

Abstract
We investigated the dynamics of autolytic damage of the cortical neurons in adult brains for 24 hours at room temperature (+20°C) after cardiac arrest. The progressive histological and ultrastructural changes were documented using routine and immunohistochemical staining as well as electron microscopy. Our results demonstrated that there were no autolytic damages in the ultrastructure of cerebral neurons in the first 6 hours after warm cardiac arrest, in agreement with previous studies in other mammals. Interestingly, the activation of caspase-3 was observed in a significant number of neurons of the cerebellum and neocortex 9 hours following cardiac arrest. No significant changes related to autolysis were observed using amnio-cupric acid and Nissl (thionine) staining.
Keywords: Brain death, autolysis, cerebral hypoxia, cerebral anoxia, apoptosis, caspase-3
Introduction
Death is cessation of all life (metabolic) processes. Somatic death is characterized by the discontinuance of cardiac activity and respiration, which eventually leads to death of all body cells due to oxygen deprivation. Brain death is now the legal condition in many countries, including the United States, for declaration of death [1].
There were only a few research publications about the ultrastructural changes of central nerve system neurons during prolonged cerebral hypoxia [2, 3, 4, 5]. Unfortunately, the dynamics of the autolytic damages of the CNS neurons was not investigated in these published studies. The purpose of our study was to investigate the progressive changes of cortical neurons during prolonged global cerebral anoxia via cardiac arrest at room temperature. The parameters evaluated included morphologic (light microscopic and ultrastructural) and biochemical alterations. Although some of these parameters have been previously investigated in several animal model systems, they were either performed on neonatal brains [6], under hypothermic conditions [7, 8] or involving application of reperfusion prior to dissection [9]. Our study appears to be the first in investigating multiple parameters in adult rodent brains under normal room temperature.
Material and Methods
Animals
The animals (18 Wistar rats, male, 94–112 days, 375–399g) were purchased from Harlan Inc. (Indianapolis, IN). The animals were euthanized with Halothane (Sigma-Aldrich; Cat. #B4388) and their cadavers were then kept at the room temperature (RT) (+20°C). The animals’ brains were dissected1, 3, 6, 9, 12 and 24 hours at room temperature following cardiac arrest. The research protocol was approved by the Alcor Institutional Animal Care and Use Committee.
Electron Microscopy
The brain cortex specimens (3×3×3mm) were cut from the frontal lobe of the brain and fixed in 2.5% glutaraldehyde in 0.1M Sorensen's sodium buffer (Electron Microscopy Sciences; Cat. #15980). Samples were post-fixed with 1% osmium tetroxide in the same buffer for 1 hr at room temperature, and then washed 3 times with the same buffer and 3 times with distilled water. Samples were stained en bloc with 0.5% uranyl acetate for 2 hrs at room temperature, then washed 3 times with distilled water and gradually dehydrated with ethanol before being transferred to acetone. Spurr's epoxy resin was used for infiltration and blocks were polymerized for 48 hours at 60°C. Thin-sections were cut on a Leica Ultracut R microtome and post-stained with uranyl acetate and lead citrate. Samples were observed and recorded at 80kV accelerating voltage on a Philips CM-12 Scanning Transmission Electron Microscope (the Netherlands) at The Electron Microscopy and W.M. Keck Bioimaging Laboratory in Arizona State University.
Light Microscopy
The animals’ brains were fixed in 10% phosphate buffered formalin (Fisher; Cat. #SF100–4). Upon receipt, the brains were treated with 20% glycerol and 2% dimethylsulfoxide to prevent freeze-artifacts and embedded in a gelatin matrix using MultiBrainTM Technology (NeuroScience Associates, Knoxville, TN). After curing, the block was rapidly frozen by immersion in isopentane pre-chilled to –70°C with crushed dry ice and mounted on a freezing stage of an AO 860 sliding microtome. The MultiBrainTM block was sectioned coronally at 35µm. The brain specimens were cut in their entirety and were sequentially submitted into a 4×5 array of containers which were filled with either 10% commercial, phosphate buffered formaldehyde or Antigen Preserve solution (50% PBS pH 7.0, 50% ethylene glycol, 1% polyvinyl pyrrolidone) for sections to be used subsequently for H&E, special and immunohistochemical stains. Hematoxylin and eosin (H&E), amino cupric silver, neutral red, thionine (Nissl) and immunostain for caspase-3 were performed on about 176 individual sections at 1920 µm intervals [10]. The brain slides were read and recorded at the W.M. Kerk Bioimaging Laboratory, School of Life Sciences, Arizona State University.
