

1. Introduction
Living organisms are capable of inducing the crystallization and deposition of a wide variety of minerals1 but the vertebrates mainly utilize the calcium phosphates in constructing their mineral phases in both normal circumstances in bone, dentin and tooth enamel and in pathological ectopic mineral deposits. The predominant form of the mineral in all situations is as carbonated apatite. However, the extent of mineralization in a particular tissue or organ is quite variable and crystallite size, crystal shape, and the packing and organization of the mineral crystals may also be variable. It is clear that the same physical chemical principles must apply to all, but it is equally clear that the organism must tightly regulate the local environment where the mineral is formed. This is an intrinsically complex problem because the mineral crystals of bone and dentin form in the extracellular matrix, external to the cells which are the ultimate regulators of the process.
Years of study of this complex set of problems has led to the general consensus that the cell-controlled processes of mineralization begin with the manufacture of an organic structure within which, or a compartment surface upon which, the mineral crystals may be initiated. The cells secrete and organize macromolecular structures which determine the ultimate character and orientations of the crystals subsequently initiated and grown, but in general, these structures do not themselves have the capacity to initiate mineralization. The incipient crystal nucleation event depends upon the interaction of the structural macromolecules with another set of secreted interactive macromolecules which locate specifically on the structural framework. These interactive molecules are doubly interactive, binding specifically to the framework on the one hand, and attracting the requisite mineral ions on the other to initiate the mineral ion clustering to a critical size that nucleates crystal growth. The biological fluids are supersaturated in mineral ion content with respect to the solubility of their biogenic mineral crystal phase, but unrestricted and uncontrolled crystal growth are not permissible in the vertebrate animal so there is a third line of macromolecular components interactive with the growing crystal surfaces that can limit growth rates, and do so selectively, controlling the crystal shape as well as size. A final component of the regulation is the availability of the crystal microions, pumped in controlled fashion from the cell into the extracellular matrix. The situation is shown schematically in Figure 1, wherein the solid, heavy arrows trace the construction of the structural matrix (a), the addition of the interactive macromolecules that activate the structural matrix (b) to bind the mineral ions (c), leading to growth of the oriented crystals (d), and finally, the addition of further macromolecules (e) to shape and delimit the crystal size to attain the mature mineralized matrix form. Note that in this paragraph, and in Figure 1 the nature of the macromolecules and mineral phase has not been specified. This is intentional to emphasize the point that in the majority of mineralizing tissues in animal systems the same overall scheme of events is likely to apply, although the structural scaffolds and mineral types can vary greatly.
Figure 1.
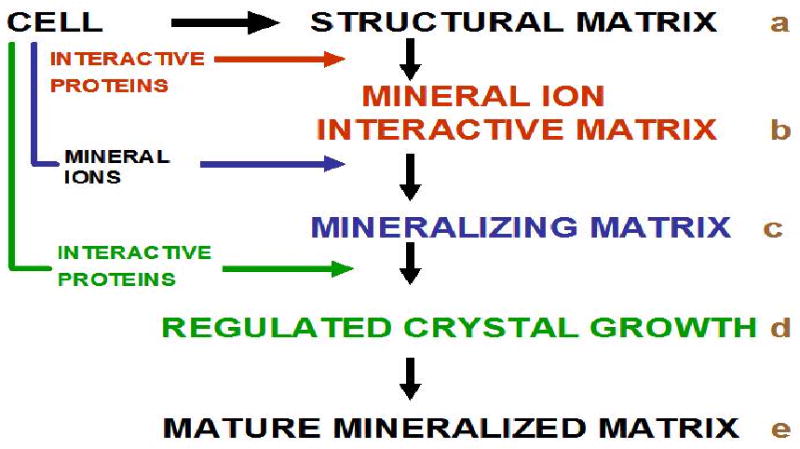
A proposed generalized scheme for matrix-regulated mineralization reactions. Modified and reprinted with permission from Reference 280 [Veis, Reviews in Mineralogy & Geochemistry, V. 54]. Copyright 2003 Mineralogical Society of America.
The calcium and phosphate ion product (Ca × P) in the extracellular fluids (ECF) of the vertebrates are greater than the solubility products of most crystalline forms of calcium phosphate, with hydroxylapatite (HA) being the major least soluble form of calcium phosphate. Hence, HA would precipitate spontaneously if it were not for the inhibition of crystal formation by the reduction of the activity of these ions, and their sequestration, by various macromolecular components. Thus, the ECF proteins in Ca-rich milk and blood, for example, play important roles as inhibitors of apatite precipitation. The initial form of the Ca – P solid phase deposits in vitro have been shown to be disordered and exhibit a diffuse X-ray diffraction pattern, indicating that initially only a very short range ordering of calcium and phosphate ions relative to each other takes place, producing a structure called “amorphous calcium phosphate” (ACP) 2,3,4. Depending upon the conditions of temperature, pH, and ion concentrations, the solid phase may grow through a variety of forms from the ACP to (in order of decreasing solubility) brushite [CaHPO4·2H2O], whitlockite [Ca3(PO4)2 ·3H20], octacalcium phosphate [Ca8H2(PO4)6·5H2O], and hydroxyapatite [Ca10(PO4)6(OH)2]. In bone, dentin, cementum and enamel, in vivo, where the nucleation process and the final crystal composition are determined by the local environment created by the combination of the collagen or amelogenin structural matrix and particular NCP components, it is evident that the transition from ion aggregate to crystalline form is tightly regulated. A question of great importance for those interested in development of biomimetic models of bone is how the NCP might template the desired crystal forms.
The principal focus of this review, however, will be on the influence of the phosphoproteins of the matrix on the formation and growth of crystals in two systems: the small, plate-like crystals and crystal aggregates of carbonated apatite found in bone, dentin and cementum; and the large, high axial ratio rod-like crystal aggregates found in dental enamel. As pointed out many years ago5, the dentin and enamel are formed in apposition to each other and provide direct contrasts between the nature of the compartments created by the cells, the structural matrices produced, and the extracellular matrix macromolecules that control the nucleation, growth and structure of their carbonated apatite mineral phases. Here we consider these two systems separately.
2. Collagen Based Mineralization Systems
Bone, dentin and cementum are produced by osteoblasts, odontoblasts and cementoblasts, respectively. Each of these cells produces a distinct set of extracellular matrix macromolecules, but produce similar structural matrices predominantly constructed from Type I collagen fibrils.
2.1 The Type I collagen matrix
2.1.1 The gap –overlap model
Although the aim of this review is to discuss the role of the phosphorylated proteins of the ECM, so much of the discussion relates to how the non-collagenous ECM proteins might interact with the collagen matrix that an introductory discussion of the particulars of the type I collagen fibril organization and structure is necessary, as well as consideration of whether the collagen itself might induce mineral deposition. Type I collagen molecules, 300 nm in contour length, are best considered as semi-flexible rods with an axial ratio, if fully elongated, of about 200/16. When assembled into fibrils, the triple helical collagen I monomer molecules pack into ∼ 70 nm D-periodic fibrils7,8,9 readily visualized at the electron microscopic level. A comparison of the molecular length of 3000 Å with the fibril periodicity of 670 Å and, led to the familiar Hodge-Petruska “quarter-stagger, overlap model” 8,9 in which all molecules are aligned in the same N- to C- direction, and each molecule is a staggered by a 1 D period with respect to its nearest neighbors in one plane. However, to accommodate the fact that the rod-like molecule has a length equivalent to 4.4 D, the next molecule with the same axial orientation thus must have its N-terminal begin at a distance of about 0.6 D from the C-terminal of the first molecule, leaving a gap of about 0.6 D in length. In the assembled fibril, there is thus a periodic set of gaps or holes along each filament or fibril axis, and in a fully assembled 3 dimensional native D-periodic fibril, a regular series of gaps or channels.
2.1.2. Where does the mineral go?
This same periodicity can be observed in unstained mineralized fibrils at the electron microscopic level, indicating that the mineral deposits have the same disposition as the collagen “gaps/holes”. In spite of an enormous amount of work since this relationship was first determined [see Glimcher10 for a review of all the early work in this area] it is not entirely established as to how much of the mineral formed is retained within the fibrils. In bone it does appear as if the holes create channels through each fibril, perhaps allowing for mineral crystals periodically running continuously through the fibril. A crystal filling such a channel would have dimensions of 3 - 6Å in width and 400 Å in height in the axial direction, with a length dependent on the fibril size. These dimensions are compatible with the thin plate-like crystals seen by electron microscopy and x-ray crystallographic studies of early crystal formation11,12,13,14,15. Earlier Katz and Li 16,17 had explored the concept that there were spaces of two kinds, the holes or gaps themselves, and pores that consisted of the spaces (undoubtedly hydrated) between the molecules packed in the collagen overlap regions. They made the observation that the pore size was dependent upon the particular collagen, with the pore size larger, almost double, in mineralized collagen fibrils than in those of the soft tissue rat tail tendon collagen. Thus it seemed likely that, in the gap region of mineralized collagens, the pore-hole combination could certainly provide accessible channels for growth of plate-like crystals transversely though the fibrils. The crystals are not confined to the internal spaces within a fibril, but may mature and grow along the fibril surface or expand between layers of collagen molecules within a fibril12,15. Moreover, the surface of a D-periodic fibril displays both the overlap and gap regions along its entire surface so that crystal nucleation could either initiate along the fibril surface, from which crystal growth could proceed internally in the channels, or alternatively, the crystals could initiate internally within the channels and then spread to the fibril surface.
Crystallographic studies of the three-dimensional molecular packing arrangement of type I collagen show that the triple helical molecules are arranged on a quasihexagonal lattice with a minimum microfibril structure represented by five 1 D staggered molecules18. The most recent X-ray diffraction studies by Orgel et al.19,20 determined the structure of the collagen molecule in situ at a ∼5-10 Å resolution, and, as depicted in Figure 2, shows that the unit cell 1D period in length contains the chain segments from each molecule such that the unit cell contains the equivalent of a single collagen molecule. The neighboring molecules form a supertwisted right-handed microfibril that interdigitates with neighboring microfibrils, establishing the crystallographic superlattice of quasi-hexagonally packed collagen molecules.
Figure 2.
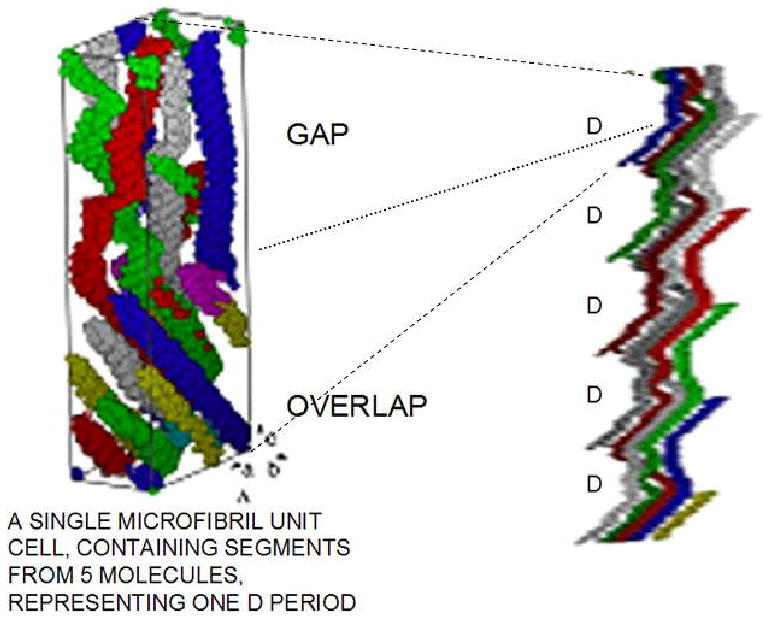
Three dimensional models of a putative collagen microfibril (five molecules in the quarter-stagger-overlap arrangement). Left: A unit cell, containing the equivalent of all segments from a single molecule in a single D-period. The gap region is less dense, with only four molecules, as compared to the five strands within the overlap zone. Right: A 5-stranded microfibril with the molecules traced through 5 D-periods showing the helical entwinement and reorientation of each single 4.4 D-period long molecule along the microfibril. The differences in molecular flexibility within the gap zones, as compared to the overlap zones are evident. Modified and reprinted with the permission from Figure 3 B and 3E of Reference 19 [Orgel et al. PNAS 103: 9001-9005 (2006)]. Copyright 2006, National Academy of Sciences, USA.
2.1.3 The nature of the gap environment
In the context of mineralization, the important point is that the gap region chain segments present a very different chain conformation and pattern of side chain residues so that one might expect the two repeating (0.4D and 0.6D) regions to interact differently with other molecules and perhaps mineral ions. Since the complete sequences of type I collagen chains have been determined, it is easy to determine the exact nature of the charge distribution, and the relative polarity of each molecular domain. Chapman21,22 was the first to do this. Using Chapman's alignment of the three chains in type I collagen, Veis et al.23 showed that in a 1 D segment, the boundaries of the gap region and the central region of the gap zone had particularly intense groupings of both positively and negatively charged groups along the five molecule microfibril, Figure 3a, is aligned with an electron micrograph in Figure 3b, positively stained to reveal the band pattern. The staining and schematic representation of the positive and negative amino acid side chains clearly match. Although many studies showed that pure collagen fibrils did not bind any significant numbers of Ca and phosphate ions, even at high micro ion concentrations24,25,26, Silver et al.27, and Landis and Silver28, commenting on the same Chapman analysis of the side chain ionic group distributions proposed that the juxtaposition of anionic groups on all three chains could be important on localizing both cations and anions for specific binding to collagen that could lead to nucleation of mineral deposition within the collagen fibril. Their arguments describing the arrangements of the groups as shown in Figure 3c, are quite logical, but do not seem to be supported by any specific data. However, recent discussions29 have suggested that phosphate ion localization on the collagen framework may also play an important role in mineral phase nucleation.
Figure 3.
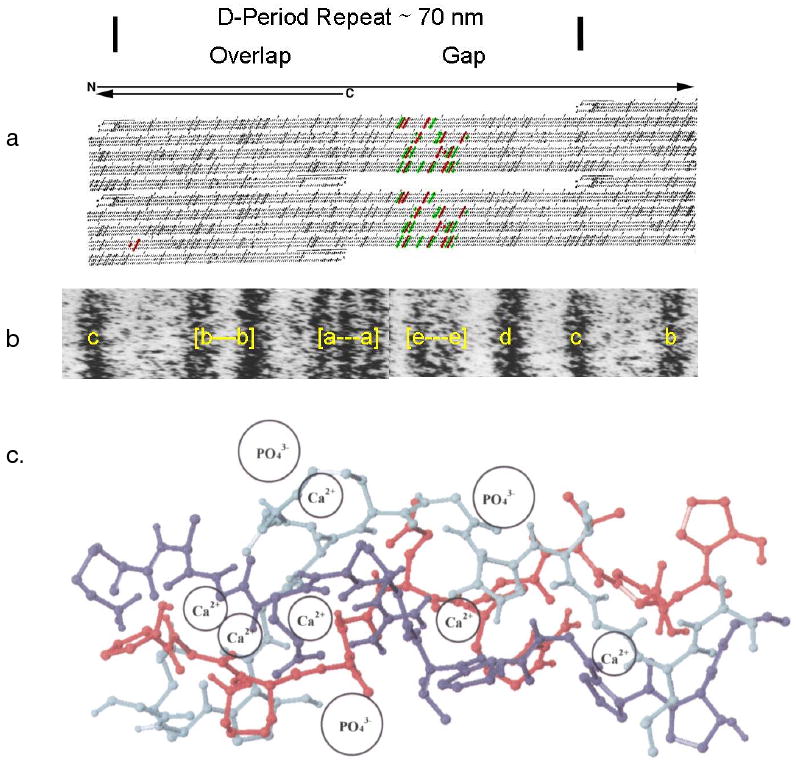
Correlation of collagen packing and sequence in the gap and overlap segments of 1 D period. a. Schematic showing charged residues as dark tilted stripes across all three chains, the segment correlated, in b, with the EM e-band, shown in the positively labeled 1 D period of a fibril. c. A section of type I collagen triple helix with a proline and hydroxyproline deficient region, residues 414 to 424. According to Silver et al27 this is a flexible region capable of binding both Ca and PO4 ions, and changing the conformation of the helix, based on the use of the SYBYL molecular modeling program and energy minimization after the addition of the mineral ions at the sites proposed. a. and b. Modified and reprinted with permission from Reference 280 [Veis, Reviews in Mineralogy & Geochemistry, V. 54]. Copyright 2003 Mineralogical Society of America. c. Modified and reprinted with permission from Reference 27 [Silver, et.al. Biomacromolecules]. Copyright 2001 American Chemical Society.
