

Abstract
Parkinson's disease (PD) has long been considered to be a sporadic entity, perhaps with an environmental etiology. However, recent genetic discoveries have challenged this view, as there are many families with diseases of Mendelian inheritance that clinically resemble PD. Here, we will review in detail the neuropathological data relating to familial cases of PD. We will discuss the complicated relationships between the genetically defined cases and the two key pathological events seen in PD, namely loss of dopaminergic neurons in the substantia nigra pars compacta and the formation of protein inclusions, Lewy bodies, in the neurons that survive to the end stage of the disease course. These observations will be synthesized into an overall scheme that emphasizes the two key aspects of the neuropathology as distinct events and suggest that each gene tells us something a little different about the neuropathology of PD.
Keywords: Parkinson's disease, parkinsonism, α-synuclein, LRRK2, parkin, tau, neuropathology
Introduction
From the neuropathological viewpoint, Parkinson's disease (PD) is a reasonably defined disease entity. Pathological diagnosis requires loss of melanized dopaminergic neurons in the substantia nigra pars compacta (SNpc) with an intact striatum, the area to which the SNpc normally projects. Other sets of neurons are also lost, but the loss of SNpc neurons is extremely dramatic and is the major pathological event linked to the movement disorder seen in PD clinically. However, cell death alone is not sufficient for pathological diagnosis. Most definitions also require the presence of Lewy bodies [1], which are intraneuronal deposits of lipids and proteins, principally a small acidic protein called α-synuclein. Lewy bodies themselves are not truly diagnostic for PD as they are found in other disorders. For example, Lewy bodies are seen in cognitive disorders such as Alzheimer's disease and, more germane for the discussion here, diffuse Lewy body disease (DLBD). Several previous reviews have discussed Lewy body diseases as part of a broader family of synucleinopathies, with the same protein deposited [2–4]. Therefore, we have a two-part, additive logic for the neuropathology of PD; Lewy bodies and nigral cell loss are both required (Figure 1). Although there are overlap syndromes, the definitional approach cleanly limits what PD is from what it isn’t.
Figure 1.
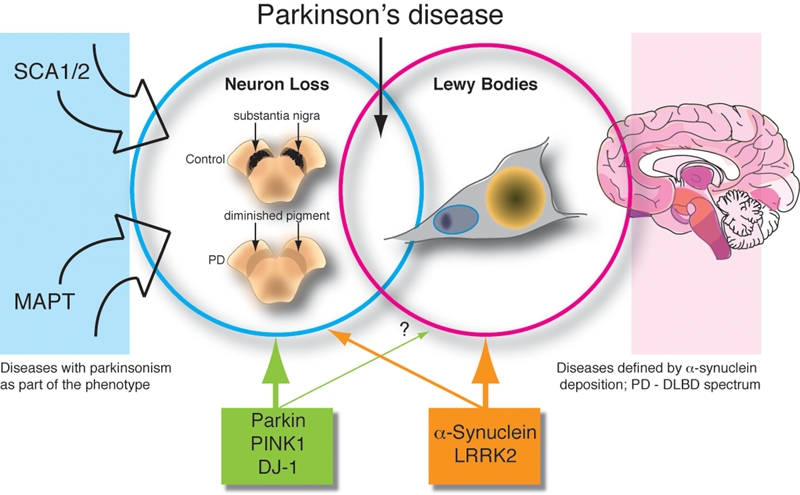
The relationships between cell death and Lewy body formation with respect to inherited Parkinson's disease. In this schematic, we emphasize that there are two aspects to the pathology of PD; cell loss (blue circle) epitomized by, but not limited to, loss of neurons in the substantia nigra pars compacta and Lewy body formation (magenta circle), which are perinuclear neuronal aggregates of α-synuclein and other proteins, represented here by the orange structure. On the left are other genetic disorders discussed in the text that can have parkinsonism (ie nigral cell loss) as part of their phenotype. On the right is a cartoon (adapted from figure 4 of [69]) suggesting the regional distribution of Lewy bodies in PD where darker colors indicate higher concentrations of α-synuclein pathology. This data suggests that PD is part of a spectrum of disorders that includes diffuse Lewy body disease (DLBD), for example. Finally, underneath the figure are the five PARK genes that are well-described genetically. We suggest here that the recessive genes parkin, PINK1 and DJ-1 (green box) are more informative for cell loss and the dominant genes α-synuclein and LRRK2 (orange box) tell us something about both cell loss and Lewy body formation.
In contrast, PD is very poorly defined from an etiological viewpoint. Thinking on the root cause of the disorder has shifted over time, with genetic and environmental hypotheses falling in and out of fashion and each having their own dedicated followers. For example, PD was commonly considered to be “simply” an environmentally caused disorder in the 1970s and 1980s, although in fact its familiality has been recognized since the time of the great French physician, Charcot. The controversy is ongoing, but some statements are generally accepted. Firstly, there are families throughout the world who have inherited forms of PD. These are rare and the mode of inheritance is mixed, with both recessive and dominant (often with incomplete penetrance) genes reported [5]. At a population level, the contribution of Mendelian forms was therefore probably missed for many years [6]. True single causes from the environment are probably rarer still, limited to a small number of cases of poisonings that have parkinsonism. This leaves the majority of PD cases with an unclear etiology, neither having a strong genetic or environmental cause. Weaker acting risk factors are therefore postulated to be important in lifetime risk of PD, by analogy to many other common diseases. A genetic-environmental “interaction” model is often invoked, although direct evidence for this is absent.
One of the difficulties in tying together neuropathology and etiology is that, by current definitions, PD has no known causes. For example, a strong family history is an exclusionary criterion in the London Brain Bank criteria [1]. The purpose of this is to exclude heredodegenerative conditions that can have parkinsonism as a symptom (see below) which we assume are etiologically distinct. Likewise, secondary parkinsonism due to exposure to drugs is excluded. This puts us in a logical conundrum: can we infer pathogenic pathways from the neuropathology of cases where we know the cause of a disease if we have excluded all cases with known causes? Specifically, if we have genetic diseases that are full or partial mimics of a generally sporadic disease like PD, can we reasonably assume that they share pathogenic pathways if the neuropathology is variable?
The purpose of this article is to discuss the neuropathology of sporadic PD/parkinsonism. To achieve this, we will first describe the different genetic diseases with a special emphasis on evaluation of which mutations are likely pathogenic and which are ambiguous. This is an important distinction for a common sporadic disease as misidentification of a genetic variant as pathogenic may lead to an association with ‘typical’ Lewy body PD that is spurious because the disease is common and the mutation is innocuous. We will then describe the neuropathological observations that have been made in cases with a definitive Mendelian inheritance. Finally, we will discuss how (or in fact whether) the genetic and neuropathological data can be tied together.
