

Abstract
Hippocampal sclerosis (HS) continues to be the most common pathology identified in patients with refractory temporal lobe epilepsy undergoing surgery. Wilhelm Sommer described this characteristic pattern of neuronal loss over 120 years ago through his post‐mortem studies on patients with epilepsy. Neuropathological post‐mortem studies in the 20th century proceeded to contribute significantly to the understanding of this disease process, with regard to the varying patterns of HS and involvement of adjacent limbic structures. From studies of surgical temporal lobe specimens from the 1950s onwards it was recognized that an early cerebral injury could act as the precipitant for the sclerosis and epilepsy. Modern neuropathological studies have focused on aspects of neuronal injury, loss of specific neuronal groups and cellular reorganization to address mechanisms of epileptogenesis and the enigma of how specific hippocampal neuronal vulnerabilities and glial proliferation are both the effect and the cause of seizures.
Keywords: hippocampal sclerosis, epilepsy, neuropathology, historical perspectives
INTRODUCTION
Hardening or sclerosis of the hippocampus in patients with epilepsy has been recognized for at least 180 years. Early neuropathologists were able to give detailed anatomical descriptions of the selective patterns of neuronal loss and gliosis based on post‐mortem material. These observations form the foundation of current neuropathological diagnostic criteria for hippocampal sclerosis (HS). What has caused the most debate in the interim, continuing into the modern era, is the unresolved question of “cause or effect” of seizures. Trying to dissect the cellular processes responsible for epileptogensis in this region of scarring has proved a more daunting quest with a wealth of published literature. Reviewing some of the historically most noteworthy studies devoted to HS allows us an overview of the progress made in understanding this enigmatic pathology.
AMMON'S HORN SCLEROSIS (AHS): EARLY DESCRIPTIVE NEUROPATHOLOGY
One of the earliest description of HS or AHS as it was then termed, appears in 1825. Bouchet and Cazauvieilh (7) described lesions of increased consistency involving the hippocampus in 8 of 18 post‐mortems among patients with epilepsy and insanity, although they ascribed little importance to this finding at that time. It is Sommer's name that is more generally associated with this pathology as he was the author of a landmark paper recognizing a potential importance of this lesion. In 1880 he reviewed the accumulating data regarding the association of abnormalities involving the Ammon's horn in patients with epilepsy. His studies were based both on his own observations and the reports of others (including Bergmann, Bouchet and Cazauvieilh, Hemkes, Pfleger and Meynert) from a total of 90 post‐mortems that he carefully tabulated (46). At macroscopic examination, the Ammon's horn generally appeared of diminished size, grey, “rigid” or with a leathery cartilaginous consistency. In fewer cases softening of this region was noted. Sommer provided the first detailed histological descriptions based on hematoxylin and carmine‐stained sections with great anatomical precision. He considered the extensive loss of neurons in the pyramidal cell layer as the most important finding: “All pyramidal cells. . . . which are doubtlessly of greatest importance for the function of Ammon's horn are missing.” He localized the neuronal loss to a segment adjacent to the medial wall of the lower horn of the ventricle, describing the anatomical boundaries of the affected region (Figure 1) that was to become known as “Sommer's sector,” our modern day CA1. He appreciated that some regions of Ammon's horn were spared but noted a reduction in the number of neurons in the region within the dentate fascia or the end plate (what we now term CA4). Regarding this later observation he curiously states “I wouldn't want to attach importance to this by the way” although he doesn't disclose his reasons.
Figure 1.
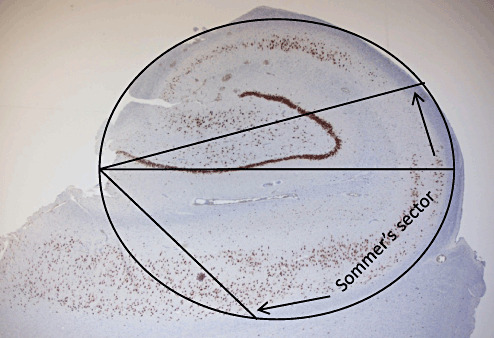
Sommer's description of the vulnerable region of neuronal loss in Ammon's horn sclerosis that became known as Sommer's sector. In his original paper he describes this as “The whole part of the grey matter that belongs to the medial wall of the lower horn, approximately from the fold‐over locus of the ventricular endothelium over the middle of the curvature . . . or if one considers the Ammon's horn as an ellipsoid, whose biggest axis goes parallel to the horizontal plane, in a sector in which the lower exterior quadrant (calculated from the medial plane of the brain) covers approximately 60 degrees of the horizontal axis towards the smaller one, and 20 degrees in the upper exterior.” In the figure these parameters have been superimposed on a surgical resection specimen immunostained with neuronal nuclear antigen to highlight Sommer's description.