Strong evidence that nucleation of mineralization in collagen I fibrils could be quite collagen structure specific was demonstrated by the electron microscopic observation that in vitro in turkey tendon fibrils, the first mineral deposited from a calcium phosphate solution appeared at the electron microscopic e-band30, which, as shown in Figure 3a and b is directly correlated with a region of putative high charge density, bounded by sequences of lower polarity20. Earlier neutron diffraction studies31 had also shown that in mineralizing turkey tendon, the mineral density began to increase within the gap zone of the collagen fibrils.
3.0 Phosphoproteins as mediators of collagen mineralization
3.1 Extraction protocols
In the early 1960's the presence of organic phosphorous, as serine phosphate, was detected in bone collagen32, and in dental enamel proteins33. In parallel studies it was shown that dentin collagen contained covalently bound phosphate, attributed to the presence of both directly collagen-bound phosphate34 and to the presence of one or more unique non-collagenous proteins35,36,37,38,39,40,41 which were also shown to be strongly associated to the mineralized collagen. The unique dentin phosphorylated NCP were found to be present in the dentin from all species examined, but variable in molecular size and degree of phosphorylation, and in amino acid composition42. As the methods for demineralizing the dentin matrix and isolating these proteins were developed43 they were applied to mineralized bone matrix44. Termine et al. 45 improved the extraction process by using a prior treatment of the mineralized matrix with 4.0 M Guanidine·HCl to dissociate cellular and other accessible protein components not bound to the mineral phase before EDTA demineralization. Termine and colleagues46,47,48,49,50,51,52,53,54 applied these techniques to a variety of vertebrate mineralized tissues and isolated a number of other mineral-related phosphorylated NCP-proteins. A second dissociative extraction, after demineralization was added to this protocol55 to remove tightly bound mineral-protected NCP molecules from those bound to the structural matrix. The three extracts are generally labeled as G1(First 4.0 M guanidinium·HCl extraction); E (0.5 M EDTA extraction); and G2 (Second 4.0 M guanidinium·HCl extraction). The G2 extracted proteins are tightly bound to the demineralized collagen matrix, but it is not clear if these are covalently linked and freed by hydrolysis or by some very strong non-covalent interaction. One of the proteins of a combined EG2 extract was found to be a phosphorylated form of the NH2-terminal propeptide of the alpha 1 chain of type I collagen, cleaved from the collagen during secretion and fibril assembly50. The others, however, were non-collagenous proteins.
3.2 The SIBLING Proteins
The major phosphoproteins of this group are now known as the SIBLING (Small Integrin-Binding Ligand, N-linked Glycoprotein) family56. Each of the proteins is described separately in detail below. The linkage between them resides in the facts that their genes are all clustered within an approximately 375 kb span of nucleotides on human chromosome 4q21 (mouse 5q21). Further, the genes of each protein have similar intron-exon structures, with sequences of similar character located in similar exons57 and show a bias for the use of the AGC and AGT codons for serine relative to the average use of those codons in other proteins58. At the protein level, they are all acidic proteins with poorly defined, more or less random structures. However, in spite of those similarities, when compared at the sequence homology level the proteins are all distinctly different, with few long-range sequence alignments55. Thus, while displaying strong gene synteny the SIBLINGS (osteopontin [OPN], matrix extracellular phosphoglycoprotein [MEPE], bone sialoprotein [BSP], dentin matrix protein 1 [DMP1], dentin sialophosphoprotein [DSPP]) have different tissue distributions and functions. OPN and MEPE act as inhibitors of mineralization and are found widely in mineralized and non-mineralizing tissues, whereas BSP, DMP1, and DSPP (and its subdomains DSP [dentin sialoprotein], DGP [dentin glycoprotein59] and DPP [dentin phosphoprotein, also known as phosphophoryn]) are heavily engaged in bone and dentin mineralization. Once thought to be quite tissue specific, DMP1, DSP and DPP are present in non-dentin tissues. Similarly, BSP is not exclusively confined to bone, and all have been shown to have both structural and signaling activities.
The acidic SIBLINGS are obviously rich in acidic amino acids, Asp and Glu, but also contain basic residues, Arg and Lys. When ionized, these residues, along with Gln, Gly, Pro and Ser are considered to be structure breaking, unfavorable for long range ordering of the backbone protein conformations60,61. All are also post-translationally phosphorylated and display microheterogeniety in phosphorylation.
3.2.1 Osteopontin phosphorylation
OPN is the most thoroughly studied example. Sorensen and colleagues62,63 have shown that the Ser or Thr residues in OPN reside in sequences that are substrates for the specific mammary gland casein kinase (MGCK, also known as the Golgi kinase) S*/T*-X-E/S(P)/D, or for casein kinase II, CKII [S*-X-X-E/S(P)/D]. Of the 36 Ser residues in human OPN 29 are in positions susceptible to MGCK and 6 are substrates for CKII. It is unusual however, for all of these sites to be phosphorylated, so that in any given preparation of OPN there may be an average number of charged phosphate residues less than the maximum, but distributed over a number of individual molecular possibilities so that the preparation is microheterogeneous. Rat bone OPN, with 29 potential phosphorylation sites and a homologous amino acid sequence has only about 13 of these sites phosphorylated64. The degree of phosphorylation has important implications for the effectiveness of mineralization inhibition65 and mineral induction66. The phosphorylation sites in human milk OPN are mainly Ser residues, only 2 Thr residues are phosphorylated, whereas 7 Thr sites are O-glycosylated, the 2 potential Asn residue N-glycosylation sites are not glycosylated63.
3.2.2 Dentin Sialophosphoprotein, DSPP, phosphorylation, structural consequences and calcium ion binding
DSPP has also been studied in some detail with respect to phosphorylation. As indicated earlier, the molecule is processed into 2 major segments: DSP and DPP. The DSP is a sialoprotein found in two forms, based on the sites where the post-translational endopeptidase cleavage takes place67. Depending on the species and form the DSP may have from 5 to 10 potential phosphorylation sites and extensive glycosylation68. The DSP portion may also contain chondroitin 6-sulfate proteoglycan attachments69. DPP the main form remaining closely associated with mature mineralized dentin, is unique in composition with respect to all other SIBLINGS. As shown in Figure 4, the DPP domain of human DSPP has an extensive set of repetitive DSS repeats. Bovine DPP, when isolated from dentin with careful attention to possible degradation, has a molecular size greater than that of the human, with an apparent mass of 155,000 Da based on primary analytical ultracentrifuge data and indirect gradient gel electrophoretic analysis70, and was found to be phosphorylated on about 95% of its Ser residues70. The mass and extent of phosphorylation were confirmed by MALDI mass spectrometry71. The DSS repeat sequence region in the bovine dentin must be considerably greater in extent than in the human, shown in Figure 4, and the rat, where the molecular mass is on the order of 70 kDa72. The DSS repeat regions fit the model of intrinsically unstructured proteins58,73. The presence of the phosphates modulates this to some extent, on the basis that the neighboring electrostatic group repulsions might stiffen the protein backbone somewhat and increase the polypeptide chain persistence length74,75,76,77. However, this stiffness is essentially local, and the long chains still have the characteristics of a random or unordered polymer chain60,61. Dunker et al.61 have argued that the protein backbone flexibility can convey important properties which enable a particular sequence in a poorly ordered chain conformation to adapt to a variety of substrates or interaction partners with which it might interact. In a protein with a mixed sequence this permits interaction with a variety of different partners in different domains. For example, a cell surface integrin sequence may interact with a SIBLING protein, which, utilizing another domain, may in turn bind and bridge to another ECM component. In the specific case of the phosphophoryn DS*S* repeats (in this notation S* signifies phosphorylation), these have an avid affinity for binding Ca2+ ions, inducing the DPP to locally assume a more extended and ordered β-chain-like conformation. Phosphorylated DPP shows an affinity for collagen and at the same time maintains the ability to sequester Ca2+ ions, even at high concentrations of monovalent Na+ and K+ ions26. Similarly, OPN, BSP and DMP1 have sequences such as SSSEE and SGSSEE which can be phosphorylated. Although not phosphorylated, many proteins with extended polyAsp or polyGlu sequences are not uncommon78. Interestingly, proteins of highly acidic character, with extensive stretches of Asp and/or Glu are common in many types of mineralizing systems, such as those involving silicates and calcium carbonates.
Figure 4.

The amino acid sequence of human DSPP [PubMed ID: Q9NZW4]. Green: Signal Sequence; Red: Endopeptidase cleavage sites; Black: DSP Sequence; Blue: DPP(DMP2) sequence, including, in Dark Red the DSS Repeat Region, with a few DSS motif interruptions.
Phosphorous 31P-nuclear magnetic resonance studies showed that all of the organic phosphate of DPP was in the monoester form, with a pK a = 6.8076. The addition of Ca2+ to an aqueous DPP solution progressively broadened the 31P-NMR peak and diminished its intensity, indicating that the phosphates interacted with the Ca ions, and this was confirmed by infrared spectroscopy. Infrared spectroscopy also showed that the carboxyl groups of the DPP were involved in binding to Ca ions. In fact, at low DPP concentrations where intramolecular interactions were favored, Ca binding data indicated that total Ca uptake per mole of DPP was equivalent to the sum of Ser P and Asp groups79, that is, the Ca ions might bridge between acidic residues on the distant portions of the same polypeptide chain. At higher DPP concentrations the DPP would form large intermolecular aggregates and precipitate. This observation formed the basis for the use of high Ca ion concentrations in the isolation of purified DPP80,81,82. NMR studies of DPP in the presence and absence Ca ion confirmed the supposition that DPP is intrinsically disordered, but that in the presence of Ca the DPP backbone becomes more ordered, or of restricted activity74.
3.3 SIBLING Protein Collagen Interactions
All of the SIBLING proteins share in the two properties hypothesized as crucial in Figure 1, namely the binding of the SIBLING to some specific component of the matrix, collagen fibrils or cells, and having the capacity to interact with and accumulate Ca2+ ions in their vicinity. This dual behavior is entirely compatible with the flexible, multi-domain character discussed above. Several important tests of this hypothesis were carried out by Crenshaw and associates83,84,85. DPP was immobilized by covalent linking to agarose beads, these beads were then incubated at 37°C in metastable calcium phosphate solutions that did not spontaneously precipitate. The control was the DPP soaked on the beads without cross-linking immobilization83 and placed in the mineralizing solution. Calcium phosphate crystals developed on the immobilized DPP, and were shown by X-ray crystallography to be hydroxyapatite. The same treatment of the DPP-soaked agarose beads did not yield any calcium phosphate precipitates in the time frame examined. Free soluble phosphophoryn in the mineralizing buffer also failed to yield apatite precipitates. These experiments definitively showed that surface- immobilized DPP could nucleate apatite crystallization.
Collagen fibrils could substitute for the insoluble agarose beads in these experiments. Reconstituted collagen fibrils alone did not induce mineral formation in the mineralization buffer. Purified soluble type I collagen from skin was reconstituted as fibrils and then reacted with bovine DPP under conditions allowing reversible non-covalent binding, or irreversible covalent cross-linking. Phosvitin from egg yolk, an unrelated highly phosphorylated, intrinsically unstructured soluble protein was also bound to reconstituted skin type I collagen fibrils by adsorption or by covalent cross-linking. Collagen fibrils with covalently bound DPP or phosvitin induced apatite mineralization of the fibrils, while under the conditions used, the same amounts of non-covalently linked DPP and phosvitin did not induce any mineral. If the proteins were dephosphorylated by acid phosphatase before binding and immobilization on collagen neither protein induced mineral deposition84. Removal of the ester phosphate by reaction with acid phosphatase after the proteins had been covalently cross-linked to the collagen matrix also destroyed the ability of the complex to precipitate apatite. The mineral induced by the cross-linked phosphoproteins was apatite, and the crystals were confined to the surface of the collagen fibrils. With decreasing medium saturation the time required for mineral induction increased. The interfacial tensions calculated for apatite formation by either phosphoprotein cross-linked to collagen were about the same as that for phosphatidic acid liposomes and hydroxyapatite. This similarity in values indicated that the nucleation potential of these highly phosphorylated surfaces was about the same. From these experiments it was concluded that phosphoproteins must be irreversibly bound to collagen fibrils for the in vitro mineralization of the collagen network in solutions that do not spontaneously precipitate. The phosphate esters of phosphoproteins are required for mineral induction, and the carboxylate groups alone are not sufficient.
3.3.1 Covalent Collagen-SIBLING interactions
From the very earliest studies of dentin it was suggested that the mineralized tissue collagens contained covalently bound phosphoprotein attachments34,35,36,37,38,39,40,41. Fujisawa et al. 80 subsequently determined that DPP was covalently associated with collagen and proposed that transglutaminases led to the cross-linking of asparagines or glutamines of the DPP with lysyl residues of collagen. More recently Kaartinen et al. 86,87 demonstrated that transglutaminase 2 (TG2), a general protein cross-linking enzyme specifically joining the γ-carboxamide group of a glutamine residue to the ε-amino group of a lysyl residue to form a stable isopeptide cross-linkage, readily catalyzed the formation of high molecular weight forms of OPN, BSP, DMP1 and DPP in various bone and dentin extracts. Direct studies of bone and dentin E-extracts showed that they contained an abundant, active TG2 activity, which could catalyze the formation of γ-glutamyl-ε-amino lysyl isopeptide bonds. Asparagine is not a substrate for the TG2 activity, yet DPP, which does not contain glutamine, formed high molecular weight aggregates87. Examination of the DPP aggregates showed that some contained bound TG2, but that could not account for all the DPP aggregation. It was suggested that the ε-amino groups of lysyl residues in the DPP were reacting with glutamines on other molecules, with the large size of the aggregates evidence that several DPP molecules were linked together. That is, the DPP is effectively multivalent, with several of its Lys residues available to react with the TG2 itself, or with glutamines on collagen or other NCP components. DPP was primarily present in the E extract of dentin but some was clearly present in a high molecular weight aggregate in the G2 extract87. Purified monomeric DPP was converted to aggregate form, detected by anti-DPP, upon reaction with TG2. The same aggregate stained with anti-TG2. The aggregate band for the initial G2 extract (formed in the presence of the endogenous TG2) was broader than that seen when the purified DPP was reacted with purified TG2. Thus, while DPP cannot form homotypic DPP-DPP aggregates via TG2-catalyzed cross-linking, TG2 enables the formation of insolublized DPP in combination with other NCPs, or directly by association with the collagen. These observations help to rationalize the requirement for immobilization of the bound DPP for initiation of mineralization in the Crenshaw work in vitro, and the hypothesis that collagen-bound DPP is the in vivo nucleator of mineralization in dentin. It is interesting that, as shown in Figure 4, human DPP contains a single glutamine residue, Q1224, near the C-terminus, whereas that residue is not present in the bovine, rat and mouse sequences.