The Genetics of Parkinson's Disease
As stated above, there are several genes now known to cause PD, at least if defined clinically and with the exception to the rule that PD does not have a genetic etiology. One might expect that the protein products of these genes would map onto a single biochemical pathway as they appear to do in the case of Alzheimer's disease (AD). However, AD is explicitly defined by its pathology whereas the diagnosis of Parkinson's disease is less clear, and thus one cannot be sure whether all of the cases grouped as PD actually represent the outcome of a single pathogenic process or more than one. Most cases with clinically diagnosed PD have Lewy body (α-synuclein) pathology and a reasonable supposition is that Lewy body Parkinson's disease represents a single pathogenic process. However, this is a complex issue, even if one defines the disorder through pathology. With this background, here we have grouped the genes involved in the disorder into 3 groups: dominant mutant genes which cause Lewy body Parkinson's disease, recessive mutant genes which cause parkinsonism but whose relationship to Lewy body disease is unclear, and dominant mutant genes whose phenotypes mimic Parkinson's disease.
Dominant Genes Whose Phenotype Include Lewy Body Parkinson's Disease
This includes two genes: α-synuclein and LRRK2. Mutations in both genes cause Lewy body Parkinson's disease. In both cases, however, there are complications with the straightforward relationship.
SNCA/α-Synuclein
A key moment in our understanding of both the genetics and the neuropathology of PD was the discovery of the A53T mutation in the gene for α-synuclein (SNCA) linked to this disease [7]. Subsequently, A30P [8] and E46K [9] point mutations were described in families from Germany and Spain, respectively. Families where the SNCA gene is triplicated on one chromosome [10], leading to a doubling in the amount of protein [11], or duplicated [12], leading to a 50% increase in protein, have also been described. In most cases with SNCA mutations (either point mutations or gene multiplications), the disease starts as PD but can involve dementia later. However, in some cases, the disease starts with cognitive changes and the parkinsonism is later and can be quite mild. Although the number of cases with these mutations is quite small, the general picture is that duplication families have a more brainstem restricted damage and a movement disorder but in the triplication families there is a more pronounced dementing illness [13]. The point mutations have characteristics of both clinical syndromes, even within the same family.
The SNCA gene encodes a small lipid-binding protein, α-synuclein, that has unclear function but may be involved in synaptic dynamics [14]. Importantly, α-synuclein is the major protein building block of Lewy bodies. In all cases with SNCA mutations, Lewy body pathology is the distinctive pathology (see below). Furthermore, genetic variability at the α-synuclein locus contributes to the etiology of sporadic PD [e.g., 15, 16]. These findings collectively put α-synuclein at the center of the pathogenesis of PD and DLBD.
LRRK2/Dardarin
Mutations in the LRRK2 gene have been described in many families throughout the world [17–19]. All of the mutations known to date are point mutations and are spread throughout the protein, which is a large and complex protein kinase. Some of these are quite rare, several are shared by a few families and one mutation (G2019S) is relatively common [reviewed in 20]. In fact, the frequency of the G2019S mutation varies between 1% and 40% of PD cases depending on the type of survey done (specifically whether known familial cases are included or excluded) and the population studied. The highest known frequencies of G2019S are seen in people of Ashkenazi or North African descent [21]. If one takes a low estimate of frequency of about 1.5% of all PD cases in Caucasians, with 1 million people living with PD in the US alone, this predicts about 15,000 cases with a single point mutation that causes their disease. Such a figure is astounding for a ‘non-genetic’ disease and might raise a question as to whether G2019S is pathogenic. The answer is that the penetrance of the mutation, i.e. the proportion of mutation carriers that express the clinical signs of PD, is age-dependent and incomplete. Thus, there are reports of G2019S carriers that do not get PD even until late in life [22]. There are many more PD than controls with the mutation, and the overall penetrance of the G2019S allele is less than 50%, although this varies with age [reviewed in 23]. This means that if one had a theoretical family with a PD patient with the mutation where one parent had died before the age of 55 and one sibling was clinically unaffected, then the case might look ‘sporadic’ but would in fact have a genetic etiology. Therefore, G2019S cases should be referred to as ‘apparently sporadic’ PD.
The major difficulty with LRRK2 is that the pathology is very variable. Most cases that have been autopsied have Lewy body PD but an appreciable proportion has nigral cell loss or unusual protein inclusions. Therefore, although clinically similar to sporadic PD, LRRK2 mutation patients represent a range of pathological outcomes, which blurs the correlation between etiology and disease expression. This data will be discussed in more depth in the later parts of this review.
Genes for Recessive Parkinsonism
Parkin and PINK1
Parkin was the first identified gene for autosomal recessive juvenile parkinsonism [24], and is numerically the most important of this class. Because of the relatively high frequency, parkin is the only one of the recessive parkinsonism genes with autopsy information that allows for the relationship to Lewy body disease to be clarified, which will be discussed below. Although parkin is the frequent cause of early onset parkinsonism (onset between the ages of 20 and 50) and because such early onset cases account for less than 10% of all clinical PD, parkin makes a relatively small contribution to PD at the population level. There have been suggestions that parkin may make a more subtle contribution to PD in that heterozygous mutations may contribute to an increased lifetime risk for sporadic PD [25]. However, these arguments are complicated by the fact that PD is common and there are a number of variants of parkin that are rare, but not pathogenic. Therefore, there may be people with sporadic PD with a rare variant by chance alone.
The parkin protein is a ubiquitin-E3 ligase that has roles in control of protein degradation [14]. Many mutations have been reported, including truncations, exon rearrangements and point mutations. Most of the convincing mutations are recessive and associated with a loss of parkin function.
PINK1, which encodes a mitochondrial kinase, again has a number of loss of function mutations that cause a phenotype very similar to parkin [26]. Because no PINK1 cases have yet been autopsied, the relationship between mutation and pathology cannot be discerned and we will not discuss these cases further. However, recent data from model organisms where PINK1 has been knocked out suggest that there is a genetic relationship with parkin [reviewed in 27], and we might therefore assume that PINK1 and parkin cases have similar phenotypes.
Other Rare Genes for Recessive Parkinsonism
For the sake of completeness, there are two more genes associated with recessive early onset parkinsonism. DJ-1 is an oxidative stress response protein and mutations have been found in a very small number of cases with early onset parkinsonism [28] or, in one family with amyotrophy and dementia [29]. ATP13A2 is a lysosomal ATPase associated with Kufor-Rakeb syndrome, a complex phenotype that includes parkinsonism amongst other symptoms [30]. However, ATP13A2 mutations are also found in cases with early onset parkinsonism, perhaps justifying a PARK locus description [31]. However, neither DJ-1 nor ATP13A2 cases have yet been autopsied and we will not discuss them further in this article.