These observations of this first review of AHS was largely confirmed in a similar study by Bratz (8) that followed a few years later in 1899. He published a series of findings in 70 patients with epilepsy, providing further detail of the pathology of AHS in addition to one of the earliest histological illustrations (Figure 2). He described the sharp interface of neuronal loss between CA1 and the preserved subiculum. Similar to Sommer he confirmed a “less complete” loss of neurons in the hilus or end folium, a preservation of “spindle cells” in what we now term the stratum oriens (likely describing preservation of interneurons), in addition to the recognition of a resistant sector (corresponding to what we now term CA2). Bratz also commented on the resistance or remarkable preservation of the granule cells of the dentate gyrus. Interestingly, both Bratz and Sommer drew attention to abnormalities of the vasculature within the sclerotic region, the vessels being described as “thick walled and glossy” with Bratz alluding to a proliferation of microvasculature. This pathological feature has been somewhat overlooked until very recently with emerging evidence that angiopathology may be of significance through local dysfunction of the blood brain barrier promoting epileptogenesis in HS as well as other focal epilepsies 24, 39, 55.
Figure 2.

One of the first illustrations of hippocampal sclerosis from an original paper published in 1987 by Bratz (8) depicting the regions of neuronal loss and reduction in volume in the hippocampus compared with a control. Note that the granule cell layer also appears somewhat broader than in the control although the phenomena of granule cell dispersion in the dentate gyrus remained to be described until 1990.
The early literature also made evident that the severity of the sclerosing process and distribution of neuronal loss could vary between cases. On this matter, Sommer himself had been careful to report that he could identify subtle histological changes of AHS where he was unable to confirm macroscopic atrophy, concluding that this could “constitute earlier stages of a similar process”(46). Spielmeyer around 1927 was the first to note that in severe cases all the sectors of the pyramidal cell layer in the hippocampus could be affected, even the dentate gyrus and the resistant sector: we now refer to this pattern as total HS (48). In a post‐mortem study of 55 epilepsy patients, Margerison and Corsellis (1966) delineated a further pattern of cell loss and gliosis localized to the hilus with complete sparing of Sommer's sector: This was termed “end folium sclerosis” as distinct from classical AHS; however, they regarded both as forms of HS (29). In recent years a further less frequent pattern with restricted damage of CA1 sector has been recognized 6, 15, 16, 40.
HS: ASSOCIATION WITH TEMPORAL LOBE EPILEPSY (TLE) AND THE SURGICAL ERA
The first descriptions of TLE were given by Hughlings Jackson (1889), these episodes being characterized by olfactory, gustatory or visceral aura, déjà vu or a dream‐like state (26). In cases with this symptomatology, small focal lesions were found in the anterior temporal lobe, and in particular, in the hippocampus, but he did not go as far as to link AHS with TLE. Stauder (1935) was the first to correlate AHS with seizures of temporal lobe semiology (49). In his autopsy study he showed a clear correlation of the clinical signs of TLE and AHS that was identified in 33 of 36 cases. With the dawn of electroencephalography, the diagnosis of what became termed “psychomotor epilepsy” was better established (22). Sano and Malamud (1953) went further and were able to confirm a relationship between the presence of AHS (as confirmed histologically at autopsy) and electroencephalogram evidence of psychomotor or temporal lobe seizures (41). These observations led to the conclusion that AHS was the cause of TLE.
The 1950s were notable as the era of the expansion of surgical programs and treatments for TLE, allowing the unique opportunity for neuropathological analysis of the resected tissues. Some of the earliest surgical series findings were published by Penfield et al in Montreal 17, 18. They identified what they called “incisural sclerosis,” variably involving the temporal lobe, hippocampus, uncus and amygdala, as the most common lesion, although detailed pathology was lacking mainly because the temporal lobe was sucked out in fragments during this procedure, making subsequent anatomical and pathological assessment difficult. Systematic histological studies on surgical temporal lobectomy specimens, however, proceeded from the Institute of Psychiatry, London, under the auspices of Meyer, then Professor of Neuropathology, working closely with the neurosurgeon Falconer in the early 1950s. The standard procedure of their surgical approach included removal of the anterior two‐thirds of the temporal lobe intact together with the hippocampus, uncus and part of the amygdala. What emerged from this group's studies of these tissues (1954–1956) was the first clear delineation of the pathological changes in TLE, and it was possible to confirm AHS as the most frequent pathology in lobectomy specimens 10, 34, 35. Bruton (1988), in his Maudsley monograph, emphasized these findings through the documentation of the extensive surgical series of 249 cases operated by Falconer over a 25‐year period, with 49% showing AHS (9).