4.0 Individual Sibling Details
As indicated above, the SIBLING proteins are considered to be related because they all have been implicated in mineralization processes in vivo and because their genes are clustered on chromosome 4q21-2354 within a 375 kb region (Figure 5). BSP and OPN are found in higher concentrations in bone while DMP1, DSP and DPP are found in greater amounts in dentin88,89,90. Their acidic domains can function in mineralization by nucleating hydroxyapatite crystallization, inhibiting crystal formation, or modulating crystal growth to obtain particular crystal shapes. The SIBLING proteins also contain domains that mediate cell adhesion. In spite of adhering very strongly to hydroxyapatite crystal surfaces, their flexibility leaves the rest of the molecule free to interact with other proteins or facilitate cell attachment with the exposed RGD integrin-binding domain.
Figure 5.
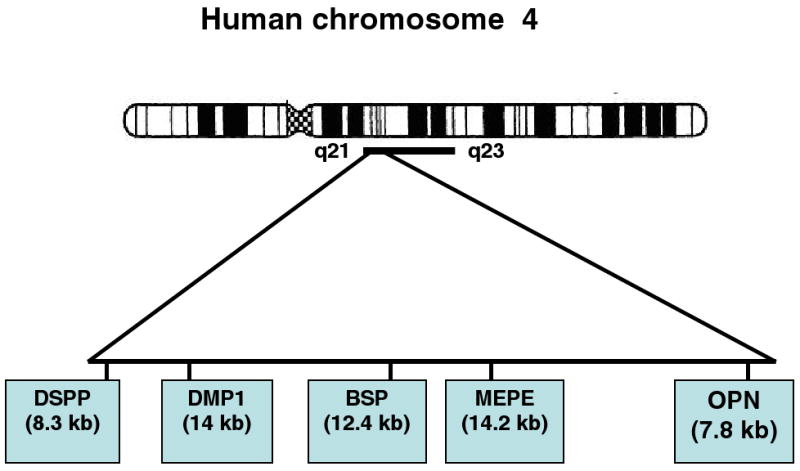
The tandem arrangement of the genes comprising the SIBLING family members on human chromosome 4q21-23 region.
4.1 Osteopontin (OPN)
Osteopontin is an arginine-glycine-aspartate (RGD) containing adhesive glycoprotein that was first identified as a major sialoprotein in bone, dentin, hypertrophic cartilage and subsequently found to be expressed in kidney, brain, macrophages, vascular smooth muscle cells91 and many cells of epithelial linings92,93,94,95,96,97. OPN is also enriched in biological fluids such as milk, urine, bile and seminal fluids having elevated levels of calcium salts which is indicative of a role in preventing spontaneous precipitation of calcium salts98,99,100,101.
4.1.1 Structural features of OPN
OPN is an acidic glycoprotein with a molecular mass of ∼44 kDa and has a high content of serine, asparagine and glutamate90,102,103,104. Analysis of the deduced amino acid sequence has indicated that OPN has several structural domains105 (Fig 6). Its ubiquitous expression pattern suggests that OPN may have different functions based on the use of various domains under different conditions and cell-type specificity. The amino and carboxyl terminal ends contain 8 alpha helices that constitute a large portion of the molecule. The middle of the molecule contains a very acidic region with a typical RGD cell attachment domain flanked on both sides by two segments of β-sheet structures94. At the amino terminus there is a highly acidic region that contains nine consecutive aspartic acid residues106. Other features of the molecule include a calcium binding loop and two heparin binding sites. A number of isoforms exist which differ mainly in the level of post-translational modification, particularly phosphorylation. It is rich in aspartic acid, glutamic acid and serine and contains ∼30 monosaccharides including 10 sialic acids. Carbohydrate is variable in extent107, with approximately 28 sites distributed throughout the molecule62. Kinases found to be responsible for OPN phosphorylation are casein kinase II, cAMP-dependent protein kinase, Golgi casein kinase and protein kinase C108. The high number of phosphorylation sites on OPN may be a function of the protein's multiple phosphate-dependent functions63,64,109,110, 111,112,113. OPN has a protease hypersensitive site that separates the integrin and CD44-binding domains. The thrombin cleavage motif in this region has a conserved sequence RSK present in most species. The C-terminal fragment of OPN binds directly to CD44v6, while the RGD –containing phosphorylated N-terminal domain obtained by thrombin cleavage is a prerequisite for efficient engagement of OPN to the αvβ3 integrin receptor 114. Thus, thrombin cleavage of secreted OPN releases two receptor binding domains which carry out distinct signaling functions.
Figure 6.
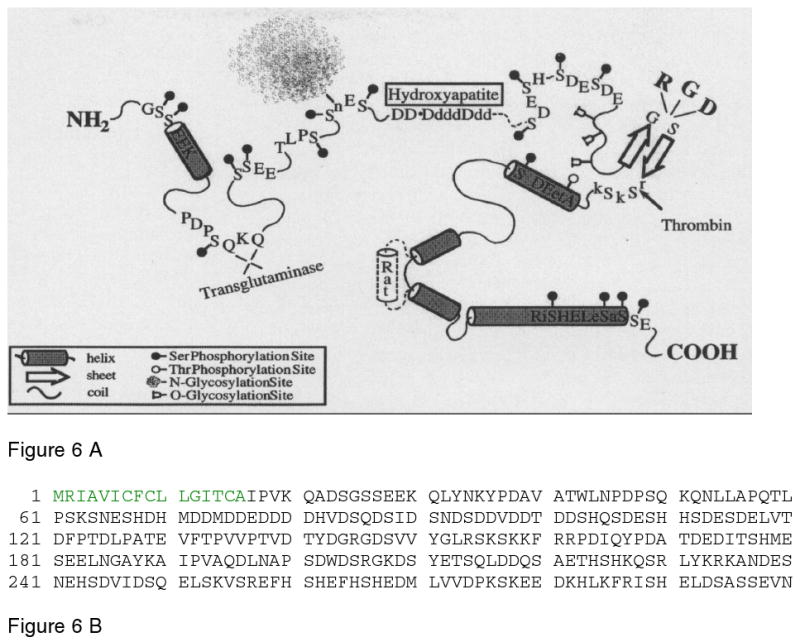
A. Schematic representation of the structural model for osteopontin adapted from Sodek et al.93. The model highlights the presence of serine and threonine phosphorylation sites; region containing the poly aspartic acid residue; glycosylation sites, thrombin cleavage site and the integrin binding domain. B. The sequence of human osteopontin, taken from Pub. Med. Protein data bank, ID AAA59974. The signal peptide sequence is in green. Reprinted with permission from Reference 93. Copyright 2000, International Association of Dental Research.
4.1.2 Localization of OPN during development
The OPN cDNAs from various mammalian species exhibit a high degree of sequence homology. In mineralizing tissues of rat, OPN has been shown by light microscopy immunostaining to be present in the osteoid, in mineralized bone matrix and in mineralizing cartilage115. In sections of the epiphyseal plate in tibiae, OPN was present in the mineralizing cartilage and in the primary and secondary spongiosa. In fetal porcine bone, expression was observed in osteoblastic cells and associated osteoid of newly-forming trabecular bone as well as in the mineralized matrix of calvarial bone116,117. Immunogold labeling experiments localized OPN with patches of diffuse, electron-dense material present between the collagen fibrils in the mineralized tissue and at sites of mineralization containing organic matrix in the osteoid93,118. In the tibiae, presence of OPN was reported to be observed on the surface of mineralized cartilage, and in the newly formed bone deposited on the surface of mineralized cartilage.
During development, OPN was expressed early on in developing bone and achieved maximum expression in 14 day old rats and declined rapidly thereafter. In the developing tooth, OPN has been shown to be distributed focally and not uniformly, however, it is expressed in a well regulated manner during tooth development119.
Although it was considered to be a principal component of bone, OPN has been found in non-mineralizing tissues such as kidney and mammary epithelium, which are involved in Ca2+ ion transport120,121.
4.1.3 OPN and collagen binding
Like other members of the SIBLING family OPN binds stably to collagen fibrils. A collagen binding motif (CBM) in OPN was identified by chymotrypsin digestion and comprised the peptide sequence from 150-177, GLRSKSKKFRRPDIQYPDATDEDITSHM. This sequence is highly conserved between species in human, mouse, rabbit, bovine and porcine122. Analysis of this sequence shows that it consists predominantly of positively charged amino acids such as Arg, Lys, His, as well as hydrophobic Met, Phe, Tyr, alternating with Asp. Amino acids such as Phe, Tyr and Arg are an essential component to create self-assembly. BIAcore technology was used to examine the binding kinetics of this peptide to collagen, and the binding constant, KD, was determined to be 5μM. Ionic interactions between the basic amino groups in CBM and the collagen fibrils might be responsible for the high affinity since the binding affinity decreased as the ionic concentration increased and when the pH was higher or lower than 5122.
4.1.4 OPN and Mineralization
OPN is involved in the regulation of both physiological and pathological mineralization 123,124,125. It is abundant in highly supersaturated physiological fluids, especially abundant in calcium oxalate containing kidney stones126. OPN is not normally expressed in blood vessels, but found in atherosclerotic plaque127,128. OPN is also expressed in soft tissues and probably functions to protect against dystrophic calcification129. This is further supported by the fact that calcification of vascular smooth muscle cells is inhibited by OPN130,131,132. Recent studies have shown that the function of OPN is dependent on the extent of post-translational modifications133. In-vitro experiments have demonstrated that OPN is a potent inhibitor of de novo HAP formation134,135,136,137. Analysis of the amino acid sequence in terms of sequences responsible for mineralization has identified conserved acidic domains such as an extended stretch of aspartic acid residues, DDDDDDDDD in the rat and DDEDDDD in humans63. There are also conserved domains in OPN in which Ser residues are adjacent to or embedded within acidic regions and could be phosphorylated by MGCK or CKII, e.g. in humans138: S8GS10S11EE, S45S46EE, S59NS61NES64HD, ES84DES87VDS90DES93DES96HHS99DES102. OPN molecules in different extracellular matrix tissues are differentially phosphorylated and dephosphorylated, therefore OPN phosphorylation regulates its interaction with HAP and OPN mediates HAP nucleation and growth137.
Based on published reports, phosphorylation of OPN appears to be vital for its ability to inhibit mineral growth139,140,141. Dephosphorylated OPN decreased its ability to inhibit HAP. In-vitro nucleation experiments with several synthetic peptides corresponding to the acidic domains in OPN concluded that the presence of phosphate groups, carboxylate groups and negative-charge density were all important components in inhibiting HAP formation 65,133,137,142,143. This was confirmed in the OPN-null mice which had a greater mineral content and presence of large mineral crystals144,145. The transglutaminase cross-linked bovine milk OPN was also an effective inhibitor of seeded growth. Neither the recombinant OPN which had no phosphorylation, nor the chemically dephosphorylated OPN affected de novo HAP formation. However, the highly phosphorylated milk OPN promoted HAP formation and growth in the in vitro gelatin-gel system, but at high concentrations (50-100μg/ml) HAP formation was inhibited. This anomalous behavior of OPN in milk was attributed to its function in stabilizing amorphous calcium phosphate (ACP)146. The ACP granules in milk are localized within micelles coated with casein, which would prevent the conversion of ACP to HAP65. Thus, in vivo as well as in vitro data suggested that OPN is a modulator of bone crystal maturation.
OPN is present in non-mineralizing tissues such as kidney and mammary epithelium which are involved in Ca 2+ ion transport. In these tissues, the acidic domains of OPN can bind tightly to hydroxyapatite surface and inhibit crystal growth.
FTIR (Fourier Transform Infrared Spectroscopy) experiments determined that highly phosphorylated OPN underwent a slight conformational change indicating that this change was brought about by phosphate interactions with the cationic charge on the HAP surface. Overall, the protein remained largely unstructured and these results were well corroborated with nuclear magnetic resonance (NMR) studies of the nonphosphorylated OPN which demonstrated that the protein had a flexible random coil structure in which no structural change was observed in the presence of calcium56,106,147.
The possible mechanism by which OPN inhibits HAP formation is by the adsorption of phosphate groups to the surface of HAP by replacing orthophosphate ions within the crystal lattice, thus sterically preventing further deposition of lattice ions148. The acidic amino acid groups further contribute to this inhibitory effect by producing an electrostatic repulsion of inorganic phosphate ions once the protein is adsorbed to the surface of the crystal. The high flexibility of the OPN structure might facilitate efficient binding to the HAP surface and inhibit mineral growth.
4.2 Bone sialoprotein (BSP)
BSP was the first and the most abundant (15% of the NCP) noncollagenous protein identified from the mineral compartment of bone. There is considerable homology among the BSPs from various species. With their mass of about 70 kDa, the BSPs are highly glycosylated proteins with a high sialic acid content52. Most of the carbohydrate moieties are present as O-linked oligosaccharides. The BSPs are also phosphoproteins, but not as highly phosphorylated as osteopontin. Human BSP is encoded by a single copy 15-kb gene located on chromosome 4q28-q31 57,105,149,150,151,152,153.
4.2.1 Structural features of BSP
The nascent BSP polypeptide has a size of 34 kDa and undergoes extensive post-translational modification151 (Fig. 7). Principal modifications are the glycosylation of the O- and N-linked sites, O-phosphorylation primarily at serines, and sulphation which occurs primarily at tyrosine – O-SO4. Analysis of the primary sequences for both human and rat demonstrate the paucity of hydrophobic amino acids153. The carboxyl terminal domain is particularly conserved among species and this region contains 14 tyrosines of which 9 are retained within the recognition sequences for tyrosine sulphation. The amino-terminus contains tyrosines and phenylalanines embedded within highly basic residues. It also contains 5 prolines which might render structural stability to the N-terminal region. The middle segment of the BSP molecule is rich in acidic amino acids and includes the two polyglutamic acid segments, and serines which have the potential to be phosphorylated. BSP contains an integrin binding RGD domain68,149,154.
Figure 7.
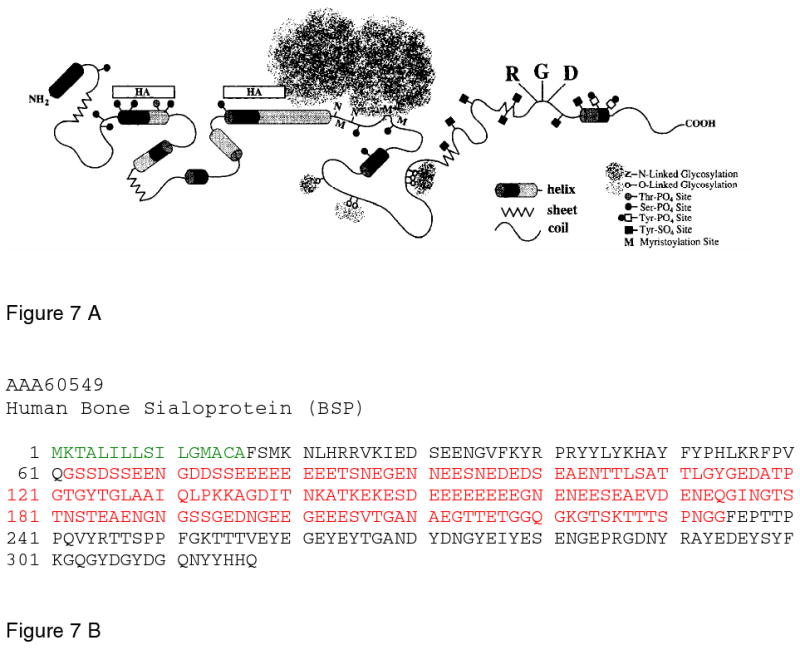
A. Schematic representation of the structural model for bone sialoprotein adapted from Ganss et al.185. The model highlights the highly flexible structure, the presence of an hydroxyapatite binding domain at the N-terminus, and O and N-linked glycosylation sites. The Integrin binding RGD domain is located at the carboxyl end of the molecule. B. The sequence of human BSP, taken from the Pub. Med Protein Data Bank, ID AAA60549. The signal sequence is in green, the very acidic, mineral bindng domain is in red. Reprinted with permission from Reference 185. Copyright 1999, International Association of Dental Research.
