML pipeline. Two possible connections between Reactome and GPML are shown. Each wave block is a data type; each parallelogram is a software application. One connection uses an EBI created comma-separated text format and a novel converter to create a GPML file, a format suitable for WikiPathways and GenMAPP. Additionally, Reactome offers the option to export all its pathways and reactions as BioPAX level 2 compliant files, which can then be directly imported in the java-based Cytoscape application and converted to GPML.
Only one connection between Reactome and GPML is readily available, implementing an intermediary EBI created comma-separated text format. There is another suggested connection through the BioPAX level 2 format [27]. BioPAX Level 2 is a suitable exchange format for biological pathway content, but at the moment does not store the additional graphical layout information that is required for transparent conversion of pathway representations from pathway analysis tools such as GenMAPP and Cytoscape. Reactome offers the option to export all its pathways and reactions as BioPAX level 2 compliant files, which can then be directly imported into the java-based Cytoscape application and converted to GPML.
Pathway curation is a Sisyphean task, because new discoveries constantly lead to novel additions to and adaptations of a pathway. Also, the ‘beginning’ and ‘end’ of a pathway are arbitrary definitions. This latter difficulty can be overcome by creating so-called meta-pathways, covering several separately defined pathways and their respective biochemical and biological connections, similar to the Reactome sky map. The former challenge can be met by the adaptation of standard exchange formats for biological pathways, enabling automated pathway data acquisition and comparison in a network analysis tool. With such additions in place, the road to curated pathways will become less long and winding.
Acknowledgments
We would like to thank Sander Kersten and Jochum Plat, whom we consulted as experts in the field of fatty acid metabolism; Marjan van Erk, Rachel van Haaften, Peter d’Eustachio and Bernard de Bono for their valuable advice and support.
References
- 1.Cary MP, et al. Pathway information for systems biology. FEBS Lett. 2005;579:1815–1820. doi: 10.1016/j.febslet.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Bader GD, et al. Pathguide: a pathway resource list. Nucleic Acids Res. 2006;34 (Database issue):D504–D506. doi: 10.1093/nar/gkj126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galperin MY. The molecular biology database collection: 2008 update. Nucleic Acids Res. 2008;36 (Database issue):D2. doi: 10.1093/nar/gkm1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vastrik I, et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol. 2007;8:R39. doi: 10.1186/gb-2007-8-3-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanehisa M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36 (Database issue):D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekins S, et al. Pathway mapping tools for analysis of high content data. Methods Mol Biol. 2007;356:319–350. doi: 10.1385/1-59745-217-3:319. [DOI] [PubMed] [Google Scholar]
- 7.Salomonis N, et al. GenMAPP. 2 New features and resources for pathway analysis. BMC Bioinform. 2007;8:217. doi: 10.1186/1471-2105-8-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavalieri D, De Filippo C. Bioinformatic methods for integrating whole-genome expression results into cellular networks. Drug Discov Today. 2005;10:727–734. doi: 10.1016/S1359-6446(05)03433-1. [DOI] [PubMed] [Google Scholar]
- 9.Doniger SW, et al. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cline MS, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandey R, et al. Pathway Miner: extracting gene association networks from molecular pathways for predicting the biological significance of gene expression microarray data. Bioinformatics. 2004;20:2156–2158. doi: 10.1093/bioinformatics/bth215. [DOI] [PubMed] [Google Scholar]
- 12.Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. 2000;16:202–208. doi: 10.1016/s0899-9007(99)00266-x. [DOI] [PubMed] [Google Scholar]
- 13.Garrett RH, Grisham CM, editors. Biochemistry. Brooks & Cole; 1999. [Google Scholar]
- 14.Vockley J, Whiteman AH. Defects of mitochondrial β-oxidation: a growing group of disorders. Neuromuscul Disord. 2002;12:235–246. doi: 10.1016/s0960-8966(01)00308-x. [DOI] [PubMed] [Google Scholar]
- 15.Rubio-Gozalbo ME, et al. Carnitine-acylcarnitine translocase deficiency, clinical biochemical and genetic aspects. Mol Aspects Med. 2004;25:521–532. doi: 10.1016/j.mam.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 16.McClelland GB. Fat to the fire: the regulation of fatty acid oxidation with exercise and environmental stress. Comp Biochem Physiol B: Biochem Mol Biol. 2004;139:443–460. doi: 10.1016/j.cbpc.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Wanders RJA. Peroxisomes, fatty acid metabolism, and peroxisomal disorders. Mol Genet Metab. 2004;83:16–27. doi: 10.1016/j.ymgme.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 18.Joseph SB, Tontonoz P. LXRs: new therapeutic targets in atherosclerosis? Curr Opin Pharmacol. 2003;3:192–197. doi: 10.1016/s1471-4892(03)00009-2. [DOI] [PubMed] [Google Scholar]
- 19.Dentin R, et al. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): two key regulators of glucose metabolism and fatty acid synthesis inliver. Biochimie. 2005;87:81–86. doi: 10.1016/j.biochi.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Uyeda K, et al. Carbohydrate responsive element-binding protein (ChREBP): a key regulator of glucose metabolism and fat storage. Biochem Pharmacol. 2002;63:2075–2080. doi: 10.1016/s0006-2952(02)01012-2. [DOI] [PubMed] [Google Scholar]
- 21.Matuoka K, Chen KY. Transcriptional regulation of cellular ageing by the CCAAT box-binding factor CBF/NF-Y. Ageing Res Rev. 2002;1:639–651. doi: 10.1016/s1568-1637(02)00026-0. [DOI] [PubMed] [Google Scholar]
- 22.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 23.Edwards PA, et al. Biochim Biophys Acta – Mol Cell Biol Fatty Acids. 2000. Regulation of gene expression by SREBP and SCAP; pp. 1–3.pp. 103–113. [DOI] [PubMed] [Google Scholar]
- 24.Kota BP, et al. An overview on biological mechanisms of PPARs. Pharmacol Res. 2005;51:85–94. doi: 10.1016/j.phrs.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 25.Mandard S, et al. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci. 2004;61:393–416. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rastinejad F. Retinoid X receptor and its partners in the nuclear receptor family. Curr Opin Struct Biol. 2001;11:33–38. doi: 10.1016/s0959-440x(00)00165-2. [DOI] [PubMed] [Google Scholar]
- 27.Strömbäck L, Lambrix P. Representations of molecular pathways: an evaluation of SBML, PSI MI and BioPAX. Bioinformatics. 2005;21:4401–4407. doi: 10.1093/bioinformatics/bti718. [DOI] [PubMed] [Google Scholar]