Caspase-3 Immunohistochemistry
For caspase-3 immunochemistry, the sections were stained free-floating. After a hydrogen peroxide treatment and blocking serum, the sections were immunostained with a 1:600 dilution of primary anti-caspase-3 polyclonal antibody (Pharmingen, Catalog #557035), a secondary antibody, and an avidin-biotin-HRP complex (Vectastain ABC Elite Kit, Catalog #PK-6101). Incubation times were 1 hour for the blocking antibody, 24 hours for the primary antibody, 1 hour for the secondary antibody, and 2 hours for the avidin-biotin-HRP complex. All of the incubations were done at room temperature. Sections were treated with hydrogen peroxide-diaminobenzidine tetra-hydrachloride to visualize the antibody binding sites and mounted on gelatinized (subbed) glass slides.
Results
A deeply placed esophageal temperature probe measured the temperature of the animals. The graph showed temperature of the animals’ carcasses at different time points after cardiac arrest (Figure 1).
Figure 1.
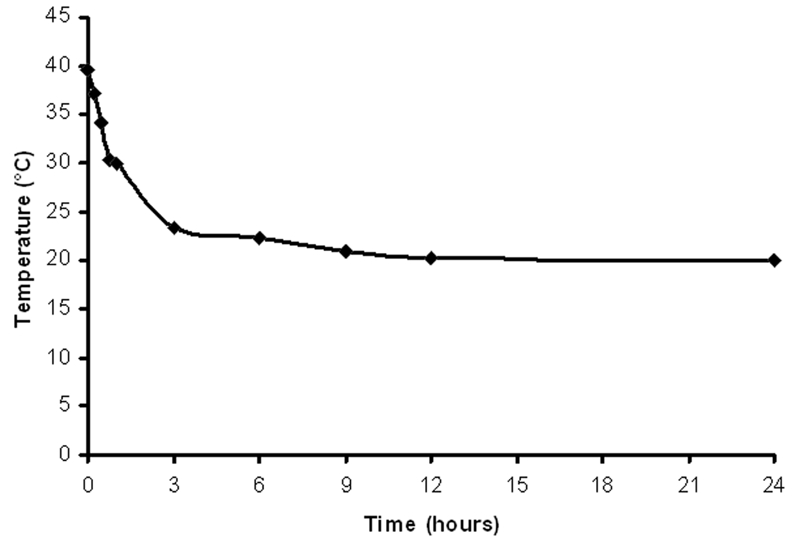
Postmortem temperature changes of the animals’ carcasses.
Electron Microscopy
The electron microscopy data show no obvious autolytic changes of the cortical neurons in the first 6 hours following cardiac arrest at room temperature (Figure 2A). Mitochondrial swelling, which may be the result of membrane permeability, was detected 3 hours after global normothermic cerebral anoxia. (Figure 2B). Vacuolization of the rough endoplasmic reticulum (RER) and swelling of the lysosomes were only observed 6 hours after cardiac arrest (Figure 2C). The ultrastructure of the mitochondrial crista was intact for 6 hours following warm cerebral anoxia. There were no signs of injury in the form of condensation, increased matrix density, and deposits of electron-dense material in mitochondria. In some neurons, the perinuclear mitochondria demonstrated evidence of mild edema with matrix and intracristal swelling. The neurons exhibited evidence of chromatin clumping at 6 hours (Figure 2C). The first feature of autolysis, disappearance of ribosomes, was only observed at 9 hours after warm cardiac arrest, and occurred in about 55% of neurons (Figure 2D). The complete chromatolysis was observed at 24 hours following warm cerebral anoxia and occurred in about 72% of the rat's cortical neurons (Figure 2F).
Figure 2.