HS AND THE BROADER CONCEPT OF MESIAL TEMPORAL LOBE SCLEROSIS
Although the Ammon's horn shows the most striking atrophy in TLE, it has long been noted from neuropathological studies that the sclerotic process may be more widespread, involving adjacent mesial temporal and limbic structures. Sommer and Bratz both reported cases of AHS where the atrophic process extended to involve “the convolutions of the temporal lobes,” the parahippocampal gyrus, as well as more widespread “hemispheric atrophy”8, 46. Meyer and Beck (1955) disclosed histological evidence of sclerosis of the amygdala involving the paraventricular region of the basal nucleus (34) (Figure 3). Meyer commented on the potential functional significance of this finding in the light of recent experimental work by Gastaut at that time (21), supporting the amygdala as the region from which experimental TLE could be most easily elicited.
Figure 3.

Illustrations taken from the 2nd edition of Greenfield (33) in the chapter on epilepsy authored by Alfred Meyer showing (A) severe degeneration in the amygdala (arrows), (B) Ammon's horn sclerosis involving Sommer's sector and (C) temporal neocortex with severe laminar neuronal loss from mainly cortical layer three (indicated by arrows). This pattern of temporal lobe neocortical sclerosis, with loss of neurons from the superficial cortical layers, is more easily visible in lobectomy specimens with neuronal nuclear antigen immunohistochemistry, as illustrated in (D). The reproduction of figures from Greenfield's Neuropathology, 2nd Edn., is with permission from Hodder Education.
Meyer together with Cavanagh (10) published the matter of the “diffuse and disseminated” lesions associated with AHS in more detail. In a group of 19 cases with AHS, laminar nerve cell loss in layers II and III of the temporal neocortex was seen in the majority, as illustrated in Meyer's chapter on epilepsy in the early editions of Greenfield's Neuropathology (33) (Figure 3). In addition, gliosis of the white matter, subpial layer of the neocortex, the parahippocampal gyrus as well as amygdala sclerosis of the basal nuclear groups were seen in around half of the cases. These features were more prominent in AHS cases than patients where epilepsy was secondary to a tumor. It is from these observations that the term “medial temporal sclerosis” was adopted in preference to HS or AHS to convey the more widespread sclerosis involving limbic structures of which the hippocampus appeared to be the epicenter. Margerison and Corsellis (29) also recognized significant sclerosis of the amygdala in their post‐mortem series of patients with TLE, being present in 15 of 55 cases with AHS. They noted however, that “the identification of slight nerve cell loss was particularly difficult” because of the complex anatomy of this region. Despite these early landmark papers, there is relatively sparse neuropathology literature devoted to surgical pathology of the amygdala proceeding from the modern era, with a handful of publications as reviewed in (37), compared with the abundance of literature devoted to the hippocampus. This is possibly a result of its complex anatomy and incomplete and fragmentary removal during temporal lobe surgery. As such the incidence, diagnostic criteria and significance of amygdala sclerosis remains a somewhat controversial area inviting further study (57). Similar arguments also apply to the presence and definition of temporal lobe sclerosis and involvement of the parahippocampal gyrus, where there are somewhat conflicting pathological data 4, 13, 16, 58.
HS: THEORIES AS TO ITS CAUSE
Although the neuropathological diagnosis of AHS remained largely unchanged over the last century, diverse theories have evolved as to its cause and relationship to seizures. Erkrankung des Ammonshornes als aetiologisches Moment der Epilepsie, as his title suggests, gives the clear impression that Sommer considered Ammon's horn disorders as the likely cause of epilepsy. However, following very shortly on his work, Pfleger published his own observations of acute changes to the Ammon's horn following status epilepticus (36) with the logical conclusion that this process was all secondary to seizures, contradicting Sommer's theory and kicking off the ongoing debate. Spielmeyer and his school of followers including Scholz (1951) theorized that disturbances of vasomotor regulation arising during a major convulsion resulted in the necrosis of the hippocampus (42). Stauder (1935), a pupil of Spielmeyer, who linked the pathology of AHS with TLE, suggested that frequent falls during a seizure could result in temporal lobe injury and this pattern of sclerosis (49). Controversial theories for the time were proposed by (Cecile and Oscar) Vogt in works published in 1925 and 1937 claiming that the specific patterns of hippocampal neuronal loss were a result of “physicochemical” properties common to groups of neurons in their “pathoclisis” theory (56). Based on their surgical observations, Penfield et al promoted the idea that “incisural sclerosis,” as they termed AHS, was a result of temporal lobe compression or tentorial herniation (including cerebral vessels), as a result of head molding during childbirth 17, 18. They considered these regions to be poorly developed and susceptible in the neonate. It was postulated that such acute lesions would “ripen” into scar tissue, eventually disrupting cerebral activity, resulting in a tendency for epileptic attacks. Margerison and Corsellis argued against a traumatic event antedating HS and suggested that if traumatic events were identified at post‐mortem, they were more likely a result of a seizure (29). Beginning the era of quantitative neuropathology studies, Babb (1984) proposed that the focal loss of neurons along the axis of the hippocampus could not support the early theories of vasomotor hypoxia or circulatory insufficiency (4). Furthermore, the lack of correlation between hippocampal cell density and seizure history suggested to him that most of the neuronal loss had occurred prior to onset of habitual seizures rather than a result of it.