Mature human BSP has 301 amino acids that can be divided into 3 domains Figure 7 B. The amino (residues 1-45) and carboxyl (residues 219-301) domain contain the majority of the bulky aromatic groups in the protein. The central domain (residues 46-218) contains three acidic-rich sequences; each characterized by polyglutamic acid repeats (residues 52-87, 130-157 and 179-188. These repeats are highly conserved among mammalian BSPs. Interestingly, the two largest acidic domains are separated by a hydrophobic cluster. Overall, it is a charged molecule with the amino terminal domain comparatively rich in basic residues, while the carboxyl domain is mostly acidic155.
Analysis of the structure by one-dimensional NMR has indicated that BSP in solution is unstructured and has a flexible conformation56,156. Circular dichroism spectra of BSP revealed a low proportion of alpha helix and β-sheet elements and a high content of random coil structure, however it can acquire appropriate structure upon binding to specific ligands. Wuttke et al.157 demonstrated that recombinant BSP could adopt a globular structure linked to a thread-like structure of total length of 35 ± 7 nm by rotary shadowing techniques. As the C-terminus of the protein was devoid of glycans they suggested that the C-terminus formed a globular structure while the highly glycosylated N-terminus adopted the thread like structure due to the lack of secondary structure. Sulfation of tyrosines normally occurs in β-turn regions adjacent to acidic residues.
4.2.2 localization of BSP in mineralized tissue
BSP transcripts are predominantly expressed at high levels by differentiated osteoblasts involved in de novo bone formation and at more modest levels by odontoblasts, cementoblasts and hypertrophic cartilage cells158,159,160,161. Localization on de novo bone formation indicated a specific role in the initial mineralization of the collagenous matrix. However, BSP message levels were shown to decrease rapidly with age. Immunohistochemical analysis localized the protein to the sites of mineralization in the osteoid in rats, with cellular staining limited to the periphery of the osteoblasts and in mineralizing cartilage160,161,162,163,164. Pulse-chase experiments of bone formation by fetal calvarial cells in vitro indicated that most of the BSP bound rapidly to the mineral but a significant portion bound to the collagenous matrix165. Immunogold localization of BSP revealed that the gold particles were associated with amorphous, electron-dense material in the mineralized matrix of the tibiae166,167. However, in the osteoid, label was localized within the foci of mineralization containing organic crystallites.
Based on its tissue distribution, BSP has been proposed to function as an apatite nucleator as it is found at the mineralizing boundaries of bone, dentin and calcifying cartilage tissues, colocalizing with the smallest detectable foci of newly forming mineralized matrix in osteoid. BSP is expressed prior to and during the active mineralization periods in osteoblastic cultures. BSP has also been implicated as a key molecule in the formation and remodeling of bone133,168,169,170.
4.2.3 Post-translational modifications
Post-translational modifications account for 50% of the mass of BSP. Molecular weights for BSP have been reported in the range from 52,000-82,000 Da. This heterogeneity of the apparent size has been attributed to the extensive and heterogeneous post-translational modifications, particularly the variations in the N- and O-glycan content171. The numerous phosphorylated serine and threonine residues, the sialylated N and O-linked oligosaccharides and the sulfated tyrosine residues172 all contribute to the anionic nature of BSP.
It has been suggested that the post-translational modifications, especially the phosphorylation of BSP may be important for mineralization and bone resorption. The large numbers of covalently bound phosphates on BSP have been implicated in such biological functions. The presence of phosphate groups on BSP led to the speculation that phosphorylation may be regulating mechanism for hydroxyapatite nucleation78,173. The in vivo phosphorylation sites were determined by derivatization and proteolytic digestion of BSP, followed by MALDI-TOF-MS analysis of the tryptic peptides. These studies confirmed that there were approximately 5.8 mol P-Ser/mol of BSP66,173,174. There was no overlap between the phosphorylation and glycosylation sites. The major kinases responsible for phosphorylation in vivo were found to be CKII (4.1 mol), protein kinase C (0.9mol) and cGMP-dependent kinase (0.8mol). Phosphorylated residues (as indicated schematically in Fig. 7, were predominantly on the N-terminal half of the protein,109,111,113,175. In mature bovine BSP, 11 in-vivo phosphorylation sites were identified, namely in the following peptide sequences (numbering beginning from the N-terminal residue after cleavage of the signal peptide)12-22 (LEDSEENGVFK), 42-62 (FAVQSSSDSSEENGNGDSS(P)EE), 80-91 (EDSDENEDEESE), 125-130 (AGAT)GK, 135-145 (EDESDEEEEEE), 250-264 (DNGYEIYESENGDPR), 282-289 (GYDSYDGQ), where the phosphorylated residues are indicated in red. It is interesting to note that not all the molecules are phosphorylated at each of the potential phosphorylation sites within a given population of molecules isolated from bone of a specific age. Bovine BSP was about 65% phosphorylated on average, and the phosphate groups were covalently bound to BSP Ser or Thr residues. This heterogeneity in phosphorylation can be attributed to the rate of phosphoprotein synthesis, the microsomal CKII activity, the available ATP concentration in the intracellular compartment and, to some extent, the possibility of dephosphorylation during the residence of BSP in the ECM. The activity and concentrations of these components vary with the developmental stage, maturation and age of bone. Interestingly, the glycosylation and phosphorylation sites do not overlap, and the glycosylation sites are concentrated in the mid-portion of the protein as shown in Fig. 7.
In vitro phosphorylation studies have identified microsomal casein kinase II as the principle protein kinase. Based on consensus sequences in the primary structure of mammalian BSP, there are 5 potential sites for serine phosphorylation by protein kinase C, 9-11 sites for Ser/Thr phosphorylation by casein kinase II, and 1-2 sites for Tyrosine phosphorylation by tyrosine kinase. Salih et al. have reported that bovine BSP can incorporate 5.8 phosphates into the protein, predominantly by casein kinase II173.
4.2.4 Interaction with collagen
In vitro studies demonstrated that BSP has an affinity to type I collagen 176, satisfying the requirement of the matrix-mediated mineralization model for a direct BSP interaction with the collagen template. The binding capacities of BSP to type I collagen vary depending on pH and ionic strength of the solution used for the binding assay. Binding of BSP to type I collagen involves long range electrostatic interactions that aid in the formation of an initial low affinity complex, prior to the formation of a high affinity complex characterized by specific short range interactions177. The sequence comprised of residues 19-46 of mature rat BSP, highly conserved in mouse, cow, pig and human, NGVFKYRPRYFLYKHAYFYPPLKRFPVQ, has been identified as the specific collagen binding domain. This region contains positively charged Lys, Arg and His residues along with hydrophobic Pro, Tyr and Phe residues. The anionic charged phosphate and carboxylate residues surrounding the collagen binding domain facilitates long range collagen binding electrostatic interactions177. An in vitro study of the binding of collagen CNBr peptides to BSP176 has shown that BSP may bind specifically to the α2 chain of type I collagen. Further, immunogold labeling studies of anti-BSP with collagen fibrils showed binding of BSP at the hole zones of the collagen fibrils. These data all suggest that BSP-mediated mineralization should regulate hydroxyapatite nucleation by interaction with collagen within the exposed α2 chain surface within hole zone region of the collagen fibril, compatible with the model shown in Figure 2 and the suggestions of chain accessibility proposed by Orgel et al.19
4.2.5 BSP and mineralization
The role of BSP in biomineralization has been implicated by its temporal deposition into the ECM during development of the mineralizing tissues, its high affinity for hydroxyapatite and its function as a de-novo nucleator of hydroxyapatite crystals in–vitro178. Particularly, control of mineral deposition by BSP is intimately associated with the state of phosphorylation of BSP; the unique, extended glutamic acid –rich regions; sulfated groups on tyrosines contribute to the anionic nature of BSP; glycosylation of BSP also influence hydroxyapatite binding. Thus a synergistic activity between the charged acidic groups is responsible for calcium phosphate nucleation and binding to hydroxyapatite133,179,180,181.
Bovine BSP contains two particularly glutamic acid-rich domains DSSEENGNGDSSEEEEEEEETS) and (EDESDEEEEEEEEEE)182. The first domain is located in the N-terminus and flanked by six phosphoserines (Fig. 7). The second domain is located in the C-terminus and flanked by a phosphoserine. These phosphoserines combined with the polyglutamic acid domains probably play a role in the interaction with Ca2+ ions and binding to HAP. Post-translationally modified BSP contains phosphate, sulfate and sialic acid group contributing to the high affinity for HAP. In vitro nucleation studies using porcine recombinant BSP concluded that the N-terminus polyglutamic acid domain promotes HAP nucleation, while the C-terminus promotes nucleation only in its native state. Studies using rat recombinant BSP demonstrated that it was less potent than the native protein, emphasizing the importance of the post-translational modifications in HAP nucleation. Tye et al. performed site directed mutagenesis of the poly [E] regions with either poly [D] or poly [A]. Results demonstrated that replacement of single domains with either poly [A] or poly [D] did not alter nucleating activity. Replacement of both domains with anionic poly [D] also did not ablate nucleating activity, although replacement of both domains with poly[A] significantly decreased nucleating activity183. The conclusion from these studies was that a sequence of at least eight contiguous glutamic acid residues is required for the nucleation of HAP by BSP184. The cluster of phosphate groups in the central domain of BSP may influence the HAP binding activity of two of the polyglutamic acid repeats. Even though BSP is a phosphorylated protein, however, there are fewer phosphates than sulfates. Co-operation between the phosphate, sulphate and carboxyl side chains provide BSP with a high negative charge. These negative charges are responsible for avid binding of BSP to hydroxyapatite134,179,181,185,186.
4.3 Matrix extracellular phosphoglycoprotein (MEPE)
MEPE is a secreted 45-kDa phospho-glycoprotein that is highly charged, rich in potential serine phosphorylation sites and contains an RGD domain essential for integrin receptor interactions187. MEPE was isolated and cloned from a TIO [tumor-induced osteomalacia, also called oncogenic osteomalacia (OOM)] tumor cDNA library 188. TIO is an acquired hypophosphatemic disorder. Therefore, MEPE was thought to be a candidate for phosphatonin, the phosphaturic factor secreted by tumors that cause osteomalacia. However MEPE deficient mice have no abnormalities of serum phosphate concentrations, therefore, the role of MEPE in serum phosphate homeostasis is controversial189. The rat and mouse homologs of MEPE were isolated on the basis of their ability to regulate mineralization. The human MEPE gene is clustered in tandem with BSP and OPN on chromosome 4q21, and encodes a 525 amino acid protein. The mouse and rat MEPE proteins are 433 amino acids and 435 amino acid residues and have a 50% homology to the human MEPE.
4.3.1 Structural motifs in MEPE
MEPE appears to be a multifunctional protein with distinct domains capable of influencing the process of extracellular matrix mineralization and cell attachment (Fig. 8). The central portion of the molecule termed “dentonin” (Residues 241-263) containing the sequence TDLQERGDNDISPFSGDGQPFKD has been shown to stimulate the proliferation of human bone marrow stromal cells and their differentiation into osteoblast precursors190,191,192. It could stimulate new bone formation both in vitro and in vivo. Recently, dentonin was shown to induce cell proliferation and recruit progenitor cells during pulp repair190.
Figure 8.
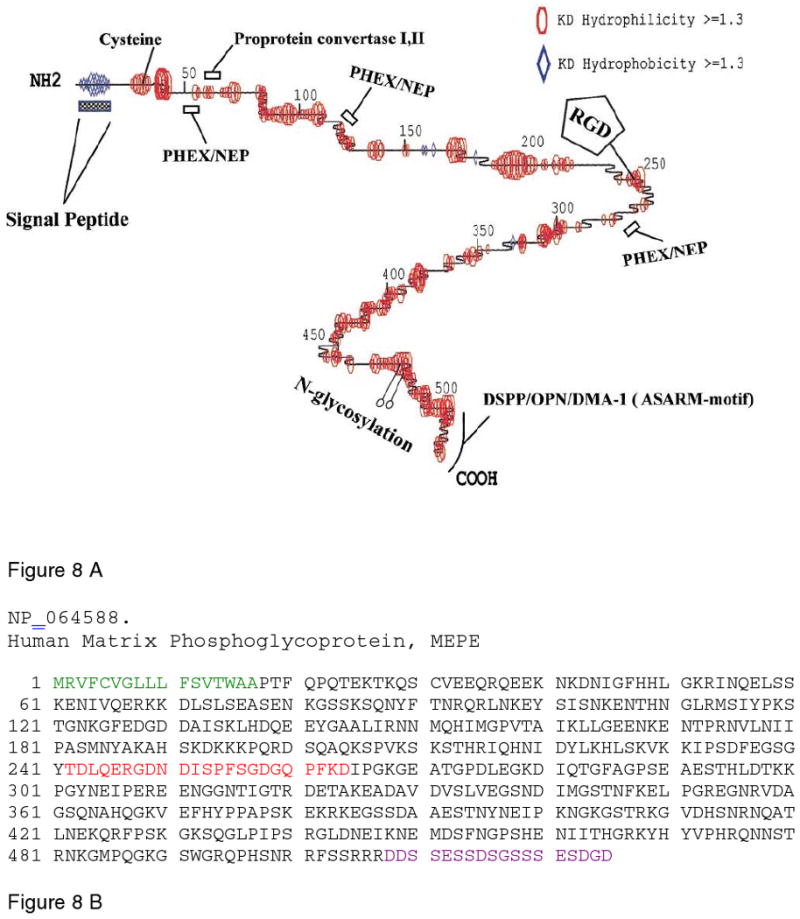
A. Schematic representation of the structural model for MEPE, depicting the highly flexible nature of the protein backbone. The model shows several PHEX cleavage sites and the hydroxyapatite inhibiting ASARM motif at the carboxyl end of the molecule. Adapted from Rowe et al.188. B. The sequence of human MEPE, taken from Pub. Med. Protein Data Bank, ID NP_064588. The signal peptide sequence is in green, the cell-proliferation stimulatory sequence is in red, and the C-terminal ASARM peptide is in purple. Reprinted with permission from Reference 188. Copyright 2000, Elsevier Limited.
Homologues of MEPE share conserved motifs, including a signal peptide, N-glycosylation sites, an SDGD glycosaminoglycan-attachment site, and an RGD cell attachment domain. A characteristic domain present at the C-terminal end is the “acidic - serine-aspartate-rich” (ASARM193) region, the last 18 amino acids (Fig.ure 8B), which functions to inhibit mineralization.
4.3.2 Localization of MEPE
MEPE is expressed in bone and teeth and like the other matrix proteins has a temporal maturation-dependent expression in osteoblast cultures, and is highly expressed within the osteocytes that are embedded within the mineralized matrix194,195. In bone, MEPE is expressed during proliferation and early-maturation by fully differentiated osteoblasts with maximum expression during mineralization196. MEPE is highly expressed in tumor-induced osteomalacia in Hyp mice, a murine model for X-linked hypophosphatemia, and in human pathology188. MEPE is also expressed at low levels in non-mineralized tissues such as brain, lung, human placenta, salivary gland and in the proximal tubules of the adult kidney197 In the developing tooth, MEPE is expressed during odontogenesis in odontoblasts, but is down-regulated as dental pulp stem cells differentiate198.