Mutant Genes Whose Phenotype Can Mimic Parkinson's Disease
Three genes fit this category: microtubule-associated protein tau (MAPT), spinocerebellar ataxia (SCA) 2 and SCA3.
MAPT
Mutations in the MAPT gene cause Frontal Temporal Dementia with Parkinsonism Linked to Chromosome 17 with Tau Pathology [32]. The MAPT gene encodes a major microtubule binding protein, Tau, that is hyperphosphorylated when deposited in neurons of several related diseases. Most cases have a dementia phenotype with mild parkinsonism, but some, especially the pallido-ponto-nigral degeneration (PPND) family, are clinically similar to sporadic PD [33].
SCA2
As the name implies, most cases with SCA2 mutations have a pure ataxia phenotype; however, many families of Asian background have a phenotype which expands to and includes a purely parkinsonian disorder [34]. This also occurs, but much more rarely, in Europeans [e.g., 35].
SCA3
It has long been recognized that SCA3/Machado-Joseph disease included parkinsonism in some kindreds. However, with the assessment of kindreds of sub-Saharan African descent it became clear that this was the predominant phenotype in that race and that the phenotype extends to include typical clinical Parkinson's disease [36].
Pathology of Genetic Forms of PD/Parkinsonism
SNCA/α-Synuclein
Since the description of PD cases with SNCA mutations in 1997, pathology reports for A53T or E46K point mutations have been published, as well as several cases with pathogenic triplications. The first autopsy report from the A53T kindred predated the identification of the mutation and therefore relied on classical staining techniques rather than immunohistochemical (IHC) analysis [37]. In the one case subjected to comprehensive autopsy, severe neuronal loss was observed in the SNpc, with abundant Lewy bodies and apparently extracellular/axonal Lewy body-like structures. Interestingly, given the late stage dementia described in this family, there was some cortical Lewy body pathology. Incomplete archive tissue from a second case was examined and revealed a similar picture with cell loss and Lewy bodies/Lewy body-like structures. According to the authors, these cases represented “pathologically typical Lewy body parkinsonism (ie PD as usually defined)”. Subsequent to the identification of the A53T mutation, two cases from an Australian family of Greek extraction were autopsied and their brains examined using both classical staining and IHC [38]. These cases had extensive de-pigmentation, neuronal loss and gliosis in the SNpc, with prominent Lewy body pathology. The observed Lewy bodies were labeled with antibodies directed against both α-synuclein and ubiquitin, with the α-synuclein antibody also staining extensive deposition in neurites within the SNpc and the cortex. The cortical pathology was characterized by vacuolar degeneration associated with Lewy neurites, suggesting that the pathological course in this region and in this case may have been particularly aggressive.
Following on from this work, brain sections from the original A53T family were re-examined in 2002 using antibodies directed against both α-synuclein and Tau [39]. Similar to the pathology reported in the Australian cases, there were extensive α-synuclein positive neuritic pathology and Lewy bodies. Additionally, widespread Tau deposition was reported in several regions of the brain and inclusions containing both α-synuclein and Tau were found.
Although there are no pathological reports from patients A30P mutation yet, an autopsy has been carried out on a patient carrying the E46K mutation [9]. The index case was diagnosed with DLBD, having presented with parkinsonism and dementia. Autopsy revealed, in addition to widespread cell loss and gliosis, extensive α-synuclein pathology in several regions of the brain, with a concentration of Lewy bodies and Lewy neurites in the SNpc, amygdala, parahippocampus and cingular cortex. There was no evidence of Tau deposition in this case.
As in the case of the A53T mutations, neuropathological reports from kindreds with SNCA gene multiplication predated the recognition of α-synuclein as the key player in the pathology of PD and so were carried out using classical staining methods [40, 41]. These revealed nerve cell loss in the SNpc, locus ceruleus and the hippocampus, noted as being unusually severe and accompanied by gliosis. There were also widespread cortical and sub-cortical Lewy bodies, with vacuolar degeneration also observed in the cortex. These initial reports were followed up in 2000 by a detailed autopsy examination of a 47yr old member of the triplication kindred by directly comparing brain sections to several sporadic cases of DLBD and a case of multiple system atrophy (MSA) [42]. As in the previous cases from the same family, there was widespread vacuolar degeneration of the cortex (described as ‘spongiosis’), closely associated with widespread α-synuclein staining manifesting as dystrophic cell processes and as Lewy bodies. The SNpc showed massive neuronal loss and an abundance of both Lewy neurites and Lewy bodies in the remaining cells. In addition to the neuronal pathology, there were also α-synuclein positive inclusions observed in glia, similar to those observed in MSA, and scattered thread-like Tau pathology. In summary, the pathology in this case is reminiscent of several diseases with α-synuclein deposition.
A final case report, from a Swedish-American kindred with a triplication, yielded a similar pathological picture, with widespread Lewy body pathology and neuronal loss [43]. No pathological data has yet been reported for patients with duplications in α-synuclein, although the later age of onset and symptoms similar to those of idiopathic PD suggest that it is likely that the neuropathology will be closer to that of sporadic PD.
LRRK2
Mutations in LRRK2, are much more common than SNCA and because of this a number of autopsy reports have been published for several of the LRRK2 mutants (summarized in Table 1). There are four published autopsy reports relating to the R1441C mutation [18, 44]. These are all from the same family, a multigenerational kindred from western Nebraska. Despite having a relatively uniform clinical phenotype of parkinsonism, the pathology in these cases is quite variable. All four patients had cell loss, de-pigmentation and gliosis in the SNpc, with two cases (III-14, who died in 1955 and whose archived brain tissue was examined, and III-20) having Lewy body pathology. In case III-14 this was localized to the brain stem, with case III-20 exhibiting a pattern closer to that of DLBD with some tau pathology in the subthalamic nucleus and a limited number of senile plaques. Case III-21, a younger sibling of III-20, had no α-synuclein pathology but did display tau-positive neurofibrillary tangles (NFTs). Although there was a clinical presentation of supranuclear gaze palsy in this patient in addition to parkinsonism, the authors described the pathology as not being consistent with that observed in progressive supranuclear palsy or frontal temporal dementia with Parkinsonism linked to chromosome 17, with a final diagnosis of parkinsonism due to tauopathy. The final case, IV-20, had no distinctive neuropathology associated other than the nigral degeneration.
Table 1.