Since the early post‐mortem studies by Sommer and Bratz, it has been known that HS may frequently be a bilateral disease process 8, 46. Sommer interestingly states, “since both organs [hippocampi] are usually affected. . . . unilateral localization may only be due to the differential advancement of disorganization of both”, implying that he considered it a bilateral, if sometimes asymmetrical disease, process. This possibility still persists in the contemporary literature (57), and indeed milder pathology in the remaining hippocampus post‐surgery could be one cause of failure to control seizures. Corsellis documented a variety of symmetrical and asymmetrical bilateral patterns in patients with TLE syndromes at post‐mortem (29). In the most recent PM study from Meencke and Veith (31) regarding a series of 650 cases, AHS was bilateral in nearly a half of cases with this pathology, although less frequent in TLE compared with other epilepsy syndromes. They argued that bilateral HS supports the concept of a “secondary epileptogenesis” with discharges from one hippocampus inducing or “kindling” changes in the contralateral hippocampus. This draws comparisons with the hippocampal neuronal loss that has been observed ipsilateral to a cortical lesional pathology causing epilepsy in studies published by Babb and Mathern (so‐called dual pathology) 4, 30 promoting the idea that there could be “primary” and “secondary” forms of HS (57).
Sommer, in his discussion, also entertained a “developmental defect” as the cause of AHS, although thought this improbable based on what he considered “a progressive decay . . . that develops intra vitam and probably continues until death.” Developmental or dysmaturational abnormalities of the hippocampus underlying a susceptibility to develop AHS have, however, recently gained some popularity, as reviewed by Blumcke (2002) (5). Cytoarchitectural aberrations, particularly dispersion of the granule cells of the dentate gyrus, are often prominent findings in HS. This observation, first published by Houser (1990) (25) and which was probably previously overlooked (Figures 2, 4c and 7b in (35)), appears unique to epilepsy and shown to correlate with the severity of hippocampal neuronal loss 20, 51as well as early onset of seizures, although its significance remains uncertain. Recent studies have addressed the possibility of a disturbance of ongoing neurogenesis in this region as a result of seizures, a local abnormality of reelin signaling and proliferation of glial cells forming a scaffold for cell migration as potentially contributing to this cytoarchitectural change 20, 52.
HS AND EARLY SEIZURES: MEYER'S HYPOTHESIS
Sommer considered the possibility of an initiating event that could “prime” the hippocampus for sclerosis but lacked supportive evidence: “I am unable to confirm the frequent preceding of an acute disease with symptoms . . . but I do not want to put too much stress on this”(46). Early post‐mortem studies highlighted the effect of early age of onset of seizures as a risk factor for the development of AHS (36), and Corsellis (1957) (11) noted—in a post‐mortem series of 200 cases—that when the onset of seizures occurred in infancy, the majority showed AHS. This led to the impression that early seizures were a risk factor for AHS. It was Meyer, however, who synthesized these observations. From his series published with Cavanagh (10) with more detailed clinical–pathological data, the striking finding was that in over 60% of their patients with AHS, there was an early history of status epilepticus, sometimes associated with a febrile illness. “Meyer's hypothesis” was widely debated at the Marseille Colloquium of 1955, which proposed that an initial event of a prolonged complex febrile convulsion, occurring between 0.5 and 5 years could result in AHS and eventually lead to habitual TLE. In essence seizures caused sclerosis and vice versa. Meyer considered that the theory of anoxia or birth injury as the cause of HS was an “over‐simplification,” but that events occurring in early infancy, childhood and early adolescence played a equal, if not greater, part in this process (35). This association between AHS and early febrile seizures has been confirmed in numerous subsequent clinical–pathological studies since, as reviewed by Meldrum (32). Recently, Mathern (30) readdressed Meyer's hypothesis in a large series of over 500 TLE patients operated on over a 40‐year period, expanding Meyer's concept of a brain insult to include any significant medical event prior to habitual seizure onset [an “initial precipitating injury” (IPI)]. He confirmed a strong association of an IPI, particularly a seizure‐IPI, and AHS compared with patients without sclerosis, with the majority occurring before 4 years of age.