4.3.3 Role of MEPE in mineralization
MEPE inhibits phosphate uptake and mineralization in vivo and in vitro193. Observations from the MEPE deficient mice provided an insight into the role of MEPE during mineralization. In these mice, increased bone mass was observed due to increased osteoblast activity resulting from an increased osteoblast number, however, the osteoclast number was unaltered189. In vitro cell culture of the osteoblasts derived from these knock-out animals produce significantly higher mineralized nodules when compared with the wild type. Inhibition of mineralization was localized to a small (2 kDa) cathepsin B released carboxyl-terminal MEPE peptide (protease-resistant) containing the acidic serine-aspartate-rich motif (ASARM peptide). Based on these results the function of MEPE might be to inhibit mineralization, hence termed “minhibin”193. Cathepsin B cleavage sites have been identified in MEPE, and cathepsin B has been shown to cleave MEPE in vitro releasing the ASARM peptide. This peptide may also inhibit phosphate uptake199.
4.3.4 Role of ASARM peptide in mineralization
Phosphorous is an important component of hydroxyapatite. Deficiency of phosphorous and inorganic phosphate results in impaired mineralization of bone resulting in osteomalacia or rickets. The amount of nonmineralized matrix is significantly increased in osteomalacia. MEPE inhibits phosphate uptake and mineralization in vivo and in vitro. The ASARM peptide is the functional domain responsible for phosphate and calcium regulation during mineralization process200. As noted above, in vitro inhibition of mineralization is mediated by the short 2 kDa cathepsin B-released carboxyl-terminal ASARM peptide, which is protease resistant. This peptide might inhibit phosphate uptake. Large quantities of the ASARM peptide (SSRRRDDSSESSDSGSSSESDGD) have been found to accumulate in HYP (hypophosphatemia) mice as a consequence of loss of PHEX, a proteolytic enzyme. Under normal conditions, PHEX prevents proteolysis of MEPE and release of protease-resistant ASARM peptide. This peptide has been implicated in inhibiting calcification. Physiochemical properties suggest that the released ASARM peptide is highly acidic, with a pI of 2.3 and a net charge of -6.9 at pH 7. There are three casein kinase sites that could result in phosphorylation of three serine residues and further increase the negativity of the molecule. The kinases responsible for phosphorylation of MEPE are PKC, tyrosine kinase, and cAMP dependent kinases. The proposed mechanism by which ASARM peptide inhibits mineralization is by sequestering free calcium with high avidity and inhibit mineralization193,199,200,201,202,203. The proposed function of ASARM peptide is consistent with the phenotype observed with MEPE knock-out mouse model which resulted in increased bone formation and bone mass. Like DPP, the ASARM peptide is resistant to most proteases, is phosphorylated, with a low pI, high charge and low molecular weight and is responsible for the mineralization related function of MEPE201.
4.4 Dentin Matrix Protein 1 (DMP1)
Dentin Matrix Protein 1 (DMP1) initially named AG1 was the first of the acidic proteins cloned from the dentin matrix76 DMP1 is remarkably hydrophilic. Although initially isolated from the dentin matrix, and thought to be unique to dentin and named accordingly, it has now been found to be present in all mineralized tissues of the vertebrate system. The localization of the DMP1 gene to human chromosome 4q21204,205 was the first indication that this region might represent a “mineralized tissue” gene locus, borne out by all of the subsequent studies on the related SIBLING proteins.
4.4.1 Salient features of DMP1
Analysis of the protein sequence of DMP1 showed three important facts: 1) except for a short-classical hydrophobic signal sequence of 16 residues and a very short domain at the COOH-terminal end, the entire 473 residues of the secreted protein is hydrophilic and does not show any preferred domains of β-sheet or α-helix; 2) DMP1 has a high content of glutamic acid and aspartic acid residues and the acidic nature of the molecule is markedly enhanced when the potential for phosphorylation of serine and threonine residues is considered. These acidic domains are interspersed throughout the molecule with two or more consecutive Asp (D) or Glu (E) residues, and these domains frequently also contain one or more Ser (S); 3) it appears that 55 of the 107 serine residues in the secreted DMP1 could be phosphorylated. If this were the case and with the Ser(P) dissociation constant at pKa 6.8, DMP1 would carry a charge of -88 from the phosphate groups alone and a net charge of -175/molecule at physiological pH. Such a molecule would have a very high capacity for binding divalent cations such as calcium. Of the total of 107 S residues, 55 are in regions which are consensus sequences for post-translational modification to phosphoserine by messenger independent casein kinases76,206. Other post-translational modifications that could occur are O-glycosylation at several sites and N-glycosylation at a single site. There is one RGD sequence, a potential cell-surface integrin binding sequence207. In vivo DMP1 is cleaved to a 57 kDa C-terminal polypeptide and a 37 kDa N-terminal polypeptide208. The C-terminal polypeptide contains both the HAP and collagen binding domains.
4.4.2 Localization of DMP1 in bones and teeth
RNA expression analysis on tissue sections by in-situ hybridization demonstrated that DMP1 undergoes tight transcriptional regulation during tooth and bone development209. The earliest expression analysis of mouse DMP1 mRNA in mineralized tissues was in the osteoblasts of the alveolar bone and calvaria at E18 (E = embryonic day) and was continuously expressed during development210,211. In mature bone, DMP1 was expressed constitutively, as bone undergoes constant remodeling209,212.
In the developing dentin, the message was first observed at E20 in the young secretory odontoblasts of the incisors. In a typical developing molar, the expression of DMP1 is restricted to early differentiating odontoblasts and polarized odontoblasts. As the initial matrix mineralizes, there is down regulation of the DMP1 message and it completely disappears when dentin formation is complete213. The timing and the quantitative expression level of DMP1 mRNA depends upon the appropriate tissue-specific regulatory factors that act on the DMP1 gene214.
4.4.3 Structure of DMP1
Analysis of the protein sequence of DMP1 (Fig. 9) showed that the secreted protein adopts a random structure. Circular dichroism analysis has shown that recombinant DMP1 exists as a random coil with no distinct fold215,216. However, once calcium binds, the C-terminal domain adopts a β-sheet secondary structure and gradually self-assembles into oligomers and microfibrils. To gain further insight into the molecular shape and self-assembly of DMP1 in the presence of calcium, low-resolution protein structures were generated by the ab-initio program GASBOR using the data from the small angle scattering X-ray experiments217 (Fig 9a). The low resolution models depicts the molecular shape of DMP1 as an elongated molecule, however, in the presence of calcium, the conformation depicted an extended structure at one end of the molecule with a compact globular domain at the other. With further calcium binding, DMP1 was shown to undergo extensive dimerization. This was facilitated through the C-terminal domain, yielding a symmetric two-domain structure. The low-resolution model obtained in the presence of calcium depicts a dumb bell-shaped structure with two elongated C-terminal domains wound together and the globular N-termini located at both ends217. The dimerization may be initiated by conformational changes from a random coil to β-sheet structures and are likely to be formed by intermolecular assembly of two C-terminal domains. Presumably, binding of calcium to phosphate and carboxyl groups on DMP1 weakens the mutual repulsion of negatively charged domains and permits formation of β-sheets. Thus, the oligomerization property of DMP1 might be essential for the formation of ordered templates for binding calcium phosphate nanoparticles216.
Figure 9.
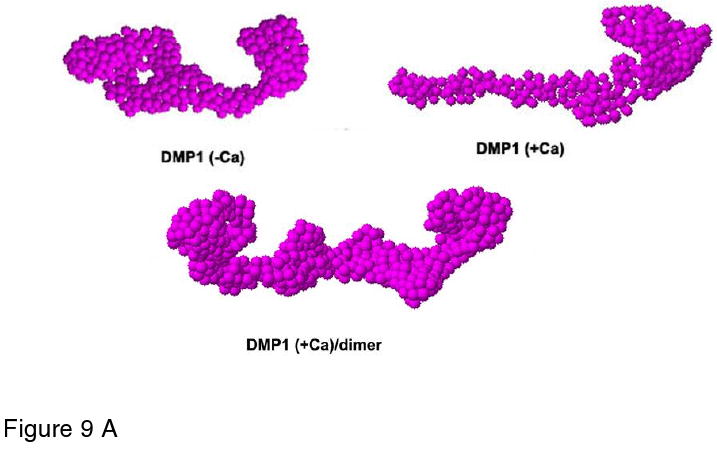

A. Structural analysis of DMP1 as deduced from small angle X-ray scattering experiments. Low resolution models of DMP1 determined using the ab initio program GASBOR depicting the structure in the absence and presence of calcium. In the absence of calcium DMP1 adopts an elongated structure. Within 1 minute of calcium binding DMP1 adopts an extended structure at one end of the molecule with a compact globular After incubation for 3 minutes DMP1 undergoes extensive dimerization. Reprinted with permission from Reference 217. Copyright 2005, American Chemical Society.
B. Proposed model for the dual functional role of DMP1. Model demonstrates that DMP1 can inhibit spontaneous mineral precipitation and promote controlled mineral nucleation on a collagenous template. Reprinted with permission from Reference 217. Copyright 2005, American Chemical Society.
C. The sequence of rat DMP1 taken from Pub. Med. Protein Data Bank, ID NP_987089. The signal peptide sequence is in green, the mineral binding domains are in red, and the cell attachment domains are in purple.
4.4.4 DMP1 and collagen binding
One of the requirements for DMP1-mediated spatial regulation of matrix-mediated mineralization is that specific collagen fibril-DMP1 interaction is necessary for mineral induction. He et al. have demonstrated that rat DMP1 interacts with type I collagen with a KD of 3.8 ± 1.7 μM218. Two peptide clusters at the C-terminal end of DMP1 349DSESSEEDR357 and 424SEENRDSDSQDSSR437 were identified as collagen – interactive peptides by peptide mapping218. These binding domains contain clusters of acidic residues, which suggest that the binding between DMP1 and collagen are driven by electrostatic interactions. Such strong binding of DMP1 on the collagen substrate might aid DMP1 to adopt an appropriate conformation conducive for mineral induction. When DMP1-collagen complex was incubated in a pseudo-physiological buffer and tested for its mineralization property in vitro, it was observed that only DMP1-collagen complex initiated biomineral deposition on the collagen surface, whereas there was no calcium phosphate deposition on the controlled collagen surface. On the collagen fibril, the N-telopeptide region was identified as the DMP1 binding site. The N-telopeptide region of the α1(I) chain consists of 16 amino acids, which have been highly conserved in a variety of species. This agrees well with the neutron scattering studies of calcifying collagen in turkey leg tendons which showed an initial nucleation site located toward the N-terminus of the gap region, which led to the axial propagation of the mineral deposit along the gap region31.
4.4.5 DMP1 and mineral nucleation
The recombinant and non-phosphorylated form of DMP1 can nucleate the formation of HAP in-vitro in a multi-step process that begins by DMP1 binding calcium ions and initiating mineral deposition215,216,219. A remarkable feature observed by SEM was that that the HAP mediated by DMP1 grew by a sequential stepwise process216. During the nucleation and growth process, the intermediates had highly ordered structures. Formation of nanometer-sized amorphous calcium phosphate was the initial precursor phase which then transformed into c-axis oriented hydroxyapatite crystals. After the formation of the initial amorphous phase, structural rearrangement of the dispersed ionic cluster then takes place into more ordered structures dictated by matrix interactions. Two acidic peptides at the C-terminal domain, namely 386ESQES390 and 415QESQSEQDS422 were shown to play a precise molecular role in mineral surface recognition216. Circular dichroism studies showed that these two peptides did form an intermolecular β-sheet structure in the presence of Ca2+. Intra- or intermolecular assembly of these peptides might provide the localized structured surface for c-axis-oriented calcium phosphate crystal growth216.
In the presence of type I collagen and recombinant DMP1, needle shaped HAP crystals with high crystallinity were obtained220. Post-translationally modified native C-terminal DMP1 comprising residues 170-489 also promoted HAP nucleation and growth. It appears that the charged residues namely serine and glutamic acid present at the C-terminal end of DMP1 are spatially organized in a form that specifically complements the crystallography of the calcium phosphate nuclei220. Interestingly, the N-terminal domain of DMP1 can stabilize the amorphous phase220. The high charge density created by the aspartic acid residues at the N-terminus of DMP1 can bind calcium ions very strongly favoring formation and stabilization of the amorphous nuclei. The stabilization of amorphous precursors during the growth phase is important in biomineralization because transient amorphous minerals play an important role in many organisms. Transition of amorphous phase to crystalline phase is carried out in a controlled manner by organisms. However, amorphous calcium phosphates may play a significant role in pathological calcifications. Thus, matrix proteins like DMP1 are responsible for regulating the transformation of amorphous calcium phosphate to crystalline HAP as well as stabilizing the ACP phase when required.
In solution, DMP1 could sequester and stabilize newly formed calcium phosphate clusters from supersaturated solution. DMP1 could facilitate this stabilization by forming a structured template with the presence of a hydrophilic surface comprised of negatively charged aspartic acid and glutamic acid residues at the C-terminus217. Such an arrangement facilitates sequestration of calcium phosphate nuclei present in a supersaturated solution and forms fibrils of the protein-mineral complex. Therefore, this negatively charged fibril forms a stable structure by binding with calcium phosphate nanoparticles and protects the nascent mineral nuclei from further growth and precipitation. Specific recognition between DMP1 and the apatite surface might be necessary for the molecular construction of discrete nanoparticle precursors. Such a molecular recognition mechanism could aid in the controlled growth of the nuclei with crystallographic orientation that could play a vital role in controlled bone and dentin calcification. Thus, DMP1 could serve dual roles during bone and dentin formation: crystal growth inhibition and prevention of further calcium phosphate nucleus growth by stabilizing these nanoclusters and at the same time promoting controlled mineral nucleation when immobilized specifically on a self-assembled collagen template (Fig 5b)
Decreased mineral content and increased crystal size in DMP1 knock-out animals demonstrate that depletion of DMP1 in vivo decreases bone mineralization and reveal the importance of DMP1 in regulating mineralization221.
4.5 Dentin Phosphophoryn (DPP, DMP2)
A key interactive protein of the dentin matrix is the acidic Asp-Ser-rich protein known as dentin phosphophoryn (DPP)70,222,223,224. The molecule is called thus as it is considered to be a “phosphate carrier”. DPP as isolated from dentin has a unique composition with aspartyl and seryl residues comprising at least 75% of the amino acid residues and with 85-90% of the seryl residues phosphorylated225,226,227,228,229,230,231,232. Characterization of this protein has proved difficult due to the high degree of phosphorylation, extreme negative charge (pI 1.1) and redundant amino acid composition233. DPP has been considered to be the archetype of macromolecules which might regulate the biomineralization process as it is highly and evenly negatively charged.