Reported mutations in LRRK2 and their associated pathology
| Mutant | Domain | Case | Pathology | References |
|---|---|---|---|---|
| I1371V | ROC | 1x | Nigral cell loss, extensive Lewy body pathology in the substantia nigra and cortex, limited Tau pathology in the hippocampus and amygdala | Giordana et al |
| R1441C | ROC | III-14 | Nigral cell loss, brain stem Lewy body disease | Zimprich et al |
| R1441C | ROC | III-20 | Nigral cell loss, diffuse Lewy body disease | Zimprich et al |
| R1441C | ROC | III-21 | Nigral cell loss, Tau NFTs focused in the subthalamic nucleus | Zimprich et al |
| R1441C | ROC | IV-20 | Nigral cell loss, no specific pathology | Zimprich et al |
| G2019S | Kinase | 1x | Nigral cell loss, typical Lewy body pathology | Gilks et al |
| G2019S | Kinase | 1x | Nigral cell loss, diffuse senile plaques and NFTs | Gilks et al |
| G2019S | Kinase | 3x | Nigral cell loss, transitional Lewy body disease | Ross et al |
| G2019S | Kinase | Nigral cell loss, diffuse Lewy body disease | Ross et al | |
| G2019S | Kinase | 4x | Nigral cell loss, brainstem focused Lewy body disease | Ross et al |
| G2019S | Kinase | 1x | Alzheimer's pathology | Ross et al |
| G2019S | Kinase | Case A | Lewy bodies, Lewy neurites, α-synuclein spheroids plus α-synuclein cortical pathology. Hippocampal senile plaques and NFTs | Giasson et al |
| G2019S | Kinase | Case B | Nigral cell loss, cortical senile plaques and NFTs. Lewy bodies and neurites in the SNpc, brainstem and neocortex | Giasson et al |
| G2019S | Kinase | Case C | Nigral cell loss, limited NFTs and neuritic plaques | Giasson et al |
| G2019S | Kinase | 1x | Abundant NFTs, similar to PSP | Rajput et al |
| G2019S | Kinase | 1x | Mild nigral degeneration, no pathology | Gaig et al |
| G2019S | Kinase | 1x | Ubiquitin positive inclusions, similar to FTD-U | Dachsel et al |
| I2020T | Kinase | 4x | Nigral degeneration, no distinctive pathology | Hasegawa et al |
NFTs, neurofibrillary tangles; FTD-U, frontotemporal dementia, Tau-negative, ubiquitin-positive; PSP, progressive supranuclear palsy, SNpc, substantia nigra pars compacta.
The pathology of the last of these cases was similar to that described in the original Park 8 family, the Samigahara kindred from Japan, now recognized as being due to an I2020T mutation in LRRK2 [45]. Four patients from this family have come to autopsy, all of which were described as having pure nigral degeneration in the absence of Lewy bodies or NFTs although a detailed pathology report has not yet been published.
Because of the frequency with which the mutation occurs, several G2019S cases have been autopsied. Of the papers that originally described this mutation, Gilks et al reported two unrelated cases with Lewy body pathology typical of PD and one case with diffuse senile plaques and neurofibrillary tangle pathology, although the paper did not include a detailed description of the neuropathology [46]. In another study, eight archive cases of G2019S linked PD were found to consistently present with Lewy body pathology – with four cases localized predominantly to the brainstem and the others presenting with either transitional (3) or diffuse (1) Lewy body pathology [47]. Several of the cases also presented with limited AD pathology (senile plaques), with one case exhibiting full-blown AD pathology. An additional patient in this series who had a clinical presentation and pathology characteristic of AD with limited Lewy body pathology in the amygdala was also found to carry the G2019S mutation, although the authors noted that this may be a coincidence. Giasson and colleagues described three further cases, again unrelated, with a clinical presentation of PD harboring the G2019S mutation. These displayed a mixture of pathology: case A had Lewy bodies, Lewy neurites and α-synuclein spheroids in the SNpc, with limited α-synuclein pathology in the cortex, plus senile plaques and NFTs in the hippocampus and neocortex [48]. Case B had senile plaques and NFTs in the cortex and hippocampus with Lewy bodies and Lewy neurites in the SNpc, brainstem and neocortex – the case as a whole being described as consistent with the Lewy body variant of AD. Case C had no Lewy body/α-synuclein pathology and only limited NFTs and neuritic plaques. When examined with an antibody directed against LRRK2, an accumulation of this protein was observed in dystrophic neurites within the SNpc.
In addition to the multi-case reports, there have recently been several single-case pathological studies published describing atypical pathology in G2019S cases. Rajput and colleagues described a male with PD who died at the age of 85 with the G2019S mutation [49]. Upon autopsy, no Lewy bodies were observed; the predominant pathology being Tau reactive, similar to that seen in progressive supranuclear palsy (PSP) with abundant NFTs and some AD pathology. Notably, the patient did not present with supranuclear palsy and, also atypically, his PD did not respond to L-Dopa treatment. A second case, a woman from Barcelona who died at age 77 from PD, showed no observable histological pathology and only mild degeneration of the SNpc [50]. Finally, a patient diagnosed with familial frontotemporal dementia (FTD), and displaying pathology similar to that seen in FTD with ubiquitin positive inclusions, was found to be negative for mutations associated with FTD (Tau, PRGN, CHMP2B and VCP), but carried the G2019S mutation [51]. No α-synuclein, Tau or Aβ pathology was observed in this case.
Three patients carrying the Y1699C mutation in LRRK2 have been autopsied. One, from a family in Lincolnshire, England, had extensive cell loss, de-pigmentation and gliosis in the SNpc along with scattered Lewy bodies and Lewy neurites [52]. The locus ceruleus and cortex also exhibited Lewy body pathology, with the cortical inclusions consistent with brainstem predominant Lewy body disease. In addition, there were extensive NFTs in the hippocampus, sibiculum, entorhinal cortex, transentorhinal cortex and olfactory bulb, although no neuritic plaques were observed. Two cases from family A, a German/Canadian family, displayed severe cell loss and de-pigmentation in the SNpc but no Lewy body pathology [44, 53]. There were nuclear and cytoplasmic ubiquitin-positive inclusions in both cases, with the nuclear inclusions being described as reminiscent of Marinesco bodies [54]. One of the patients had NFTs, senile plaques and diffuse amyloid deposits in the hippocampus, amygdala, entorhinal cortex and neocortex consistent with a pathological diagnosis of Alzheimer's disease.
One further report has been published for a patient carrying an I1371V mutation in the ROC domain of LRRK2 [55]. As for almost all of the above LRRK2 case reports, there was extensive cell loss in the SNpc. This case had extensive Lewy body pathology along with Lewy neurites in the substantia nigra and limited Tau pathology in the hippocampus and amygdala. The authors described the case as being “indistinguishable from that of typical PD”, and that the distribution of cortical Lewy bodies was consistent with transitional Lewy body disease.