THE LAST 20 YEARS: EXTRICATION OF THE CRITICAL CELLULAR EVENTS IN HS
A steady increase in the study of HS in the modern era has continued, with over 62% of all scientific papers on this topic since 1950 published since 2000. Many of the main research themes have steered towards elucidating the mechanisms underlying epileptogenesis in the hippocampus, which are still largely unresolved, particularly the question of how seizures continue to be maintained in a region of neuronal devastation (14). Indeed, the major challenges when examining the advanced stage of the disease in human tissues are to try to distinguish alterations that could represent pre‐existing structural abnormalities from adaptive reorganization as a result of seizures or neuronal loss.
Abnormal connectivity and reorganization of the mossy fiber pathway was first observed in animal models of epilepsy and was subsequently demonstrated in human HS specimens in a landmark paper by Sutula (50). This synaptic reorganization occurring in the epileptic hippocampus continues to be argued as highlighting a potentially functionally significant alteration through promoting synchronization of discharges. Demonstration of abnormal mossy fiber sprouting, either with Timms' method or dynorphin immunohistochemistry, is therefore of diagnostic value in current neuropathology practice (15) (Figure 4). To address possible imbalances of neuronal excitability vs. inhibition as to the cause of seizures, several studies have considered the vulnerabilities of specific excitatory and interneuronal populations in hippocampal subfields. For example, relative rates of depletion of hilar excitatory mossy cells, neuropeptide Y‐secreting neurons and GABAergic neuronal subsets with reorganization of neurotransmitter receptor systems have been extensively studied in different subfields mainly using immunohistochemistry methods (see (53) for overview). For example, one widely debated theory proposed by Sloviter based on experimental systems suggested that early loss of hilar mossy cells resulted in understimulation of surviving inhibitory basket cells, rendering the region hyperexcitable (45). Understanding the mechanisms of hippocampal cell injury and death in epilepsy, including activation of apoptotic pathways, has also been a further important focus of studies with view to the development of neuroprotection treatment strategies (23).
Figure 4.

Mossy fiber sprouting, as described by Sutula in 1989, can be seen in the molecular layer of the dentate gyrus in hippocampal sclerosis using (A) Timms method or (B) dynorphin immunohistochemistry. Dispersion of the granule cells of the dentate gyrus in hippocampal sclerosis is also a common finding and was first described by Houser in 1990 and is illustrated here with neuronal nuclear antigen staining (C). Abbreviations: GCL = granule cell layer; IML = inner molecular layer.
Neuropathological studies have also played an important component in correlative studies with modern MRI observations in the presurgical evaluation of HS. Pathological qualitative grading systems—for example, the system of Wyler (1992) that utilizes a five‐grade system from absent to severe neuronal loss (12), and others employing quantitative analysis of subfield neuronal densities (6)—have been adopted. Neuronal loss has been shown to precisely correlate with hippocampal T2 signal changes (54) and it is likely that neuropathological comparisons will continue to be of value with the acquisition of newer MRI sequences aiming to give more accurate anatomical information of hippocampal structural changes (19). Studies based on large surgical series have also identified pathological criteria of prognostic value, for example, atypical patterns of HS 6, 14 that are associated with poorer long‐term, seizure‐free outcomes. Indeed, although evidence supports that seizures arise in the sclerotic hippocampus, it remains unknown why a significant proportion of patients fail to become seizure‐free following surgical removal (47), which is a future challenge for both clinicians and neuropathologists. In addition, tissue studies have also contributed towards ongoing investigations of the mechanisms underlying pharmacoresistance in patients with TLE and HS, for example abnormal local expression of drug transporter proteins could play a role 2, 44.
Among the surgical TLE series reported in the last 50 years, there is consistently a small number of patients in whom no pathological abnormality is identified, including an absence of hippocampal neuronal loss 4, 15, 30, 34; this is sometimes referred to as “paradoxical or cryptogenic TLE.” The existence of these cases has been used to argue that AHS is neither an inevitable consequence of seizures nor essential to cause temporal lobe seizures. Meyer insightfully noted that, despite the lack of neuronal loss in such cases, there was an increase in glial cells in the pyramidal layer that “might not be without functional significance.” Modern studies are beginning to address the possibility that astrogliosis in the hippocampus could be more than just a reactive “scarring” with the emergence of a less neurocentric visualization of this regional pathology. There is evidence that astrocytic glial cells may have fundamental roles in modulating seizures through glutamate regulation, altering the blood brain barrier and secretion of cytokines 14, 27, 43. In addition, work by Aronica and others has highlighted the potential importance of activation of microglia and specific inflammatory pathways, which, as well as having roles in neuronal injury, could modulate neuronal function and play a key role in epileptogenesis 3, 38.