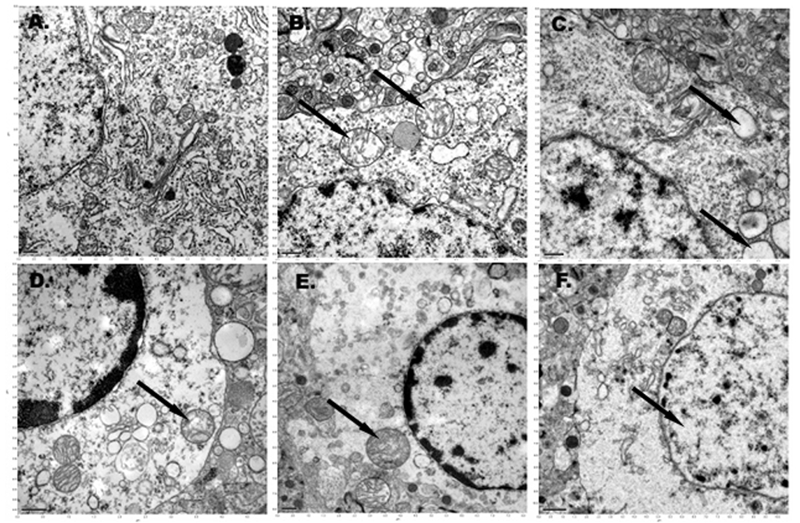
A. Rat cortical neuron at1 hour after warm cardiac arrest. Intact cellular ultra-structures (scale bar — 0.5µm). B. Rat cortical neuron at 3 hours after warm cardiac arrest. The mitochondria are intact in general but some signs of its swelling just appeared (black arrows) (scale bar —0.5µm). C. Rat cortical neuron at 6 hours after warm cardiac arrest with vacuolization of RER (black arrows) (scale bar — 0.5µm). D. Rat cortical neuron at 9 hours after warm cardiac arrest. Mitochondria and nuclear membrane of the neuron are intact. Vacuolization of RER., disappearance of ribosomes, initial signs of autolysis and welling of the lysosome (black arrow) are seen (scale bar — 1.0µm). E. Rat cortical neuron at 12 hours after warm cardiac arrest. Autolysis, swelling of the lysosome (black arrow) are present (scale bar — 0.5µm). F. Rat cortical neuron at 24 hours after warm cardiac arrest. Cell autolysis, chromatolysis (black arrow) are seen (scale bar — 1.0µm).
Light Microscopy
Routine staining with H&E revealed no appreciable histologic changes of the cortical, cerebellar, and hippocampal neurons within the study period (Figures 3A and 3B).
Figure 3.
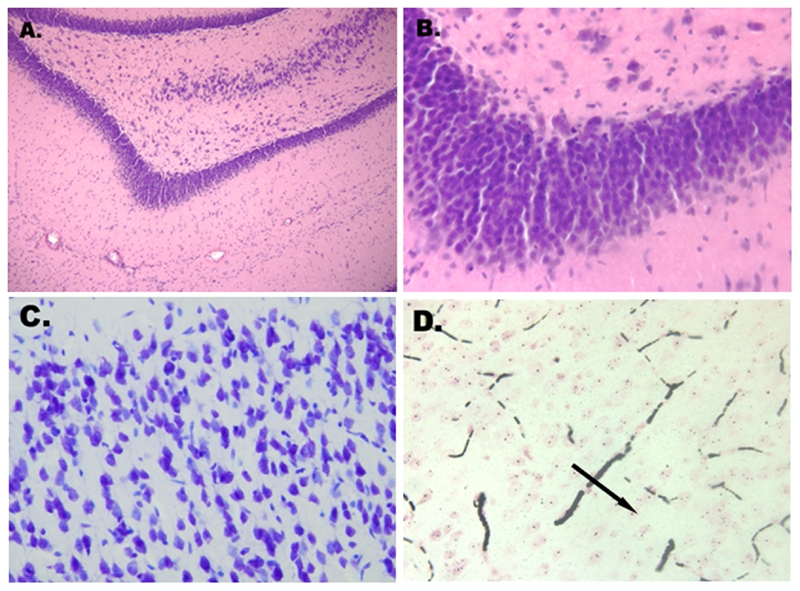
A. and B. Absence of appreciable histological changes of the hippocampal neurons at 12 hours after warm cardiac arrest (H&E, ×100; ×400). C. Absence of the picnotic neurons at 9 hours after warm cardiac arrest (Nissl, ×400). D. Rouleaux formation (black arrow) of erythrocytes (blood sludge) in the cerebral cortex microcirculation at 1 hour after warm cardiac arrest (amino cupric silver method, ×400).
Thionine (Nissl) staining demonstrated no evidence of nuclear breakdown, with retained nuclear integrity, and intact Nissl substance during the entire study interval. Nissl staining only showed tigrolysis in Purkinje cells and pyramidal cells in the hippocampus 12 hours after warm cardiac arrest. No hyper-chromatosis or pyknotic neurons were seen at 9 hours following cerebral anoxia at room temperature (Figure 3C).