Recessive Parkinsonism
Of the recessive genes associated with Parkinson's disease, pathological reports are only available for parkin. Despite the large number of mutations in parkin, relatively few cases have come to autopsy. This is probably because patients with parkin mutations survive with a mild disease for 30–40 years. As with the autosomal dominant genetic forms of PD, the pathological picture presented by parkin cases is complex. The original family in which the gene was identified carried a deletion of parkin exon 4. Several cases with exon 4 deletions had autopsies, and all were from Japan. These cases displayed loss of cells in the SNpc, although the extent of cell loss varied with some patients showing only moderate loss of melanin-positive cells. Mori et al reported the case of a 62-year-old patient who died following surgery after having lived with parkinsonism for 38 years [56]. This case exhibited moderate neuronal loss in the SNpc and the locus ceruleus. No α-synuclein pathology of any kind was observed, although there were limited NFTs in the substantia nigra – and more abundant Tau pathology in the hippocampus with clusters of tangles in the frontal, temporal and parietal cortices. Tissue from this patient was later re-examined for accumulation of iron using Perls stain [57]. This revealed a build-up of iron in the substantia nigra in this case and another case with a deletion of exon 3, as compared to non-parkinsonian patients (there was no direct comparison to idiopathic PD). A second homozygous exon 4 deletion case report was published in 2000 by Hayashi and colleagues. The patient died at the age of 70 from pneumonia having suffered from PD since the age of 32. His brain exhibited substantial cell loss in the SNpc, with mild cell loss and gliosis in the substantia nigra pars reticulata. There were a few scattered NFTs, but no Lewy bodies were present.
Several cases, also from Japan, have been reported with homozygous deletions in exon 3 of parkin. One case, a patient who died after a 33-year disease course (age of onset 20), displayed pathology similar to that seen in the exon 4 deletion cases. This included loss of pigmented neurons in the SNpc, with the remaining cells decreased in size and with lowered pigmentation along with gliosis and astrocytosis. There were no Lewy bodies observed, although there were some ubiquitin positive α-synuclei-postitive dendritic inclusions [58–60]. A further exon 3 deletion homozygote came to autopsy in 2004 at the age of 70 following a disease course of 37 years [61]. The brain exhibited moderate to severe loss of neurons in the SNpc, with the more severe loss occurring in the ventrolateral and medial regions. Again, the remaining cells in the SNpc were decreased in size and pigmentation. In contrast to the exon 4 deletion cases, there was limited α-synuclein pathology, with Lewy body-like structures observed in the pedunculopontine nucleus along with ubiquitin-positive structures. No Tau pathology was present.
Pathological examination of a Tunisian patient with a homozygous two base deletion in exon 2 of parkin has been published [62]. The patient presented with resting tremor at the age of 34 and died at age 47 following fever, hemiplegia and coma. Upon autopsy, the brain showed marked cell loss and de-pigmentation in the SNpc, with the remaining cells showing decreased melanin content and pyknosis. There was spongiform degeneration and gliosis within the substantia nigra, but no Lewy body, Tau or ubiquitin-positive inclusions were observed.
The remaining published neuropathological reports for parkin describe either compound heterozygotes or heterozygote carriers. A K211N/del exon 3 case from the Netherlands who developed PD at the age of 18 and died of cardiac failure was examined by van de Warrenburg and colleagues [63]. The SNpc showed marked cell loss and de-pigmentation, with no evidence of α-synuclein pathology. There was also some cell loss in the cerebellum and in Clarks nucleus. Although there was no evidence of NFTs, there were Tau positive, thorn-shaped astrocytes in the caudate nucleus, putamen and subthalamic nucleus – with some also found in the substantia nigra. Farrer et al reported pathology for a compound heterozygote and a hemizygous case in 2001 [64]. The hemizygous case, who carried a deletion of parkin exon 3 and did not display a parkinsonism phenotype, died at an age of 93 and exhibited no pathological features in the brain. The compound heterozygote, who had an R275W mutation as well as the exon 3 deletion, died in a car crash at the age of 52 having suffered from parkinsonism from the age of 41. Upon examination, this case revealed cell loss in the SNpc and locus ceruleus, as well as Lewy body and pale body pathology. Lewy bodies were also found in the nucleus basalis of Meynert, the amygdala and the hippocampus. Overall the case was described as not differing from mild to moderate idiopathic PD. Pramstaller et al described a case in a large pedigree from the south Tyrol carrying both a deletion in exon 7 and a deletion of nucleotide T1072 [65]. The patient, who died of pulmonary embolism at age 78 and have presented with tremor at the age of 49, displayed cell loss in the SNpc and locus ceruleus, with a small number of Lewy bodies in the same region. Staining with an anti-α-synuclein antibody also revealed α- synuclein spheroids, Lewy neurites and extracellular α-synuclein deposits. α- synuclein pathology was also discovered in the dorsal vagus nucleus and the nucleus ambiguous.
A pathological report has also been published for a patient carrying a heterozygous C212Y mutation, who presented with PSP two years prior to death at age 84 from pneumonia [66]. There was severe cell loss combined with astrocytosis in the SNpc, with extensive Tau pathology including NFTs and Tau-positive astroglia. No Lewy bodies were observed.
Mutant Genes Whose Phenotype Can Mimic Parkinson's Disease
The neuropathology of SCA2 and SCA3 involves widespread changes that affect many areas of the nervous system [reviewed in 67]. Both afferent and efferent cerebellar pathways are pathologically affected, which presumably contributes to the progressive ataxia in most patients. As yet there are no pathological reports for patients with parkinsonism as the major or presenting feature. Decreased striatal dopamine function in some patients [35] suggests that they probably will have cell loss of the SNpc, but this is unproven.
Data on patients with mutations in the gene encoding tau is more plentiful [reviewed in 68]. Again, there is cell loss in the SNpc as part of the pathology, which is unsurprisingly also characterized by tau deposits of various types in various cells, depending on the mutation.
Synthesis
The various studies reviewed above leave us with a rather complex picture of the neuropathology of familial PD/parkinsonism. On the one hand, there seems to be a rather clear split between the recessive genes associated with young onset mild parkinsonism and the dominant genes associated with later onset Lewy body PD. On the other hand, there are cases that fall outside of this simple rubric where even the same mutation can be associated with either Lowy body positive PD or nigral loss without inclusion bodies, exemplified by the LRRK2 G2019S mutation. Therefore, we have to question what the relationships are between mutation, clinical phenotype and pathological outcome. Although there are no clear answers, largely due to the small number of genetic cases that have come to autopsy, we will suggest the following framework.
The key argument here is that the deposition of a-synuclein to form Lewy pathology is related to, but separable from, neuronal cell death or dysfunction (Figure 1). In this view, each gene tells us something a little different about these two aspects that, as we discussed in the beginning of this article, define sporadic PD.