The wealth of available material arising from the expansion of epilepsy surgical programs during the recent decades has provided an exceptional tissue resource for biochemical, electrophysiological, molecular and genetic studies in HS, utilizing state‐of‐the‐art methodologies. As an example, large‐scale studies of gene expression patterns have demonstrated their value in the identification of sets of genes that could function in concert and influence epileptogenesis, for example those involved in cell growth and differentiation, neuronal signaling, synaptic transmission and plasticity, transcriptional regulation and inflammation have been highlighted 1, 14, 28. The disadvantages of focusing on surgical temporal lobectomy tissue, however, is that this generally restricts studies to highly selected patients groups fulfilling stringent preoperative investigative criteria (47), confirming unilateral disease at its most advanced stage. Although post‐mortem rates have fallen worldwide, this material, where available, should not be neglected as it continues to offer possibilities in pathological studies of HS. For example, more variable patterns and severity of HS are seen at autopsy, possibly of different etiologies, and more extensive examination of adjacent limbic and cortical regions, as well as the contralateral hippocampus, becomes possible. Just as Sommer 128 years ago was able to recognize for the first time the importance of HS through his extensive post‐mortem collection, similar tissue resources are likely to be valuable in addressing some of the unanswered questions regarding this pathology in the modern era.
ACKNOWLEDGMENTS
I would like to thank Michael Gaeble, with his help in the precise translations of historical German texts, Francesco Scaravilli, for all his support and advice John Cavanagh and the staff at the Rockefeller library at the Institute of Neurology.
REFERENCES
- 1. Aronica E, Gorter JA (2007) Gene expression profile in temporal lobe epilepsy. Neuroscientist 13:100–108. [DOI] [PubMed] [Google Scholar]
- 2. Aronica E, Gorter JA, Ramkema M, Redeker S, Ozbas‐Gerceker F, Van Vliet EA et al (2004) Expression and cellular distribution of multidrug resistance‐related proteins in the hippocampus of patients with mesial temporal lobe epilepsy. Epilepsia 45:441–451. [DOI] [PubMed] [Google Scholar]
- 3. Aronica E, Boer K, Van Vliet EA, Redeker S, Baayen JC, Spliet WG et al (2007) Complement activation in experimental and human temporal lobe epilepsy. Neurobiol Dis 26:497–511. [DOI] [PubMed] [Google Scholar]
- 4. Babb TL, Brown WJ, Pretorius J, Davenport C, Lieb JP, Crandall PH (1984) Temporal lobe volumetric cell densities in temporal lobe epilepsy. Epilepsia 25:729–740. [DOI] [PubMed] [Google Scholar]
- 5. Blumcke I, Thom M, Wiestler OD (2002) Ammon's horn sclerosis: a maldevelopmental disorder associated with temporal lobe epilepsy. Brain Pathol 12:199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blumcke I, Pauli E, Clusmann H, Schramm J, Becker A, Elger C et al (2007) A new clinico‐pathological classification system for mesial temporal sclerosis. Acta Neuropathol 113:235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouchet C, Cazauvieilh CA (1825) De l'épilepsie considerée dans ses rapports avec l'aliénation mentale. Recherche sur la nature et le siège de ces deux maladies. Arch Gen Med 510–542. [Google Scholar]
- 8. Bratz E (1899) Ammonshornbefunde bei epileptikern. Arch Psuchiatr Nervenkr 32:820–835. [Google Scholar]
- 9. Bruton CJ (1988) The Neuropathology of Temporal Lobe Epilepsy. Oxford University Press: New York. [Google Scholar]
- 10. Cavanagh JB, Meyer A (1956) Aetiological aspects of Ammon's horn sclerosis associated with temporal lobe epilepsy. Br Med J 2:1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corsellis JA (1957) The incidence of Ammon's horn sclerosis. Brain 80:193–208. [DOI] [PubMed] [Google Scholar]
- 12. Davies KG, Hermann BP, Dohan FC Jr, Bush KT, Foley, AJ , Wyler, AR (1996) Relationship of hippocampal sclerosis to duration and age of onset of epilepsy, and childhood febrile seizures in temporal lobectomy patients. Epilepsy Res 24:119–126. [DOI] [PubMed] [Google Scholar]