Amino cupric silver staining highlighted small, mature neurons and oligodendrocytes. No significant histopathologic changes were observed in the neurons. Rouleaux formation of peripheral blood erythrocytes (blood sludge) was seen in the microcirculation of the cerebral cortex 1 hour following cardiac arrest (Figure 3D).
Immunohistochemical staining for caspase-3 demonstrated a progressive increase of positively-stained cells, with the most significant increase in the number of positively stained axons between 6 and 9 hours. Only rare reactive axons were identified from 3 to 6 hours following cardiac arrest (Figure 4A and 4B). Beginning from 9 hours after cardiac arrest, there was a marked increase in the number of strongly-stained cells (2.5% of the rat's cortical neurons) (Figure 4C and 4D). Many scattered positive cells were easily seen at 24 hours as well. Non-specific light-brown background nuclear staining was seen, particularly at 1 hour, and the significance of this observation is unclear.
Figure 4.
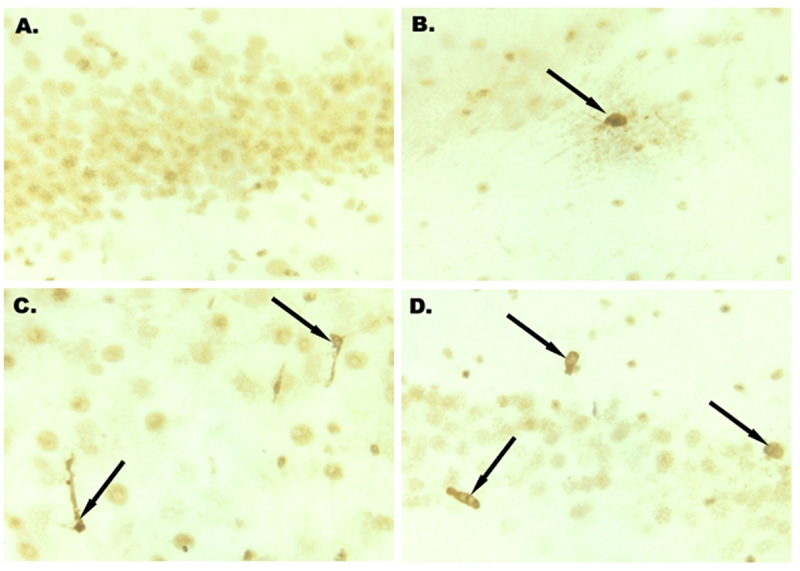
A. There is no activation of caspase-3 in the cerebellar cortex neurons’ cytoplasm 3 hours after warm cardiac arrest (caspase-3 immunostaining, ×400). B. Activation of caspase-3 in the neuron of cerebellar cortex (black arrow) at 3 hours after warm cardiac arrest (caspase-3 immunostaining, ×400). C. Activation of caspase-3 in the neuron of cerebellar cortex (black arrows) at 9 hours after warm cardiac arrest (caspase-3 immunostaining, ×400). D. Activation of caspase-3 in the neuron of cerebellar cortex (black arrows) at 12 hours after warm cardiac arrest (caspase-3 immunostaining, ×400).
Discussion
Electron Microscopy of Anoxic Brain Damage
Arsenio-Nunes et al in 1973 found the complete retrieval of ultramicroscopic structure of the cortical neurons in cats after 30 min-duration brain ischemia [2]. Hossmann & Zimmermann in 1974 found only cytoplasmic homogenization of the cortical neurons after one hour of brain anoxia in monkeys, which was reversible after retrieval of cerebral blood circulation [3]. Kleiheus et al in 1975 showed the complete retrieval of the cortical neurons’ ultrastructures after 60-min-duration complete ischemia of monkey brains [4]. In 2003, Nedzved et al also showed that the ultrastructure of human cerebral cortical neurons was intact within 4 hours of complete cerebral anoxia [11]. In their study, the authors incubated small fragments of human front lobe removed during neurosurgery for olfactory meningiomas and analyzed with electron microscopy. Our results also showed no ultrastructural changes related to autolysis of the cerebral neurons (Purkinje cells, pyramidal neurons of the hippocampus) for 6 hours after warm cardiac arrest.