α-synuclein is clearly a critical link between inherited and sporadic PD, being both a causative gene and the deposited protein in the major pathological hallmark of the disease. But, the disease is not a simple one and includes both a brainstem movement disorder (PD) and a cortical dementia (DLBD), suggesting that these disorders are etiologically related to one another. This concept is partially congruent with the Braak hypothesis [69] that Lewy body pathology can be staged, with a progression from one brain region to another. However, because the same mutation can result in either dementia or parkinsonism as the presenting syndrome the disease process is probably less stereotyped than might be expected. What causes the variation in presentation is unclear, and worth examining, although it is very likely that synuclein deposition (and attendent neuronal damage) may happen in many brain regions but at slightly different rates.
LRRK2 mutations are more difficult to understand, although their presence in such a large number of ‘sporadic’ PD cases makes this a critical problem. If all cases of LRRK2 mutations reported to date have parkinsonism as part of their clinical presentation, then LRRK2 dysfunction must tell us something about nigral cell death. As an aside, because LRRK2 is a kinase, it would seem likely that the problem caused by mutant LRRK2 involves altered cellular signaling, which may be amenable to therapeutic intervention. Because many cases with LRRK2 mutations do not have Lewy bodies, this implies that the deposition of α-synuclein is not a required part of the pathogenic process for LRRK2. However, this does not mean that α-synuclein makes no contribution to LRRK2 pathogenesis as a major hypothesis for α-synuclein mediated toxicity is that relatively soluble, partially aggregated species rather than mature Lewy bodies are the toxic molecules. In any case, as most LRRK2 mutations are associated with Lewy bodies, it seems likely that α-synuclein deposition can be a response to altered LRRK2 signaling and therefore, LRRK2 is informative about both nigral cell viability and some of the triggers for Lewy body formation.
The recessive genes, or at least parkin, probably tell us more about nigral cell death and less about Lewy body formation. The logic about LRRK2 may apply here; because Lewy bodies are not always seen does not prove that α-synuclein is not involved. However, the milder phenotypes of recessive parkinsonism, the lack of progression to dementia and the earlier onset suggest that they are different diseases from the synucleinopathies. The occasional cases with α-synuclein positive inclusions are difficult to interpret, especially given that the frequency of these lesions is often not listed. Perhaps they are chance events – Lewy bodies are seen at low frequency in the nigra of most neurologically normal individuals [70] – but the data available suggests that they are not the typical pathology seen with recessive parkinsonism.
Similar caveats hold for the tau pathology reported in a number of cases of inherited PD/parkinsonism. Although the presence of tau could be important, especially given that tau mutations cause parkinsonism as part of the disorder, it is often hard to determine whether the amount of deposition is significant enough to cause neuronal damage. Furthermore, some sporadic tauopathies are relatively common diseases themselves and might occur by chance in a person with a ‘PD’ mutation – at least one case with a G2019S LRRK2 mutation and AD pathology has been found and interpreted as a non-penetrant allele [47].
Summary
The many reports of familial PD cases discussed in this article highlight some of the complexities of the disease. First, PD has a complex phenotype that can be both a movement disorder caused by nigral cell loss and the deposition of α-synuclein in Lewy bodies. Second, although mutations in the gene for α-synuclein can be interpreted as having a simple relationship to PD, and highlighting the etiological relationship between PD and DLBD, the same statement is not easily made for other genetic variants. Rather, these genes tell us different things about PD, with parkin, PINK1 et al being more informative about survival of neurons in the SNpc and LRRK2 being somewhat informative about both neuronal maintenance and Lewy body deposition.
What is absolutely clear is that the number of cases that are well described pathologically needs to be expanded so that typical and atypical pathologies can be better separated. Given that genetic discoveries in this area are still rapidly progressing, it seems reasonable to suggest that there will be many more cases to add in the future.
Acknowledgments
This work was funded by the Intramural Research Program of the NIH, National Institute on Aging.
References
- 1.Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson's disease. Neurology. 2001;57:1497–1499. doi: 10.1212/wnl.57.8.1497. [DOI] [PubMed] [Google Scholar]
- 2.Dickson DW, Lin W, Liu WK, Yen SH. Multiple system atrophy: a sporadic synucleinopathy. Brain Pathol. 1999;9:721–732. doi: 10.1111/j.1750-3639.1999.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goedert M, Spillantini MG. Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol Psychiatry. 1998;3:462–465. doi: 10.1038/sj.mp.4000458. [DOI] [PubMed] [Google Scholar]
- 4.Trojanowski JQ, Lee VM. Transgenic models of tauopathies and synuclein-opathies. Brain Pathol. 1999;9:733–739. doi: 10.1111/j.1750-3639.1999.tb00554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan EK, Skipper LM. Pathogenic mutations in Parkinson disease. Hum Mutat. 2007;28:641–653. doi: 10.1002/humu.20507. [DOI] [PubMed] [Google Scholar]
- 6.Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson's disease and parkinsonism. Ann Neurol. 2006;60:389–398. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- 7.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 8.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 9.Zarranz J J, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 10.Singleton A B, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 11.Miller D W, Hague S M, Clarion J, Baptista M, Gwinn-Hardy K, Cookson M R, Singleton A B. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology. 2004;62:1835–1838. doi: 10.1212/01.wnl.0000127517.33208.f4. [DOI] [PubMed] [Google Scholar]