- 13. Dawodu S, Thom M (2005) Quantitative neuropathology of the entorhinal cortex region in patients with hippocampal sclerosis and temporal lobe epilepsy. Epilepsia 46:23–30. [DOI] [PubMed] [Google Scholar]
- 14. De Lanerolle NC, Lee TS (2005) New facets of the neuropathology and molecular profile of human temporal lobe epilepsy. Epilepsy Behav 7:190–203. [DOI] [PubMed] [Google Scholar]
- 15. De Lanerolle NC, Kim JH, Williamson A, Spencer SS, Zaveri HP, Eid T, Spencer DD (2003) A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia 44:677–687. [DOI] [PubMed] [Google Scholar]
- 16. Du F, Whetsell WO Jr, Blumenkopf B, Abou‐Khalil B, Lothman EW, Schwarcz, R (1993) Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Res 16:223–233. [DOI] [PubMed] [Google Scholar]
- 17. Earle KM, Baldwin M, Penfield W (1953) Incisural sclerosis and temporal lobe seizures produced by hippocampal herniation at birth. AMA Arch Neurol Psychiatry 69:27–42. [DOI] [PubMed] [Google Scholar]
- 18. Earle KM, Baldwin M, Penfield W (1953) Temporal lobe seizures; the anatomy and pathology of the probable cause. J Neuropathol Exp Neurol 12:98–99. [PubMed] [Google Scholar]
- 19. Eriksson SH, Thom M, Bartlett PA, Symms MR, McEvoy AW, Sisodiya SM, Duncan JS (2008) PROPELLER MRI visualizes detailed pathology of hippocampal sclerosis. Epilepsia 49:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fahrner A, Kann G, Flubacher A, Heinrich C, Freiman TM, Zentner J et al (2007) Granule cell dispersion is not accompanied by enhanced neurogenesis in temporal lobe epilepsy patients. Exp Neurol 203:320–332. [DOI] [PubMed] [Google Scholar]
- 21. Gastaut H, Naquet R, Vigouroux R (1953) A case of experimental amygdaloid epilepsy in cats. Electroencephalogr Clin Neurophysiol 5:291–294. [DOI] [PubMed] [Google Scholar]
- 22. Gibbs FA, Gibbs EL, Lennox WG (1937) Epilepsy : a paroxysmal cerebral dysrythmia. Brain 377–388. [DOI] [PubMed] [Google Scholar]
- 23. Henshall DC, Murphy BM (2008) Modulators of neuronal cell death in epilepsy. Curr Opin Pharmacol 8:75–81. [DOI] [PubMed] [Google Scholar]
- 24. Hildebrandt M, Amann K, Schroder R, Pieper T, Kolodziejczyk D, Holthausen H et al (2008) White matter angiopathy is common in pediatric patients with intractable focal epilepsies. Epilepsia 49:804–815. [DOI] [PubMed] [Google Scholar]
- 25. Houser CR (1990) Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res 535:195–204. [DOI] [PubMed] [Google Scholar]
- 26. Jackson H, Beevor CE (1889) Case of tumour of the right temporo‐sphenoidal lobe bearing on the localisation of the sense of smell and on the interpretation of a particular variety of epilepsy. Brain 346–357. [Google Scholar]
- 27. Lee TS, Mane S, Eid T, Zhao H, Lin A, Guan Z et al (2007) Gene expression in temporal lobe epilepsy is consistent with increased release of glutamate by astrocytes. Mol Med 13:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Majores M, Schoch S, Lie A, Becker AJ (2007) Molecular neuropathology of temporal lobe epilepsy: complementary approaches in animal models and human disease tissue. Epilepsia 48(Suppl. 2):4–12. [DOI] [PubMed] [Google Scholar]
- 29. Margerison JH, Corsellis JA (1966) Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain 89:499–530. [DOI] [PubMed] [Google Scholar]
- 30. Mathern GW, Adelson PD, Cahan LD, Leite JP (2002) Hippocampal neuron damage in human epilepsy: Meyer's hypothesis revisited. Prog Brain Res 135:237–251. [DOI] [PubMed] [Google Scholar]
- 31. Meencke HJ, Veith G, Lund S (1996) Bilateral hippocampal sclerosis and secondary epileptogenesis. Epilepsy Res Suppl 12:335–342. [PubMed] [Google Scholar]
- 32. Meldrum BS (1997) First Alfred Meyer Memorial Lecture. Epileptic brain damage: a consequence and a cause of seizures. Neuropathol Appl Neurobiol 23:185–201;discussion 201–182. [PubMed] [Google Scholar]
- 33. Meyer A (1958) Epilepsy. In: Greenfield's Neuropathology, 1st edn. Greenfield JG, Blackwood W, McMenemey WH, Meyer A, Norman RM (eds), pp. 550–567. London: Edward Arnold. [Google Scholar]