Mitochondrial swelling, as first described 80 years ago, remains one of the most universal ultrastructural changes after brain ischemia [12]. Solenski et al (2002) showed that in contrast to permanent ischemia, the re-introduction of blood flow rapidly resulted in signs of likely irreversible severe damage, including initially condensed electron-dense mitochondria with increased electron-dense deposits within their matrix, and autophagy [13]. By 24 hours of reperfusion, the mitochondria are degenerating, with complete loss of outer membrane shape and form. Neurons undergoing increasing lengths of permanent ischemia also demonstrate significant mitochondrial damage initially in the form of increased swelling with cristae disruption, intracristal dilation, and loss of matrix density. By 24 hours of permanent ischemia, in contrast to reperfused neurons at 24 hours, the perinuclear mitochondria demonstrate homogeneously increased matrix density with preservation of the typical round or tubular shape. We also observed the preservation of the typical round shape of the mitochondria at 6 hours after global cerebral anoxia at room temperature.
Brain Death and Autolysis
Ujihira et al in 1993 conducted a clinico-neuropathological study on 60 cases of brain death (29 patients died of cerebrovascular disease) [14]. The average duration of brain death was 99 hours. Histologically, the cytoplasm of neurons was pale and ghost-like. In the white matter, myelin staining was pale, and nuclei of the glial cells were shrunken and pyknotic. Autolysis of the cerebellar granular layer and the pituitary gland was evident in all cases. No reactive astrocytosis or infiltration of the cells in or around necrotic tissue could be seen. Correlation between the degree of autolysis and duration of brain death was observed, but no relationship between the degree of autolysis and the difference of underlying disease could be found. Autolysis in the cerebral cortex, thalamus, tegmentum of the brain stem, cerebellar granular layer and pituitary gland was most prominent. Purkinje cells of the cerebellum, the pyramidal cells of the CA1 subfield of the hippocampus, and the pyramidal cells in layers 3 and 5 of the cerebral cortex are extremely sensitive to the effect of cerebral anoxia.
Ogata et al in 1986 analyzed granular layer autolysis (GLA) of the cerebellar cortex in 45 patients who died of acute cerebrovascular diseases (CVDs) [15]. Twelve patients who died of causes other than intracranial disease served as controls. Tonsillar herniation occurred in all who died of acute CVDs. More advanced GLA was seen in the central folia adjacent to the central white medullary body of the cerebellum as compared with the peripheral folia. According to the author's opinion, widespread granular layer autolysis extending to the peripheral folia could be a pathological finding characteristic of brain death, where intracranial blood flow could be absent or significantly reduced. The authors concluded that brain death for less than 1 day would be necessary for granular layer autolysis to develop. In our study, the first signs of autolytic damages of the neurons were seen at 12 hours after warm cerebral anoxia by electron microscopy, while H&E sections and sections stained with Thionine were fully devoid of autolytic changes up to 24 hours. Our results demonstrated a lack of autolytic damage to the same regions on both H&E and on Nissl staining. These findings suggest a possible correlation between pre-existing vascular disease and the development of autolytic histopathology.
Brain Death and Ischemia-Reperfusion Brain Injury
Why is it almost impossible to resuscitate a patient with prolonged warm cardiac arrest? There are two possible explanations. The first one is the “no-reflow” phenomenon. The “no-reflow” phenomenon was described by Ames et al in 1968 [16]. Brain ischemia causes some degree of edema, which closes the microcirculation due to the pressure on the capillary wall from outside. In the reperfusion phase, the larger branches are perfused again, but the microcirculation remains closed due to the persistent edema.
The second one is the free radical injury. Although early reperfusion of ischemic tissue is important for the preservation of tissue viability, it is generally accepted that the re-introduction of blood flow causes the conversion of some reversibly injured cells to a state of irreversible injury. Oxygen-derived free radicals, which are formed within the first moments of reperfusion, have been implicated in the development of “ischemia-reperfusion injury”. The role of oxygen derived free radical production, and lipid peroxidation, lipid carboxilation, protein oxidation, DNA damage and neutrophil induced damage in ischemia-reperfusion injury in brain has been extensively reviewed recently [17].
Brain Death and Apoptosis
Apoptosis, or programmed cell death, plays a fundamental role in many biological processes such as morphogenesis and negative selection in the immune system as well as in multiple disease states such as cancer and neurodegenerative diseases. Apoptosis has been shown to involve mitochondrial dysfunction with sequential release of cytochrome c from mitochondria, resulting in activation of caspase-3. Caspase-3 is a central player in Fas-mediated apoptosis and is the most widely studied caspase [18]. Numerous studies have also shown that caspase-3 is a key component of cell death based on its inactivating mechanism on cell membrane calcium ion pumps in both neuronal and other somatic apoptotic cells. The caspase-induced calcium-pump failure leads to consequential ionic imbalance, calcium overload, and cell death [19].