4.5.1 Structure and post-translational modification of DPP
A very important structural feature is the presence of (DSS)n [Asp-Ser-Ser] repeats interspersed throughout the molecule (Fig. 4). SD (Ser-Asp) domains are more prominant at the C-terminal end of the molecule77. Conformational analysis suggested that the phosphorylated form of the DS*S* (S* represents phosphorylated serine) repeat sequences assume a unique structure with well-defined ridges of phosphates and carboxylates available for counter ion binding77,234 (Fig 10 a). The principal modification is that most of the serine groups in both the DSS and SD domains are phosphorylated. Phosphorylation of all potential serine residues results in a highly negatively charged molecule that can be considered as a virtual sink for the binding of calcium ions. Native extracted DPP, which is not completely phosphorylated, could be readily further phosphorylated by the casein kinases235,236. Analysis of the DPP sequences showed that there are many consensus sites for CKII phosphorylation and that phosphorylation of DPP could theoretically proceed in a hierarchical fashion. That is, initial phosphorylation at the carboxyl end of the molecule could cause the entire remaining sets of SS pairs within the DSS triplets to become secondary substrates for both CKI and CKII so that the molecule could achieve the very high degree of phosphorylation237 observed. However, it has been the experience in our laboratories that DPP produced recombinantly in bacteria can be phosphorylated by CKI and CKII only to about 10 % of the expected total. The kinases catalyzing the in vivo phosphorylation of DPP appear to be localized to the endoplasmic reticulum, microsomal compartment, and the kinases extracted from the endoplasmic reticulum do phosphorylate recombinant DPP238,239. Thus, just as milk OPN is mainly phosphorylated with the specialized mammary gland MGCK, it appears that a specialized membrane associated kinase is utilized for DPP phosphorylation in vivo240. Although it was argued from odontoblast culture studies that DPP phosphorylation might take place in the extracellular matrix after secretion of nonphosphorylated DPP protein241,242,243 this is unlikely to take place in vivo. This area requires further detailed study.
Figure 10.
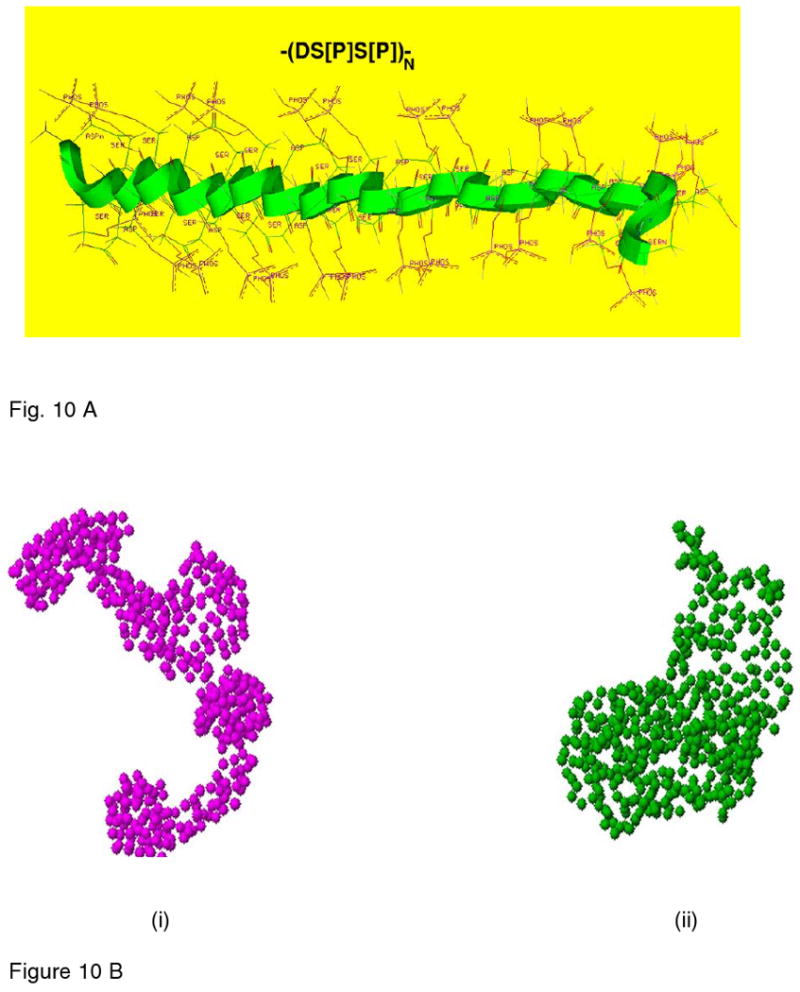
A. Molecular Modeling of the DSS domain by energy minimization. Molecular modeling shows the energy minimized lateral view of the structure showing the sommewhat extended folding of the backbone and the relative dispositions of the phosphate and the carboxyl groups relative to the backbone. A notable feature is the puckering or the mobile nature of the backbone which thus provides maximum separation between like-charged ionic groups consistent with the electrostatic repulsions and the creation of specific phosphate-carboxylate surfaces.
B. Structural analysis of DPP as deduced from SAXS experiments. Low resolution structural models were determined using the GASBOR program. (i) Model generated in the absence of calcium ion. (ii) Model generated in the presence of calcium. Reprinted with permission from Reference 255. Copyright 2005 S. Krager AG, Basel.
NMR study of bovine DPP depicted the molecule as having regions of differing mobility244,245 The mobile regions represented “hinge regions” that interrupted the longer stretches of more rigid polyanionic sequences. As noted earlier, however, even with the electrostatically induced increased persistence length along the polypeptide backbone, the entire protein backbone still acts as a random flexible chain. The binding of calcium ions may result in more rigid β-sheet structures which could serve as local templates for oriented crystal growth228.
4.5.2 Expression of DPP
Autoradiographic and immunohistochemical studies have shown that dentin phosphophoryn is secreted directly into the mineralizing dentin extracellular matrix, at the region of the mineralization front, through the odontoblast cellular processes210,246,247,248,249. The predentin matrix is free of DPP. At mouse developmental stage E20, with the differentiation of preodontoblasts to odontoblasts, abundant expression of DPP was observed in the fully polarized odontoblasts. DPP appeared as well in the undifferentiated epithelial ameloblasts at the cervical loop250. Expression of DPP disappears with the polarization of the ameloblasts, while abundant expression of DPP continues in the polarized odontoblasts. The antibody to DPP detected the expression of DPP in a number of early developmental stages in the epithelial tissues of the tooth bud, before any expression was detectable in the mesenchyme destined to become the dentin. Similarly, in other systems developing by reciprocal epithelial-mesenchymal signaling pathways, hair follicles, lung alveoli and kidney ureteric buds DPP could be detected, as early as E11 in mouse embryos. These data251 suggest the possibility that DPP may play a regulatory role in development, as well as a structural role in promoting mineralization. Another interesting feature is that the osteoblasts of the surrounding alveolar bone of post-natal 20-day-old mice expressed DPP mRNA, suggesting that this protein might also be involved in regulating later stages of osteogenesis.
4.5.3 DPP and collagen interaction
An important function attributed to DPP is its in vivo collagen binding capacity252,253. A significant fraction of DPP is cross-linked to dentin collagen in vivo75. When native DPP in solution, at low concentration, interacts in vitro with fibrillar collagen it localizes near the e-band in the hole zone of the collagen253 but at higher DPP concentrations it covers many sites on the fibrils by non-specific binding. It is difficult to expose the DPP bound to dentin collagen without etching away the surface mineral, and possibly removing some noncovalently bound DPP. However, when a careful etching was attempted246, cross-striated collagen fibrils could be seen at the mineralization front in rat incisor dentin just as they became overtaken and buried in the mineral phase. Anti-DPP labeled with gold particles could be detected on these fibrils at their junction with the mineral just before entry into the mineral. The collagen fibrils deeper into to nonmineralized predentin were not stained at all, while partial etching removal of the mineral exposed many collagen fibril-associated anti-DPP reactive sites, in about a D-periodic array. Unfortunately, the gold-conjugated anti-DPP antibody had a large footprint and it was not possible to locate the DPP precisely in relation to the collagen band pattern, except that it was in the gap-region. More deeply into the mineralized dentin, DPP was detected both in direct relation to the collagen, and without a direct association, as if the DPP had also bound to crystal surfaces246.
In in vitro collagen thermal fibrillogenesis experiments, added native DPP interacted avidly and caused rapid aggregation of the forming fibrils, resulting in asymmetric aggregates with no visible collagen D-periodicity254,255. With non-phosphorylated recombinant DPP [rDPP], the lag phase was dramatically shortened, and there was rapid growth of much longer and thinner fibrils than was the case with extracted DPP. As shown by atomic force microscopy (AFM) and TEM the thin filaments had the native collagen periodicity. The thin filaments increased in diameter with time as the rDPP induced further aggregation. These experiments suggested that in the absence of phosphorylation, rDPP bound to the monomer collagens acted to electrostatically cross-link the monomers sparsely and enhance end-to-end filament growth. The increased molecular length increased the rate of fibril nucleation and filament growth in the axial direction, but did not impair the molecular D-stagger ordering. Thus, just as the acidic glutamic acid-rich sequences in BSP, OPN and DMP1 induce collagen binding, the high content of Asp residues in rDPP are also effective.
The binding of monomeric rat DPP with monomeric type I collagen in vitro was followed by rotary shadowing TEM. At a low DPP/collagen molecular ratio, it was possible to see preferential binding at a single interaction site, approximately 210 nm from the N-terminus between collagen α-chain residues 680 and 750 where the sequence contains both positive and negative charged residues. This chain region on a single molecule appears in a D-staggered fibril at a position corresponding to the ‘c’ band of the fibril at (see Figure 3a) where the gap zone ends252. This is a very interesting region within a fibril, lying adjacent to the cross-link between the amino-telopeptide donor, K9N and the helix region cross link acceptor at position K932. This is not the position of the e-band.
In the physiological situation, as readily seen in electron micrographs246 the dentin collagen fibrils assemble and form fibrils before DPP is deposited upon them, therefore, it is unlikely that DPP plays any role in the initial fibril formation. However, electron microscopic analyses showed that the banding patterns of collagen fibrils varied in the mineralization front from that in the predentin zone256. In another study, it was determined that bovine DPP is strongly associated with a peptide derived from α2 Type I collagen75 in a peptide domain exposed at the gap zone. It would appear that the distribution of interacting groups from several collagen molecules in a D-periodic fibril might be responsible for creating the region of strong affinity for DPP. The phosphorylated DPP does not diminish in its capacity to bind calcium ions25,26.
4.5.4 DPP and Mineral Nucleation
The previous discussion makes it clear that the post-translational phosphorylation of DPP, carried out intracellularly and facilitated by a microsomal casein kinase II256 is important to its function in inducing the ordered nucleation and growth of hydroxyapatite. Computer-generated models77 of the DSS repeat domain in DPP indicated that the most probable local conformation in the repeat regions is a ribbon-like, twisting, trans-extended chain structure with repetitive arrays of carboxylate and phosphate groups on both edges of the chain (Fig 10a). Such a specific, structured, spatial arrangement could function efficiently for interaction with Ca2+ and could enhance the possibility of calcium-ion bridging between parallel chains, or interaction with a hydroxyapatite surface array of calcium ions 77,237. Specifically, the structure and orientation of the phosphate motifs on either side of the backbone would be a key factor for oriented crystal orientation. Published reports demonstrate that with bovine DPP at high concentrations (0.45mg/ml), one Ca2+ is bound for every aspartyl and phosphoseryl residue. At low concentration (∼0.05mg/ml), only one third of the amount of Ca2+ is bound at the same ionic strength 26,257. These data suggest that at a low concentration, the molecule folded back on itself, creating partially ordered, internally bridged structures, whereas at higher concentrations, intermolecular calcium-mediated aggregates formed. Recognition between DPP and the HAP surface on a certain plane may result in oriented crystal growth. The addition of even millimolar concentrations of Ca2+ to DPP was sufficient to convert DPP in dilute solution from a random structure to an ordered β-sheet like conformation258.
In-vitro nucleation studies have shown that immobilized native DPP could nucleate apatite crystals with similar morphology to that found in mineralized dentin. However, recombinant DPP mediated crystal deposits were shown to be amorphous calcium phosphate by electron diffraction patterns254. Even though both native and recombinant DPP could sequester calcium ions, rDPP failed in transforming the amorphous calcium phosphate into apatite crystals. This suggests that the ordered arrays of phosphate groups in phosphophoryn are necessary for facilitating localized apatite nucleation. Small Angle X-ray diffraction experiments unequivocally demonstrated that native DPP could bind calcium with high capacity and affinity, and upon calcium binding, the DPP molecules from an extended to a more compact structure255 (Fig. 10b). Such a calcium-mediated template might be crucial for creating an ordered Ca2+ array on the surface of DPP and facilitate apatite nucleation. Template driven apatite nucleation is impaired in the absence of phosphorylation of the serine groups on the backbone. DPP is readily adsorbed on the surfaces of HAP crystals, but binds preferentially to the (100) face of HAP, which in the HAP crystal is the ab plane, thus facilitating growth in the c-axis direction259. However, the adsorption is not a simple 1:1 binding of one phosphoprotein molecule occupying one charged binding site on the crystal surface, as would be expected from Langmuir-type monolayer adsorption isotherms260. Instead, at higher DPP concentrations, protein adsorption to the surface of HAP was highly enhanced. This was attributed to the extended β-sheet conformation where intermolecular interactions facilitated by calcium bridging between phosphate and aspartate groups on different molecules, could lead to increased availability of high-affinity calcium-binding sites on the DPP molecule. Thus, changes in the conformation would result in an increased availability of negative charged groups on the DPP to interact in an ordered fashion with binding sites on the HAP surface. It is also possible that additional cooperative interactions between DPP molecules on the surface could lead to multilayer protein assembly, thus blocking crystal growth sites. Dephosphorylation leads to reduced affinity of the protein for the HAP crystal surface. Consequently, dephosphorylated DPP was not as effective in blocking crystal growth sites on the HAP surface. As the phosphorylation state of a protein in the ECM is dynamic, it can be anticipated that DPP could be both adsorbed and desorbed from the surface, demonstrating the importance of the phosphate group. In the dentin matrix, variations in the organic matrix phosphate levels during dentin maturation have been observed261. This could possibly be a mechanism by which odontoblasts could modulate the extents of matrix protein phosphorylation, and possibly dephosphorylation, altering, in turn, the functions of phosphorylated proteins and retaining in this way a regulatory function after dentin is first formed261.
A dual functional role for DPP in the dentin matrix, similar to DMP1, can be postulated. In solution, DPP can inhibit mineralization and when immobilized on collagen fibrils can promote mineralization. The conformational state adopted by DPP might be responsible for orienting the phosphate groups and facilitating interaction with calcium as shown in Fig. 10b. Expression of DPP exclusively at the dentin mineralization front suggests that this molecule asserts well-defined control in regulating mineral deposition.
4.6 Summary
In summary, the SIBLING family members that include DMP1, DSPP, MEPE, OPN and BSP share common features such as their location on human chromosome 4q21, their multiple phosphorylation sites, making them highly acidic in nature, and the presence of an RGD integrin binding domain. Each of these proteins plays an important role in either promoting tissue mineralization or inhibiting the calcification process. A general paradigm that emerges is that macromolecules such as DMP1, DPP in solution can act as an inhibitor of crystal nucleation and growth or can act as a template for crystal nucleation when it becomes adsorbed on a solid surface. Nucleation processes usually present an activation energy barrier before allowing the formation of a solid phase from a supersaturated metastable solution. In biological micro-environments, the activation energy for nucleation can be reduced by lowering of the interfacial energy136. DPP immobilized on a collagen surface can lower the interfacial energy for HAP nucleation, indicating that DPP bound on collagen template had a high capacity to nucleate HAP136. Similar lowering of interfacial energy can be anticipated with other phosphorylated proteins. This might be a common theme for proteins containing highly anionic polyanions. Both BSP and OPN possess high affinity for calcium due to the abundance of acidic amino acids such as glutamic and aspartic acid. Acidic amino acids in OPN phosphopeptides appear to contribute to the HAP inhibiting activity by producing an electrostatic repulsion of inorganic phosphate ions once the protein is adsorbed to the crystal surface. Even during the inhibition process, it is necessary that these proteins bind to specific crystal faces. However, BSP promotes HAP nucleation when immobilized on a surface. Thus, the various acidic phosphoproteins of the extracellular matrix appear to have different crystal growth regulatory activities enabling them to present specific distinct biological activities. Co-operation of several of these proteins, would effectively promote or retard crystallization, as well as regulate the growth kinetics and size of crystals.