- 34. Meyer A, Beck E (1955) The hippocampal formation in temporal lobe epilepsy. Proc R Soc Med 48:457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meyer A, Falconer MA, Beck E (1954) Pathological findings in temporal lobe epilepsy. J Neurol Neurosurg Psychiatry 17: 276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfleger L (1880) Beobachtungen uber Schrumpfung und sclerose des Ammonshornes bei Epilepsie. Allg Z Psychiatr 36:359–365. [Google Scholar]
- 37. Pitkanen A, Tuunanen J, Kalviainen R, Partanen K, Salmenpera T (1998) Amygdala damage in experimental and human temporal lobe epilepsy. Epilepsy Res 32:233–253. [DOI] [PubMed] [Google Scholar]
- 38. Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A (2008) Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis 29:142–160. [DOI] [PubMed] [Google Scholar]
- 39. Rigau V, Morin M, Rousset MC, De Bock F, Lebrun A, Coubes P et al (2007) Angiogenesis is associated with blood‐brain barrier permeability in temporal lobe epilepsy. Brain 130:1942–1956. [DOI] [PubMed] [Google Scholar]
- 40. Sagar HJ, Oxbury JM (1987) Hippocampal neuron loss in temporal lobe epilepsy: correlation with early childhood convulsions. Ann Neurol 22:334–340. [DOI] [PubMed] [Google Scholar]
- 41. Sano K, Malamud N (1953) Clinical significance of sclerosis of the cornu ammonis. Arch Neurol Psychiatry 40–53. [DOI] [PubMed] [Google Scholar]
- 42. Scholz W (1951) Die Krampfschadigungen des Gehirns. Springer: Berlin. [Google Scholar]
- 43. Schwarcz R (2008) Early glial dysfunction in epilepsy. Epilepsia 49(Suppl. 2):1–2. [DOI] [PubMed] [Google Scholar]
- 44. Sisodiya SM, Martinian L, Scheffer GL, Van Der Valk P, Cross JH, Scheper RJ et al (2003) Major vault protein, a marker of drug resistance, is upregulated in refractory epilepsy. Epilepsia 44:1388–1396. [DOI] [PubMed] [Google Scholar]
- 45. Sloviter RS (1994) On the relationship between neuropathology and pathophysiology in the epileptic hippocampus of humans and experimental animals. Hippocampus 4:250–253. [DOI] [PubMed] [Google Scholar]
- 46. Sommer W (1880) Erkrankung des Ammonshornes als aetiologisches Moment der Epilepsie. Arch Psychiatr Nervenkr 361–375. [Google Scholar]
- 47. Spencer SS (2002) When should temporal‐lobe epilepsy be treated surgically? Lancet Neurol 1:375–382. [DOI] [PubMed] [Google Scholar]
- 48. Spielmeyer W (1927) Die pathogenese des epileptisches krampfes. Histopathologischer teil. Ztschr Neurol Psychiat 501–519. [Google Scholar]
- 49. Stauder KH (1935) Epilepsie und Schlafenlappen. Arch Psychiatr Nervenkr 104:501–520. [Google Scholar]
- 50. Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L (1989) Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol 26:321–330. [DOI] [PubMed] [Google Scholar]
- 51. Thom M, Sisodiya SM, Beckett A, Martinian L, Lin WR, Harkness W et al (2002) Cytoarchitectural abnormalities in hippocampal sclerosis. J Neuropathol Exp Neurol 61:510–519. [DOI] [PubMed] [Google Scholar]
- 52. Thom M, Martinian L, Williams G, Stoeber K, Sisodiya SM (2005) Cell proliferation and granule cell dispersion in human hippocampal sclerosis. J Neuropathol Exp Neurol 64:194–201. [DOI] [PubMed] [Google Scholar]
- 53. Thom M, Sisodiya SM, Najm I (2008) Epilepsy. In: Greenfield's Neuropathology, 8th edn. Love S, Lewis DN, Ellison DW (eds), pp. 833–887. London: Hodder‐Arnold. [Google Scholar]
- 54. Van Paesschen W, Revesz T, Duncan JS, King MD, Connelly A (1997) Quantitative neuropathology and quantitative magnetic resonance imaging of the hippocampus in temporal lobe epilepsy. Ann Neurol 42:756–766. [DOI] [PubMed] [Google Scholar]
- 55. Van Vliet EA, Da Costa Araujo S, Redeker S, Van Schaik R, Aronica E, Gorter JA (2007) Blood‐brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 130:521–534. [DOI] [PubMed] [Google Scholar]
- 56. Vogt C, Vogt O (1937) Sitz und wesen der krankheiten im lichte der topistischen hirnforschung und des variierens der tiere. J Psychol Neurol (Leipzig) 47:241–457. [Google Scholar]
- 57. Wieser HG, ILAE Commission R (2004) Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 45:695–714. [DOI] [PubMed] [Google Scholar]
- 58. Yilmazer‐Hanke DM, Wolf HK, Schramm J, Elger CE, Wiestler OD, Blumcke I (2000) Subregional pathology of the amygdala complex and entorhinal region in surgical specimens from patients with pharmacoresistant temporal lobe epilepsy. J Neuropathol Exp Neurol 59:907–920. [DOI] [PubMed] [Google Scholar]