Colbourne et al In 1999 showed that untreated global ischemic injury has necrotic, not apoptotic, morphology. However, the possibility of programmed biochemical events of the apoptotic pathway occurring before neuronal necrosis could not be completely ruled out [20]. Electron microscopy of ischemic neurons with or without post-ischemic hypothermia revealed features of necrotic, not apoptotic, neuronal death even in cells that died two months after ischemia. Dilated organelles and intranuclear vacuoles preceded necrosis. Schwab et al in 2002 showed that the degree of cellular necrosis due to brain ischemia could be reduced by therapeutic caspase inhibitors [19]. Our results showed that significant cell death was not evident until 9 hours following warm, non-reperfused cardiac arrest. These findings suggest a “window of opportunity” for caspase-inhibitor therapy to reduce neuronal damage following warm cardiac arrest.
The mechanism of cell death induced by ATP depletion has been extensively studied by many researchers [21, 22]. ATP depletion can cause either necrosis or apoptosis [22]. Ono et al in 2003 showed the ATP depletion-mediated change from apoptosis to necrosis was associated with the increase in the total volume of intracellular acidic compartments per cell (VAC) [23]. VAC, which reflects the size of lysosomes, may be linked to lysosomal disruption. Lysosomal enlargement occurring in a death program alters the membrane tension of lysosomes and therefore increases the likelihood of lysosomes to rupture. Hishita et al in 2001 found that lysosomal dysfunction induced by a reduction in ATP resulted in leakage of lysosomal enzymes into the cytosolic compartment and that lysosomal enzyme(s) may be involved in the activation of caspase-3 during apoptosis [24]. In this study, we observed initial caspase-3 activation in the neurons of the cerebellum and neocortex within 6 hours of the room temperature cerebral anoxia, though significant increase of positively-stained cells did not emerge until 9 hours. These findings can be explained by ATP depletion as the trigger for programmed cell death activation, followed by possible leakage of lysosomal enzymes as a result of lysosomal dysfunction induced by ATP depletion.
Brain Death and Hypothermia
There are many interesting cases of successful re-animation of people after long-term cardiac arrest (for 30–40 minutes; drowning in the cold water) [25, 26, 27, 28, 29, 30, 31]. Ground squirrels’ brain slices, for example, can survive at +4°C for 7 days in the buffer solution without continuous oxygenation [32]. We did not test the hypothermic conditions because we were interested in seeing the morphological hypoxic/autolytic changes during room temperature conditions. Our interest in this problem was due to the fact that many human cardiac arrests happen at the room temperature or normothermic conditions.
In conclusion, our data showed the absence of the severe apoptotic damages of the CNS neurons during the first 6 hours after cardiac arrest at room temperature. Our data can be useful for forensic medical examinations and for advancement in critical care medicine. Understanding the mechanisms of brain death will help to design new neuro-resuscitation technologies in the future.
Acknowledgments
We acknowledge Dr. Robert C. Switzer III, PhD (Neuroscience Associates, Knoxville, TN) for providing H&E, Nissl, Amino CuAg staining, and immunohistochemistry services; Dr. Robert Roberson (School of Life Sciences, Arizona State University, Tempe, AZ) for electron microscopy; Mr. Charles J. Kazilek (Life Science Visualization Group, W.M. Kerk Bioimaging Laboratory, School of Life Sciences, Arizona State University, Tempe, AZ) for providing light microscopy and recording images; Mr. Bill Voice (Alcor Life Extension Foundation, Scottsdale, AZ) for his help during the experiments; Dr. William D. Anderson (Pathology Department, St. Joseph's Hospital and Medical Center, Phoenix, AZ) for his advises. This research project was supported by Alcor Life Extension Foundation (Scottsdale, AZ 85260).