- 12.Chartier-Harlin M C, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 13.Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson EM, Schule B, Langston JW, Middleton FA, Ross OA, Hulihan M, Gasser T, Farrer MJ. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;68:916–922. doi: 10.1212/01.wnl.0000254458.17630.c5. [DOI] [PubMed] [Google Scholar]
- 14.Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- 15.Bonsch D, Lederer T, Reulbach U, Hothorn T, Kornhuber J, Bleich S. Joint analysis of the NACP-REP1 marker within the alpha synuclein gene concludes association with alcohol dependence. Hum Mol Genet. 2005;14:967–971. doi: 10.1093/hmg/ddi090. [DOI] [PubMed] [Google Scholar]
- 16.Chiba-Falek O, Nussbaum RL. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum Mol Genet. 2001;10:3101–3109. doi: 10.1093/hmg/10.26.3101. [DOI] [PubMed] [Google Scholar]
- 17.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil A M, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 18.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 19.Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, Tsuji S, Obata F. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol. 2005;57:918–921. doi: 10.1002/ana.20484. [DOI] [PubMed] [Google Scholar]
- 20.Cookson MR, Xiromerisiou G, Singleton A. How genetics research in Parkinson's disease is enhancing understanding of the common idiopathic forms of the disease. Curr Opin Neurol. 2005;18:706–711. doi: 10.1097/01.wco.0000186841.43505.e6. [DOI] [PubMed] [Google Scholar]
- 21.Lesage S, Ibanez P, Lohmann E, Pollak P, Tison F, Tazir M, Leutenegger AL, Guimaraes J, Bonnet AM, Agid Y, Durr A, Brice A. G2019S LRRK2 mutation in French and North African families with Parkinson's disease. Ann Neurol. 2005;58:784–787. doi: 10.1002/ana.20636. [DOI] [PubMed] [Google Scholar]
- 22.Kay D M, Kramer P, Higgins D, Zabetian CP, Payami H. Escaping Parkinson's disease: a neurologically healthy octogenarian with the LRRK2 G2019S mutation. Mov Disord. 2005;20:1077–1078. doi: 10.1002/mds.20618. [DOI] [PubMed] [Google Scholar]
- 23.Bonifati V. LRRK2 Low-penetrance Mutations (Gly2019Ser) and Risk Alleles (Gly2385Arg)-Linking Familial and Sporadic Parkinson's Disease. Neurochem Res. 2007 doi: 10.1007/s11064-007-9324-y. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 25.Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6:652–662. doi: 10.1016/S1474-4422(07)70174-6. [DOI] [PubMed] [Google Scholar]
- 26.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 27.Dodson MW, Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol. 2007;17:331–337. doi: 10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 29.Annesi G, Savettieri G, Pugliese P, D'Amelio M, Tarantino P, Ragonese P, La Bella V, Piccoli T, Civitelli D, Annesi F, Fierro B, Piccoli F, Arabia G, Caracciolo M, Ciro Candiano IC, Quattrone A. DJ-1 mutations and parkinsonism-dementia-amyotrophic lateral sclerosis complex. Ann Neurol. 2005;58:803–807. doi: 10.1002/ana.20666. [DOI] [PubMed] [Google Scholar]
- 30.Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 31.Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, Fabbrini G, Marconi R, Fincati E, Abbruzzese G, Marini P, Squitieri F, Horstink MW, Montagna P, Libera AD, Stocchi F, Goldwurm S, Ferreira JJ, Meco G, Martignoni E, Lopiano L, Jardim LB, Oostra BA, Barbosa ER, Bonifati V. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68:1557–1562. doi: 10.1212/01.wnl.0000260963.08711.08. [DOI] [PubMed] [Google Scholar]
- 32.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen R C, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon J M, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra B A, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 33.Reed LA, Schmidt ML, Wszolek ZK, Balin BJ, Soontornniyomkij V, Lee VM, Trojanowski JQ, Schelper RL. The neuropathology of a chromosome 17-linked autosomal dominant parkinsonism and dementia (“pallido-ponto-nigral degeneration”) J Neuropathol Exp Neurol. 1998;57:588–601. doi: 10.1097/00005072-199806000-00006. [DOI] [PubMed] [Google Scholar]
- 34.Gwinn-Hardy K, Chen JY, Liu HC, Liu TY, Boss M, Seltzer W, Adam A, Singleton A, Koroshetz W, Waters C, Hardy J, Farrer M. Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology. 2000;55:800–805. doi: 10.1212/wnl.55.6.800. [DOI] [PubMed] [Google Scholar]
- 35.Furtado S, Farrer M, Tsuboi Y, Klimek ML, de la Fuente-Fernandez R, Hussey J, Lockhart P, Calne DB, Suchowersky O, Stoessl AJ, Wszolek ZK. SCA-2 presenting as parkinsonism in an Alberta family: clinical, genetic, and PET findings. Neurology. 2002;59:1625–1627. doi: 10.1212/01.wnl.0000035625.19871.dc. [DOI] [PubMed] [Google Scholar]
- 36.Gwinn-Hardy K, Singleton A, O'Suilleabhain P, Boss M, Nicholl D, Adam A, Hussey J, Critchley P, Hardy J, Farrer M. Spinocerebellar ataxia type 3 phenotypically resembling Parkinson disease in a black family. Arch Neurol. 2001;58:296–299. doi: 10.1001/archneur.58.2.296. [DOI] [PubMed] [Google Scholar]
- 37.Golbe LI, Di Iorio G, Bonavita V, Miller DC, Duvoisin RC. A large kindred with autosomal dominant Parkinson's disease. Ann Neurol. 1990;27:276–282. doi: 10.1002/ana.410270309. [DOI] [PubMed] [Google Scholar]
- 38.Spira PJ, Sharpe DM, Halliday G, Cavanagh J, Nicholson GA. Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr alpha-synuclein mutation. Ann Neurol. 2001;49:313–319. [PubMed] [Google Scholar]
- 39.Duda JE, Giasson BI, Mabon ME, Miller DC, Golbe LI, Lee VM, Trojanowski JQ. Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol (Berl) 2002;104:7–11. doi: 10.1007/s00401-002-0563-3. [DOI] [PubMed] [Google Scholar]
- 40.Waters CH, Miller CA. Autosomal dominant Lewy body parkinsonism in a four-generation family. Ann Neurol. 1994;35:59–64. doi: 10.1002/ana.410350110. [DOI] [PubMed] [Google Scholar]
- 41.Muenter MD, Forno LS, Hornykiewicz O, Kish SJ, Maraganore DM, Caselli RJ, Okazaki H, Howard FM, Jr, Snow BJ, Calne DB. Hereditary form of parkinsonism–dementia. Ann Neurol. 1998;43:768–781. doi: 10.1002/ana.410430612. [DOI] [PubMed] [Google Scholar]
- 42.Gwinn-Hardy K, Mehta ND, Farrer M, Maraganore D, Muenter M, Yen SH, Hardy J, Dickson DW. Distinctive neuropathology revealed by alpha-synuclein antibodies in hereditary parkinsonism and dementia linked to chromosome 4p. Acta Neuropathol (Berl) 2000;99:663–672. doi: 10.1007/s004010051177. [DOI] [PubMed] [Google Scholar]
- 43.Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 44.Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, Strongosky AJ, Zimprich A, Muller-Myhsok B, Farrer MJ, Gasser T, Calne DB, Dickson DW. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology. 2004;62:1619–1622. doi: 10.1212/01.wnl.0000125015.06989.db. [DOI] [PubMed] [Google Scholar]