Gene knock-out models97,189,262,263,264,265 of the proteins localized on 4q21, revealed that mineralized tissue formation and growth can be compensated by other macromolecules either partially or completely as well as by other members of the SIBLING family. In vivo studies of the effects of gene knockouts of individual SIBLINGs show that they each do more than simply alter the mineralization process. They also have profound effects on development and tissue repair not directly related to mineralization as indicated below.
Targeted deletion of MEPE gene resulted in increased bone mass189. This was attributed to the increased osteoblast number and mineral apposition rate. Thus in-vivo MEPE could function as a negative regulator of osteoblast number and activity. Similarly, targeted disruption of OPN gene resulted in increased mineral content and maturity in long bones. Fourier Transform infrared microspectroscopy (FTIRM) based characterization of the mineral in bones revealed that the relative amount of mineral in the more mature areas of the bone obtained from 12 and 16 week-old OPN null mice was significantly increased145. Moreover, mineral maturity (mineral crystal size and perfection) was prominent in OPN-deficient bone. These findings are in good agreement with in-vitro data indicating that OPN is a potent inhibitor of mineral formation and mineral crystal growth and proliferation.
DMP1- null newborn mice display no gross abnormalities in mineralization indicating that there must be redundant genes that compensate for DMP1 function during early development. During post-natal development DMP1–null pups develop skeletal abnormalities like enlarged growth plate, osteomalacia and development of short limbs221,262. These defects have been proposed to be related to defective maturation of osteoblasts into osteocytes. The dentin phenotype in the null-mice show a partial failure of maturation of predentin into dentin, increased width of the predentin zone with a reduced dentin wall hypomineralization and a three-fold reduction in dentin appositional rate209,263,.
As the DSPP gene is abundantly synthesized by the odontoblasts, the tooth phenotype has been well characterized. Overall, the DSPP-null mice develop tooth defects similar to human dentinogenesis imperfecta III with enlarged pulp chambers, increased width of predentin zone, hypomineralization, and pulp exposure. Electron microscopy revealed an irregular mineralization front and a lack of calcospherites coalescence in the dentin. Interestingly, the levels of biglycan and decorin, small leucine-rich proteoglycans, were increased in the widened predentin zone and in void spaces among the calcospherites in the dentin of null teeth 264. Thus, the protein products of the DSPP gene are required for proper dentin mineralization.
BSP null mice are viable and breed normally. Bone is undermineralized in fetuses and young adults, but not in older (>12 months) BSP-/- mice. At 4 months BSP-/- mice display thinner cortical bones than wild type, but greater trabecular bone volume with very low bone formation rate. Overall, BSP deficiency impairs bone growth and mineralization.
These examples of in vivo consequences of gene ablation demonstrate both the wide range of system redundancies and the multiplicity of properties altered is evidence of the vital importance of controlled mineralization and the other effects of the SIBLINGs in both mechanical and metabolic functions.
5 Other phosphoproteins related to apatite mineralization
Protein phosphorylation is a ubiquitous reaction and many proteins depend on the reversible phosphorylation of particular groups for activation of their biological function. Most of these reactions, but not all, take place intracellularly as part of the normal metabolic cell activity. Usually in these cases only a very limited number of phosphorylation sites are involved in a particular molecule, and the resultant molecules are not directly related to the extracellular processes of matrix mineralization. Thus, they have been considered outside the realm of this review. However, there are phosphorylated extracellular matrix proteins, not part of the SIBLING family, which may be directly involved in some aspect of the matrix apatite mineralization, although not specifically the nucleation event. In this Section we consider two examples of these macromolecules, the role of a bone protein, bone acidic glycoprotein - 75 (BAG-75), and an entirely different protein, amelogenin, the principal protein linked with the formation of the large hydroxyapatite crystals in tooth enamel.
Bone acidic glycoprotein -75 and bone mineralization
BAG-75 was first described by Gorski and Shimizu266 as a new mineralized phase protein found in the 4.0 M guanidine·HCl − 0.5 M EDTA extracts of rat calvaria that, at the first stage, co-eluted from DEAE-Sephacel columns in 6.0 M urea-containing buffers with the proteoglycans at high salt concentrations (1.8 M). The BAG-75 was separated from the proteoglycans by a further round of chromatography over a calcium hydroxyl apatite column in 7.0 M urea. The relative mass, Mr, was estimated to be 75,000 by gel electrophoresis. On this basis, the molar composition (residues per mol) was: Asp/Asn 65; Glu/Gln 97; Ser 62; Organic phosphorous 44; and total basic residues Arg, Lys, His 61, clearly an acidic protein, with an additional content of 7.0% by weight of N-acetyl neuraminic acid. The N-terminal protein sequence was determined to be LPVARYQNTEEEE(D/E)-, very similar, but only 33% homologous, to that of rat OPN, LPVKVAEFGSSEEK-. The N-terminal sequence of rat BSP was not homologous with the BAG-75 sequence, but the two proteins co-eluted in the initial extractions and chromatography. It was thus suggested that BAG-75 was a component of the calvaria mineral phase, but also had interaction properties that required high concentrations of lyotropic agents such as guanidine or urea to prevent aggregation, different from both BSP and OPN. Unfortunately, the protein sequencing and the cloning of the BAG-75 gene have not yet been completed, or brought to the state of knowledge of the SIBLING proteins.
However, substantial progress has been reported. Specific BAG-75 antibodies, showed267,268 that BAG-75 is expressed in vivo early in sites of intramembranous bone formation, and is enriched in condensing osteogenic mesenchyme and in the osteoprogenitor layer of tibial periosteum, and in dentin266, 269. Strikingly, BSP was found to accumulate at the matrix sites of BAG-75 enrichment, and both BSP and BAG-75 remained within the sites after mineralization. The BAG-75 did not appear to localize in close association with the mineralized bone collagen fibrils267. Extracellular spherical structures called biomineralization foci (BMF) were found at the sites where hydroxyapatite crystals would subsequently form, and these BMF contained BAG-75, BSP and alkaline phosphatase268. Studies in mineralizing UMR osteoblastic cell cultures showed two sizes of BMF populations, 15-25 and 150-250 μm in diameter. The smaller BMF were similar to the aggregates formed in purified BAG-75 through self-association. These pure BAG-75 aggregates were able to nucleate apatite crystal formation if an organophosphate (e.g. β-glycerol phosphate) was added to the UMR cultures270. It appears that the aggregates, in the in vivo milieu specifically accumulate the BSP and alkaline phosphatase components as they increase in size, and act together to nucleate the apatite within the BMF. In a recent publication271, Midura et al. show that the mineralized BMF, called calcospherulites, seed the formation of apatite crystals within the BMF/calcospherulites, but also demonstrate that incubation of the calcospherulites in a collagen fibril hydrogel also seeds the mineralization of neighboring collagen fibrils “at distances well away from the observed calcospherulites.” Gorski et al.272 have shown that enzymatic processing of the BAG-75 and DSP takes place within the BMF releasing 45-50 kDa fragments of each protein, When the enzymatic activity is inhibited, the BMF lose their ability to nucleate apatite crystal formation in the matrix. These are very interesting data which have implications for a system for delivery of, in the case of bone, BSP and other soluble factors to the mineralization front, where they may be released to bind to the collagen fibrils and initiate the collagen-related nucleation. The mineralization front in bone has long been known as a locus of lipid accumulation, and phospholipids have been implicated as crucial components of matrix vesicles273, that is, the now designated BMF. These new data about BAG-75 and BSP in the BMF suggest that the matrix lipases may degrade the phospholipid - BAG-75 complexes and release the BSP and alkaline phosphatase activities enabling the transfer of mineral nucleating activity to the collagen fibril surface. This whole area of sequestered delivery pathways for the reactive components opens up many new directions for study of the matrix mineralization mechanisms.
Amelogenins in tooth enamel mineralization
The amelogenins are the major structural proteins of dental enamel secreted by the ameloblasts into the restricted extracellular space between the Tome's processes and the mineralized dentin at the dentino-enamel junction. The amelogenins have low molecular weights, on the order of 25-28 kDa depending on the species, but assemble in the ECM to form nanospheres, spherical assemblies of the individual molecules ranging in diameter from 30 to 50 nm. These, in turn, become linearly aligned and assist in the production of long, ribbon-like hydroxyapatite crystals274,275,276,277. The amelogenin molecules, with an intrinsically hydrophobic central region crucial to the nanosphere formation, have a proline-leucine rich C-terminal domain which terminates in a very acidic region with the sequence – TDKTKREEVD. This domain is exposed to the exterior of the nanospheres, and consequently appears to be implicated in the interaction with hydroxyapatite crystals, or the initiation of mineralization. A very thorough analysis of the phosphorylation state of the molecule showed that it was phosphorylated at a single position, Ser16, in the N-terminal region of the intact amelogenin278,279. Since, in in vitro studies, non-phosphorylated recombinant amelogenin was fully effective in assembling into ordered arrays of nanospheres and inducing mineralization275,279, it can be concluded that the phosphorylation of the single Ser residue in this system has no relevance to the mineralization process. While we had no basis for including even this brief discussion of amelogenin in this review of phosphoproteins in hydroxyapatite mineralization, we have done so to emphasize the fact that phosphoproteins are important in mineralization, and are crucial in bone, dentin and other vertebrate mesenchymal tissue mineralization processes, they are not absolutely required. What is important is the ability of the cells involved to deliver proteins bearing exposed highly negatively charged sequences to a structural matrix in the extracellular environment, so that even relatively short, but concentrated anionic sequences on an otherwise hydrophobic molecule may suffice. Such molecules are, in fact, abundant in invertebrate systems as well as vertebrates.
Acknowledgments
We are pleased to acknowledge the long term support from the National Institutes of Health. Dr. Anne George has been supported by grants DE11657 and DE13836. Dr. Arthur Veis has been supported by grants DE01374, DE014758 and AR 13921.
Biography

Anne George received her Ph.D. in Physical Chemistry from Madras University, India in 1983. She then did her Post-Doctoral work with Dr. Arthur Veis at Northwestern University on determining the structure of type I collagen in-solution using Fourier Transform Infrared Spectroscopy. She joined as an Assistant Professor in 1993 at Northwestern University where she started work on the cloning of the dentin matrix proteins. She is instrumental in identifying the family of dentin matrix proteins from the rat odontoblasts. She taught the Basic Science course to the dental students at Northwestern University and was awarded a “Teaching Excellence” award. She then moved to the University of Illinois at Chicago in 1998 as an Associate Professor and became a full Professor in 2003 and continued her work on noncollagenous proteins and their role in biomineralization. She is now an Allan G. Brodie endowed professor at the University of Illinois at Chicago. Her work was reported in Chicago Tribune “Calcium Link-Genes may solve mystery of how teeth harden” in 1994 and Scientific Year Book of Encyclopedia Britannica “Tooth Gene Studied” 1998. A documentary on her work was produced by Dallas TV. She is the recipient of the IADR Basic Research in “Pulp Biology and Regeneration” award in 2008. She is the author of over 80 papers in peer-reviewed journals.

Arthur Veis received his B. S degree in Chemical Engineering at the University of Oklahoma and his Ph. D. in Physical Chemistry from Northwestern University (1950), in the laboratory of Professor Irving M. Klotz. After a Postdoctoral year with Prof. Lloyd Swearingen, he was appointed Instructor in Physical Chemistry at the University of Oklahoma, and then joined the Physical Chemistry Division, Central Research Laboratory at Armour and Company in Chicago where in 1952 he began his basic work on collagen structure, cross-linking and properties. He also initiated work on the thermodynamics of phase separations in polyelectrolytes as related to complex coacervation. On a part-time basis he taught graduate courses in Thermodynamics at Loyola University and Physical Biochemistry at the University of Illinois Medical School. In 1960 he joined the Biochemistry Department of the Northwestern University Medical School as Associate Professor and became Professor in 1965. He held a sabbatical year John Simon Guggenheim Fellowship in the laboratory of Noel Laureate Paul J. Flory at Stanford University. Subsequently at Northwestern, he became Associate Dean of the Graduate School, for the Medical School, and Director of the MD-Ph.D. program and Associate Dean for the Honors Program in Medical Education. He founded the Department of Oral Biology in the Dental School and directed the DDS-Ph.D. program. He is now Professor Emeritus in the Department of Cell and Molecular Biology at the Feinberg School of Medicine. During all this period he continued work on collagen structure, and began his long term studies of bone and dentin mineralization mechanisms and the role of proteins of the mineralizing matrix in nucleation of mineralization. He discovered the first of the phosphorylated extracellular matrix proteins, phosphophoryn, now known as dentin sialophosphoprotein, and later, with Postdoctoral fellow, Dr. Anne George in the laboratory, cloned DMP1 and DMP2. He is a fellow of the AAAS, and a recipient of the IADR Basic Research in Biomineralization award, Marshall Urist Award for Excellence in Tissue Regeneration Research from the Orthopaedic Research Society, a Fulbright Senior International Scholar award (European Molecular Biology Laboratory for with Andrew Miller in Grenoble, France and with Wolfie Traub at the Weizmann Institute of Science, Israel), and a Case Centennial Scholar Award. He organized the First international Conference on the Chemistry and Biology of Mineralized Tissues, and became President of the Board of Trustees of that organization. Current biomineralization studies have moved on to the relationship between vertebrate and invertebrate systems, with particular interest in the proteins of the echinoderm teeth.