References
- 1.Walker EA. Cerebral Death. 3rd Ed. Baltimore, MD and Urban and Schwartzenberg GmbH: 1985. [Google Scholar]
- 2.Arsenio-Nunes ML, Hossmann KA, Farkas-Bargeton E. Ultrastructural and histochemical investigation of the cerebral cortex of cat during and after complete ischaemia. Acta Neuropathol (Berl) 1973;26:329–344. doi: 10.1007/BF00688080. [DOI] [PubMed] [Google Scholar]
- 3.Hossmann KA, Zimmermann V. Resuscitation of the monkey brain after 1h complete ischemia. I. Physiological and morphological observations. Brain Res. 1974;29:59–74. doi: 10.1016/0006-8993(74)90478-8. [DOI] [PubMed] [Google Scholar]
- 4.Kleihues P, Hossmann KA, Pegg AE, Kobayashi K, Zimmermann V. Resuscitation of the monkey brain after one hour complete ischemia. III. Indications of metabolic recovery. Brain Res. 1975;95:61–73. doi: 10.1016/0006-8993(75)90207-3. [DOI] [PubMed] [Google Scholar]
- 5.Zimmermann V, Hossmann KA. Resuscitation of the monkey brain after one hour's complete ischemia. II. Brain water and electrolytes. Brain Res. 1975;85:1–11. doi: 10.1016/0006-8993(75)90997-x. [DOI] [PubMed] [Google Scholar]
- 6.Rothstein RP, Levison SW. Gray matter oligodendrocyte progenitors and neurons die caspase-3 mediated deaths subsequent to mild perinatal hypoxic/ischemic insults. Dev Neurosci. 2005;27:149–159. doi: 10.1159/000085987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwata O, Iwata S, Tamura M, Nakamura T, Sugiura M, Ogiso Y, Takashima S. Early head cooling in newborn piglets is neuroprotective even in the absence of profound systemic hypothermia. Pediatr Int. 2003;45:522–529. doi: 10.1046/j.1442-200x.2003.01784.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhu H, Meloni BP, Bojarski C, Knuckey MW, Knuckey NW. Post-ischemic modest hypothermia (35°C) combined with intravenous magnesium is more effective at reducing CA1 neuronal death than either treatment used alone following global cerebral ischemia in rats. Exp Neurol. 2005;193:361–368. doi: 10.1016/j.expneurol.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Vereczki V, Martin E, Rosenthal RE, Hof PR, Hoffman GE, Fiskum G. Normoxic resuscitation after cardiac arrest protects against hippocampal oxidative stress, metabolic dysfunction, and neuronal death. J Cereb Blood Flow Metab. 2006;26:821–835. doi: 10.1038/sj.jcbfm.9600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Olmos JS, Beltramino CA, de Olmos de Lorenzo S. Use of an amino-cupric-silver technique for the detection of early and semiacute neuronal degeneration caused by neurotoxicants, hypoxia, and physical trauma. Neurotoxicol Teratol. 1994;16:545–561. doi: 10.1016/0892-0362(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 11.Nedzved MK, Sheleg SV, Gerasimova TN, Oleshkevich FV, Wolf N. [Morphologic changes of human cerebral cortex neurons under long duration anoxia] Zdravoohranenie. 2003;7:10–12. (in Russian) [Google Scholar]
- 12.Spielmeyer W. Histopathologie des Nervensystems. Berlin, Germany: Springer; 1922. pp. 74–79. [Google Scholar]
- 13.Solenski NJ, diPierro CG, Trimmer PA, Kwan AL, Helm GA. Ultrastructural changes of neuronal mitochondria after transient and permanent cerebral ischemia. Stroke. 2002;33:816–824. doi: 10.1161/hs0302.104541. [DOI] [PubMed] [Google Scholar]
- 14.Ujihira N, Hashizume Y, Takahashi A. A clinico-neuropathological study on brain death. Nagoya J Med Sci. 1993;56:89–99. [PubMed] [Google Scholar]
- 15.Ogata J, Yutani C, Imakita M, Ueda H, Waki R, Ogawa M, Yamaguchi T, Sawada T, Kikuchi H. Autolysis of the granular layer of the cerebellar cortex in brain death. Acta Neuropathol (Berl) 1986;70:75–78. doi: 10.1007/BF00689517. [DOI] [PubMed] [Google Scholar]
- 16.Ames A, 3rd, Wright RL, Kowada M, Thurston JM, Majno G. Cerebral ischemia. II. The no-reflow phenomenon. Am J Pathol. 1968;52:437–453. [PMC free article] [PubMed] [Google Scholar]
- 17.іşlekel H, іşlekel S, Güner G. Biochemical mechanism and tissue injury of cerebral ischemia and reperfusion. Part II: Tissue injury. NOROL BIL D 2000, 17 (Electronic publication). Available at: http://www.med.ege.edu.tr/norolbil/2000/NBD09200.html. Accessed January 06, 2006.