- 45.Hasegawa K, Kowa H. Autosomal dominant familial Parkinson disease: older onset of age, and good response to levodopa therapy. Eur Neurol. 1997;38(Suppl 1):39–43. doi: 10.1159/000113460. [DOI] [PubMed] [Google Scholar]
- 46.Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, Lynch J, Healy DG, Holton JL, Revesz T, Wood NW. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet. 2005;365:415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- 47.Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC, Litvan I, Gordon MF, Wszolek ZK, Farrer MJ, Dickson DW. Lrrk2 and Lewy body disease. Ann Neurol. 2006;59:388–393. doi: 10.1002/ana.20731. [DOI] [PubMed] [Google Scholar]
- 48.Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM. Biochemical and pathological characterization of Lrrk2. Ann Neurol. 2006;59:315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- 49.Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- 50.Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E. G2019S LRRK2 mutation causing Parkinson's disease without Lewy bodies. J Neurol Neurosurg Psychiatry. 2007;78:626–628. doi: 10.1136/jnnp.2006.107904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dachsel JC, Ross OA, Mata IF, Kachergus J, Toft M, Cannon A, Baker M, Adamson J, Hutton M, Dickson DW, Farrer MJ. Lrrk2 G2019S substitution in frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions. Acta Neuropathol (Berl) 2007;113:601–606. doi: 10.1007/s00401-006-0178-1. [DOI] [PubMed] [Google Scholar]
- 52.Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, Gibbons V, Gandhi S, Vaughan J, Eunson LH, Katzenschlager R, Gayton J, Lennox G, Revesz T, Nicholl D, Bhatia KP, Quinn N, Brooks D, Lees AJ, Davis MB, Piccini P, Singleton AB, Wood NW. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128:2786–2796. doi: 10.1093/brain/awh667. [DOI] [PubMed] [Google Scholar]
- 53.Wszolek Z, Vieregge P, Uitti R J, Gasser T, Yasuhara O, Mcgeer P, Berry K, Calne D, Vingerhoets F, Klein C, Pfeiffer RF. German-Canadian Family (Family A) with Parkinsonism, Amyotrophy, and Dementia – Longitudinal Observations. Parkinsonism Relat Disord. 1997;3:125–139. doi: 10.1016/s1353-8020(97)00013-8. [DOI] [PubMed] [Google Scholar]
- 54.Beach TG, Walker DG, Sue LI, Newell A, Adler CC, Joyce JN. Substantia nigra Marinesco bodies are associated with decreased striatal expression of dopaminergic markers. J Neuropathol Exp Neurol. 2004;63:329–337. doi: 10.1093/jnen/63.4.329. [DOI] [PubMed] [Google Scholar]
- 55.Giordana MT, D'Agostino C, Albani G, Mauro A, Di Fonzo A, Antonini A, Bonifati V. Neuropathology of Parkinson's disease associated with the LRRK2 Ile1371Val mutation. Mov Disord. 2007;22:275–278. doi: 10.1002/mds.21281. [DOI] [PubMed] [Google Scholar]
- 56.Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K, Mizuno Y. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology. 1998;51:890–892. doi: 10.1212/wnl.51.3.890. [DOI] [PubMed] [Google Scholar]
- 57.Takanashi M, Mochizuki H, Yokomizo K, Hattori N, Mori H, Yamamura Y, Mizuno Y. Iron accumulation in the substantia nigra of autosomal recessive juvenile parkinsonism (ARJP) Parkinsonism Relat Disord. 2001;7:311–314. doi: 10.1016/s1353-8020(00)00050-x. [DOI] [PubMed] [Google Scholar]
- 58.Yamamura Y, Arihiro K, Kohriyama T, Nakamura S. [Early-onset parkinsonism with diurnal fluctuation–clinical and pathological studies] Rinsho Shinkeigaku. 1993;33:491–496. [PubMed] [Google Scholar]
- 59.Yamamura Y, Hattori N, Matsumine H, Kuzuhara S, Mizuno Y. Autosomal recessive early-onset parkinsonism with diurnal fluctuation: clinicopathologic characteristics and molecular genetic identification. Brain Dev. 2000;22(Suppl 1):S87–S91. doi: 10.1016/s0387-7604(00)00130-3. [DOI] [PubMed] [Google Scholar]
- 60.Yamamura Y, Kuzuhara S, kiyotarou K, yanagi T, Uchida M, Matsumine H, Mizuno Y. Clinical, pathologic and genetic studies on autosomal recessive early-onset parkinsonism with diurnal fluctuation. Parkinsonism Relat Disord. 1998;4:65–72. doi: 10.1016/s1353-8020(98)00015-7. [DOI] [PubMed] [Google Scholar]
- 61.Sasaki S, Shirata A, Yamane K, Iwata M. Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology. 2004;63:678–682. doi: 10.1212/01.wnl.0000134657.25904.0b. [DOI] [PubMed] [Google Scholar]
- 62.Gouider-Khouja N, Larnaout A, Amouri R, Sfar S, Belal S, Ben Hamida C, Ben Hamida M, Hattori N, Mizuno Y, Hentati F. Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Parkinsonism Relat Disord. 2003;9:247–251. doi: 10.1016/s1353-8020(03)00016-6. [DOI] [PubMed] [Google Scholar]
- 63.van de Warrenburg BP, Lammens M, Lucking CB, Denefle P, Wesseling P, Booij J, Praamstra P, Quinn N, Brice A, Horstink MW. Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology. 2001;56:555–557. doi: 10.1212/wnl.56.4.555. [DOI] [PubMed] [Google Scholar]
- 64.Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K, Petrucelli L, Hussey J, Singleton A, Tanner C, Hardy J, Langston JW. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001;50:293–300. doi: 10.1002/ana.1132. [DOI] [PubMed] [Google Scholar]
- 65.Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I, Hedrich K, Adel S, Gonzales-McNeal M, Hilker R, Kramer PL, Klein C. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. 2005;58:411–422. doi: 10.1002/ana.20587. [DOI] [PubMed] [Google Scholar]
- 66.Sanchez MP, Gonzalo I, Avila J, De Yebenes JG. Progressive supranuclear palsy and tau hyperphosphorylation in a patient with a C212Y parkin mutation. J Alzheimers Dis. 2002;4:399–404. doi: 10.3233/jad-2002-4506. [DOI] [PubMed] [Google Scholar]
- 67.Rub U, Seidel K, Ozerden I, Gierga K, Brunt ER, Schols L, de Vos RA, den Dunnen W, Schultz C, Auburger G, Deller T. Consistent affection of the central somatosensory system in spinocerebellar ataxia type 2 and type 3 and its significance for clinical symptoms and rehabilitative therapy. Brain Res Rev. 2007;53:235–249. doi: 10.1016/j.brainresrev.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 68.Tsuboi Y. Neuropathology of familial tauopathy. Neuropathology. 2006;26:471–474. doi: 10.1111/j.1440-1789.2006.00702.x. [DOI] [PubMed] [Google Scholar]
- 69.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 70.Klos KJ, Ahlskog JE, Josephs KA, Apaydin H, Parisi JE, Boeve BF, DeLucia MW, Dickson DW. Alpha-synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology. 2006;66:1100–1102. doi: 10.1212/01.wnl.0000204179.88955.fa. [DOI] [PubMed] [Google Scholar]