- 18.Woo M, Hakem A, Elia AJ, Hakem R, Duncan GS, Patterson BJ, Mak TW. In vivo evidence that caspase-3 is required for Fas-mediated apoptosis of hepatocytes. J Immunol. 1999;163:4909–4916. [PubMed] [Google Scholar]
- 19.Schwab BL, Guerini D, Didszun C, Bano D, Ferrando-May E, Fava E, Tam J, Xu D, Xanthoudakis S, Nicholson DW, Carafoli E, Nicotera P. Cleavage of plasma membrane calcium pumps by caspases: a link between apoptosis and necrosis. Cell Death Differ. 2002;9:818–831. doi: 10.1038/sj.cdd.4401042. [DOI] [PubMed] [Google Scholar]
- 20.Colbourne F, Sutherland GR, Auer RN. Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci. 1999;19:4200–4210. doi: 10.1523/JNEUROSCI.19-11-04200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly KJ, Plotkin Z, Dagher PC. Guanosine supplementation reduces apoptosis and protects renal function in the setting of ischemic injury. J Clin Invest. 2001;108:1291–1298. doi: 10.1172/JCI13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lieberthal W, Menza SA, Levine JS. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am J Physiol. 1998;274:F315–327. doi: 10.1152/ajprenal.1998.274.2.F315. [DOI] [PubMed] [Google Scholar]
- 23.Ono K, Kim SO, Han J. Susceptibility of lysosomes to rupture is a determinant for plasma membrane disruption in tumor necrosis factor alpha-induced cell death. Mol Cell Biol. 2003;23:665–676. doi: 10.1128/MCB.23.2.665-676.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hishita T, Tada-Oikawa S, Tohyama K, Miura Y, Nishihara T, Tohyama Y, Yoshida Y, Uchiyama T, Kawanishi S. Caspase-3 activation by lysosomal enzymes in cytochrome c-independent apoptosis in myelodysplastic syndrome-derived cell line P39. Cancer Res. 2001;61:2878–2884. [PubMed] [Google Scholar]
- 25.Chochinov AH, Baydock BM, Bristow GK, Giesbrecht GG. Recovery of a 62-year-old man from prolonged cold water submersion. Ann Emerg Med. 1998;31:127–131. doi: 10.1016/s0196-0644(98)70296-3. [DOI] [PubMed] [Google Scholar]
- 26.Felix WR, Jr, MacDonnel KF, Jacobs L. Resuscitation from drowning in cold water. N Engl J Med. 1981;304:843–844. doi: 10.1056/NEJM198104023041413. [DOI] [PubMed] [Google Scholar]
- 27.Graf D, Meier P, Guse HG, Leitz KH, Bachmann H. [A drowning accident of long duration with deep hypothermia and rewarming with extracorporeal circulation. A report of 2 patients] Monatsschr Kinderheilkd. 1989;137:415–418. (in German) [PubMed] [Google Scholar]
- 28.Jacobsen JB, Nielsen H, Andersen PK. Resuscitation from drowning in cold water. N Engl J Med. 1981;305:580–581. doi: 10.1056/NEJM198109033051013. [DOI] [PubMed] [Google Scholar]
- 29.Kugler-Podelleck I, Rodewald G, Horatz K, Kugler S, Muller-Brunotte P. Resuscitation after drowning in freezing water. Ger Med Mon. 1966;11:232–236. [PubMed] [Google Scholar]
- 30.Modell JH, Idris AH, Pineda JA, Silverstein JH. Survival after prolonged submersion in freshwater in Florida. Chest. 2004;125:1948–1951. doi: 10.1378/chest.125.5.1948. [DOI] [PubMed] [Google Scholar]
- 31.Norberg WJ, Agnew RF, Brunsvold R, Sivanna P, Browdie DA, Fisher D. Successful resuscitation of a cold water submersion victim with the use of cardiopulmonary bypass. Crit Care Med. 1992;20:1355–1357. doi: 10.1097/00003246-199209000-00026. [DOI] [PubMed] [Google Scholar]
- 32.Pakhotin PI, Belousov AB, Otmakhov NA. [A comparative analysis of the functional stability (based on electrophysiological criteria) of slices of the suslik and guinea pig brains maintained under deep hypothermia] Zh Evol Biokhim Fiziol. 1991;27:479–485. (in Russian) [PubMed] [Google Scholar]