References
- 1.Lowenstam HA. Science. 1981;211:1126. doi: 10.1126/science.7008198. [DOI] [PubMed] [Google Scholar]
- 2.Eanes ED. Monog Oral Sci. 2001;18:130. doi: 10.1159/000061652. [DOI] [PubMed] [Google Scholar]
- 3.Grynpas MD, Omelon S. Bone. 2007;41:162. doi: 10.1016/j.bone.2007.04.176. [DOI] [PubMed] [Google Scholar]
- 4.Crane NJ, Popescu V, Morris MD, Steenhuis P, Ignelzi MA., Jr Bone. 2006;39:434. doi: 10.1016/j.bone.2006.02.059. [DOI] [PubMed] [Google Scholar]
- 5.Veis A, Sabsay B. Biomineralization and Biological Metal Accumulation. D Reidel Pub Co; Dordrecht, Holland: 1982. p. 273. [Google Scholar]
- 6.Veis A. Extracellular Matrix: Structure and function. Alan R. Liss, Inc.; New York: 1985. p. 351. [Google Scholar]
- 7.Schmitt FO, Gross J, Highberger JH. Proc Natl Acad Sci US. 1955;39:459. doi: 10.1073/pnas.39.6.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hodge AJ, Schmitt FO. Proc Natl Acad Sci US. 1960;45:186. doi: 10.1073/pnas.46.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodge AJ, Petruska JA. Aspects of Protein Structure and Function. Academic Press; London: 1963. p. 289. [Google Scholar]
- 10.Glimcher MJ. Handbook of Physiology-Endocrinology VII. Williams and Wilkins Co.; Baltimore: 1976. p. 25. [Google Scholar]
- 11.Landis WJ, Song MJ. J Struct Biol. 1991;107:116. doi: 10.1016/1047-8477(91)90015-o. [DOI] [PubMed] [Google Scholar]
- 12.Landis WJ, Moradian-Oldak J, Weiner S. Connect Tissue Res. 1991;25:181. doi: 10.3109/03008209109029155. [DOI] [PubMed] [Google Scholar]
- 13.Landis WJ, Song MJ, Leith A, McEwen L, McEwen B. J Struct Biol. 1993;110:39. doi: 10.1006/jsbi.1993.1003. [DOI] [PubMed] [Google Scholar]
- 14.Landis WJ, Hodgens KJ, Arena J, Song MJ, McEwen BF. Microsc Res Technol. 1996;33:192. doi: 10.1002/(SICI)1097-0029(19960201)33:2<192::AID-JEMT9>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 15.Landis WJ, Hodgens KJ, Song MJ, Arena J, Kiyonaga S, Marko M, Owen C, McEwen BF. J Struct Biol. 1996;117:24. doi: 10.1006/jsbi.1996.0066. [DOI] [PubMed] [Google Scholar]
- 16.Katz EP, Li ST. J Mol Biol. 1973;73:351. doi: 10.1016/0022-2836(73)90347-1. [DOI] [PubMed] [Google Scholar]
- 17.Katz EP, Li ST. J Mol Biol. 1973;80:1. doi: 10.1016/0022-2836(73)90230-1. [DOI] [PubMed] [Google Scholar]
- 18.Hulmes DJ, Miller A. Nature. 1979;282:878. doi: 10.1038/282878a0. [DOI] [PubMed] [Google Scholar]
- 19.Orgel JP, Irving TC, Miller A, Wess TJ. Proc Natl Acad Sci U S A. 2006;103:9001. doi: 10.1073/pnas.0502718103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orgel JP, Miller A, Irving TC, Fischetti RF, Hammersley AP, Wess TJ. Structure. 2001;9:1061. doi: 10.1016/s0969-2126(01)00669-4. [DOI] [PubMed] [Google Scholar]
- 21.Chapman JA. Connect Tissue Res. 1974;2:137. doi: 10.3109/03008207409152099. [DOI] [PubMed] [Google Scholar]
- 22.Chapman JA, Hardcastle RA. Connect Tissue Res. 1974;2:151. doi: 10.3109/03008207409152100. [DOI] [PubMed] [Google Scholar]
- 23.Dahl T, Sabsay B, Veis A. J Struct Biol. 1998;123:162. doi: 10.1006/jsbi.1998.4025. [DOI] [PubMed] [Google Scholar]
- 24.Glimcher MJ, Krane SM. Radioisotopes and Bone. 1962:392. [Google Scholar]
- 25.Stetler-Stevenson WG, Veis A. Calcif Tiss Int. 1986;38:135. doi: 10.1007/BF02556873. [DOI] [PubMed] [Google Scholar]
- 26.Stetler-Stevenson WG, Veis A. Calcif Tiss Int. 1987;40:97. doi: 10.1007/BF02555712. [DOI] [PubMed] [Google Scholar]
- 27.Silver FH, Freeman JW, Horvath I, Landis JW. Biomacromol. 2001;2:750. doi: 10.1021/bm0100615. [DOI] [PubMed] [Google Scholar]
- 28.Landis WJ, Silver FH. Comp Biochem Physiol Part A Molecular and Integrative Physiol. 2002;133:1135. doi: 10.1016/s1095-6433(02)00248-9. [DOI] [PubMed] [Google Scholar]
- 29.Mertz EL, Leikin S. Biochemistry. 2004;43:14901. doi: 10.1021/bi048788b. [DOI] [PubMed] [Google Scholar]
- 30.Traub W, Arad T, Weiner S. Matrix. 1992;12:251. doi: 10.1016/s0934-8832(11)80076-4. [DOI] [PubMed] [Google Scholar]
- 31.White SW, Hulmes DJ, Miller A, Timmins PA. Nature. 1977;266:421. doi: 10.1038/266421a0. [DOI] [PubMed] [Google Scholar]
- 32.Glimcher MJ, Francois CJ, Richards L, Krane SM. Biochim Biophys Acta. 1964;93:202. doi: 10.1016/0304-4165(64)90342-3. [DOI] [PubMed] [Google Scholar]
- 33.Glimcher MJ, Krane SM. Biochim Biophys Acta. 1964;90:477. doi: 10.1016/0304-4165(64)90227-2. [DOI] [PubMed] [Google Scholar]
- 34.Veis A, Schlueter R. Nature. 1963;197:1204. doi: 10.1038/1971204a0. [DOI] [PubMed] [Google Scholar]
- 35.Veis A, Schlueter RJ. Biochemistry. 1964;3:1650. doi: 10.1021/bi00899a009. [DOI] [PubMed] [Google Scholar]
- 36.Schlueter RJ, Veis A. Biochemistry. 1964;3:1657. doi: 10.1021/bi00899a010. [DOI] [PubMed] [Google Scholar]
- 37.Veis A, Perry A. Biochemistry. 1967;6:2409. doi: 10.1021/bi00860a017. [DOI] [PubMed] [Google Scholar]
- 38.Volpin D, Veis A. Biochemistry. 1971;10:1751. doi: 10.1021/bi00786a003. [DOI] [PubMed] [Google Scholar]
- 39.Carmichael DJ, Veis A, Wang ET. Calcif Tiss Res. 1971;7:331. doi: 10.1007/BF02062622. [DOI] [PubMed] [Google Scholar]
- 40.Volpin D, Veis A. Biochem Biophys Res Commun. 1971;44:804. doi: 10.1016/0006-291x(71)90782-0. [DOI] [PubMed] [Google Scholar]
- 41.Volpin D, Veis A. Biochemistry. 1973;12:1452. doi: 10.1021/bi00731a028. [DOI] [PubMed] [Google Scholar]
- 42.Rahima M, Veis A. Calcif Tiss Int. 1988;42:104. doi: 10.1007/BF02556342. [DOI] [PubMed] [Google Scholar]
- 43.Veis A, Spector AR, Zamoscianyk H. Biochim Biophys Acta. 1972;257:404. doi: 10.1016/0005-2795(72)90293-0. [DOI] [PubMed] [Google Scholar]
- 44.Shuttleworth A, Veis A. Biochim Biophys Acta. 1972;257:414. doi: 10.1016/0005-2795(72)90294-2. [DOI] [PubMed] [Google Scholar]
- 45.Termine JD, Belcourt AB, Christner PJ, Conn KM, Nylen MU. J Biol Chem. 1980;255:9760. [PubMed] [Google Scholar]
- 46.Termine JD, Belcourt AB, Miyamoto MS, Conn KM. J Biol Chem. 1980;255:9769. [PubMed] [Google Scholar]
- 47.Termine JD, Belcourt AB, Conn KM, Kleinman HK. J Biol Chem. 1981;256:10403. [PubMed] [Google Scholar]
- 48.Mintz KP, Grzesik WJ, Midura RJ, Robey PG, Termine JD, Fisher LW. J Bone Mineral Res. 1993;8:985. doi: 10.1002/jbmr.5650080812. [DOI] [PubMed] [Google Scholar]
- 49.Termine JD. Ciba Foundation Symp. 1988;136:178. doi: 10.1002/9780470513637.ch12. [DOI] [PubMed] [Google Scholar]
- 50.Fisher LW, Robey PG, Tuross N, Otsuka AS, Tepen DA, Esch FS, Shimasaki S, Termine JD. J Biol Chem. 1987;262:13457. [PubMed] [Google Scholar]
- 51.Fisher LW, Hawkins GR, Tuross N, Termine JD. J Biol Chem. 1987;262:9702. [PubMed] [Google Scholar]
- 52.Fisher LW, Whitson SW, Avioli LV, Termine JD. J Biol Chem. 1983;258:12723. [PubMed] [Google Scholar]
- 53.Termine JD, Kleinman HK, Whitson SW, Conn KM, McGarvey ML, Martin GR. Cell. 1981;26:99. doi: 10.1016/0092-8674(81)90037-4. [DOI] [PubMed] [Google Scholar]
- 54.Termine JD, Belcourt AB, Conn KM, Kleinman HK. J Biol Chem. 1981;256:10403. [PubMed] [Google Scholar]
- 55.Domenicucci C, Goldberg HA, Hofmann T, Isenman D, Wasi S, Sodek J. Biochem J. 1988;253:139. doi: 10.1042/bj2530139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fisher LW, Torchia DA, Fohr B, Young MF, Fedarko NS. Biochem Biophys Res Commun. 2001;280:460. doi: 10.1006/bbrc.2000.4146. [DOI] [PubMed] [Google Scholar]
- 57.Fisher LW, Fedarko NS. Connect Tissue Res. 2003;44(suppl1):33. [PubMed] [Google Scholar]
- 58.Huq NL, Cross KJ, Ung M, Reynolds EC. Arch Oral Biol. 2005;50:599. doi: 10.1016/j.archoralbio.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 59.Yamakoshi Y, Hu JC, Fukae M, Zhang H, Simmer JP. J Biol Chem. 2005;280:17472. doi: 10.1074/jbc.M413220200. [DOI] [PubMed] [Google Scholar]
- 60.Dunker AK, Lawson JD, Brown CJ, Williams RJ, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswald MD, Chiu W, Garner EC, Obradovic Z. J Mol Graph Model. 2001;19:26. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 61.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovié Z. Biochemistry. 2002;41:6573. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 62.Sorensen ES, Petersen TE. Biochem Biophys Res Commun. 1994;198:200. doi: 10.1006/bbrc.1994.1028. [DOI] [PubMed] [Google Scholar]
- 63.Christensen B, Nielsen MS, Haselmann KF, Petersen TE, Sorensen ES. Biochem J. 2005;390:285. doi: 10.1042/BJ20050341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neame PJ, Butler WT. Connect Tissue Res. 1996;35:145. doi: 10.3109/03008209609029185. [DOI] [PubMed] [Google Scholar]
- 65.Gericke A, Qin C, Spevak L, Fujimoto Y, Butler WT, Sorensen ES, Boskey AL. Calcif Tissue Int. 2005;77(1):45. doi: 10.1007/s00223-004-1288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salih E, Wang J, Mah J, Fluckiger R. Biochem J. 2002;364:465. doi: 10.1042/BJ20011272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qin C, Brunn JC, Baba O, Wygant JN, McIntyre BW, Butler WT. Eur J Oral Sci. 2003;111:235. doi: 10.1034/j.1600-0722.2003.00043.x. [DOI] [PubMed] [Google Scholar]
- 68.Qin C, Baba O, Butler WT. Crit Rev Oral Biol Med. 2004;15:126. doi: 10.1177/154411130401500302. [DOI] [PubMed] [Google Scholar]
- 69.Yamakoshi Y, Hu JCC, Fukae M, Iwata T, Kim JW, Zhang H, Simmer JP. J Biol Chem. 2005;280:1552. doi: 10.1074/jbc.M409606200. [DOI] [PubMed] [Google Scholar]
- 70.Stetler-Stevenson WG, Veis A. Biochemistry. 1983;22:4326. doi: 10.1021/bi00287a025. [DOI] [PubMed] [Google Scholar]
- 71.Huq NL, Cross KJ, Talbo GH, Riley PF, Loganathan A, Crossley MA, Perich JW, Reynolds EC. J Dent Res. 2000;79:1914. doi: 10.1177/00220345000790111701. [DOI] [PubMed] [Google Scholar]
- 72.Dimuzio MT, Veis A. Calcif Tiss Res. 1978;25:169. doi: 10.1007/BF02010765. [DOI] [PubMed] [Google Scholar]
- 73.Bright JN, Woolff TB, Hoh JH. Prog Biophys Mol Biol. 2001;76:131. doi: 10.1016/s0079-6107(01)00012-8. [DOI] [PubMed] [Google Scholar]
- 74.Cross KJ, Huq NL, Reynolds EC. J Peptide Res. 2005;66:59. doi: 10.1111/j.1399-3011.2005.00273.x. [DOI] [PubMed] [Google Scholar]
- 75.Huq N, Loganathan A, Cross K, Chen Y, Johnson N, Willetts M, Veith P, Reynolds EC. Arch of Oral Biol. 2005;50:807. doi: 10.1016/j.archoralbio.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 76.George A, Sabsay B, Simonian PAL, Veis A. J Biol Chem. 1993;268:12624. [PubMed] [Google Scholar]
- 77.George A, Bannon L, Sabsay B, Dillon JW, Malone J, Veis A, Jenkins NA, Gilbert DJ, Copeland NG. J Biol Chem. 1996;271:32869. doi: 10.1074/jbc.271.51.32869. [DOI] [PubMed] [Google Scholar]
- 78.Gorski JP. Calcif Tissue Int. 1992;50:391. doi: 10.1007/BF00296767. [DOI] [PubMed] [Google Scholar]
- 79.Lee SJ, Veis A, Glonek T. Biochemistry. 1977;16:2971. doi: 10.1021/bi00632a026. [DOI] [PubMed] [Google Scholar]
- 80.Fujisawa R, Takagi T, Kuboki Y, Sasaki S. Calcif Tissue Int. 1984;36:239. doi: 10.1007/BF02405323. [DOI] [PubMed] [Google Scholar]
- 81.Fujisawa R, Kuboki Y, Sasaki S. Calcif Tissue Int. 1986;39:248. doi: 10.1007/BF02555213. [DOI] [PubMed] [Google Scholar]
- 82.Fujisawa R, Kuboki Y, Sasaki S. Calcif Tissue Int. 1987;41:44. doi: 10.1007/BF02555130. [DOI] [PubMed] [Google Scholar]
- 83.Lussi A, Crenshaw MA, Linde A. Arch Oral Biol. 1988;33:685. doi: 10.1016/0003-9969(88)90124-0. [DOI] [PubMed] [Google Scholar]
- 84.Saito T, Arsenault AL, Yamauchi M, Kuboki Y, Crenshaw MA. Bone. 1997;21:30. doi: 10.1016/s8756-3282(97)00149-x. [DOI] [PubMed] [Google Scholar]
- 85.Saito T, Yamauchi M, Abiko Y, Matsuda K, Crenshaw MA. J Bone Mineral Res. 2000;15:1615. doi: 10.1359/jbmr.2000.15.8.1615. [DOI] [PubMed] [Google Scholar]
- 86.Kaartinen MT, Pirhonen A, Linnala-Kankkunen A, Maenpaa PH. J Biol Chem. 1999;274:1729. doi: 10.1074/jbc.274.3.1729. [DOI] [PubMed] [Google Scholar]
- 87.Kaartinen MT, Sun W, Kaipatur N, McKee MD. J Dent Res. 2005;84:607. doi: 10.1177/154405910508400705. [DOI] [PubMed] [Google Scholar]
- 88.Boskey AL. Connect Tissue Res. 1996;35:357. doi: 10.3109/03008209609029212. [DOI] [PubMed] [Google Scholar]
- 89.Young MF, Kerr JM, Ibaraki K, Heegaard AM, Robey PG. Clin Orthop Relat Res. 1992;281:275. [PubMed] [Google Scholar]
- 90.Worcester EM. J Am Soc Nephrol. 1994;5:S46. doi: 10.1681/ASN.V55s46. [DOI] [PubMed] [Google Scholar]
- 91.Denhardt DT, Lopez CA, Rollo EE, Hwang SM, An XR, Walther SE. Ann N Y Acad Sci. 1995;760:127. doi: 10.1111/j.1749-6632.1995.tb44625.x. [DOI] [PubMed] [Google Scholar]
- 92.Hijiya N, Setoguchi M, Matsuura K, Higuchi Y, Akizuki S, Yamamoto S. Biochem J. 1994;303(Pt 1):255. doi: 10.1042/bj3030255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sodek J, Ganss B, McKee MD. Crit Rev Oral Biol Med. 2000;11:279. doi: 10.1177/10454411000110030101. [DOI] [PubMed] [Google Scholar]
- 94.Yamamoto S, Hijiya N, Setoguchi M, Matsuura K, Ishida T, Higuchi Y, Akizuki S. Ann N Y Acad Sci. 1995;760:44. doi: 10.1111/j.1749-6632.1995.tb44619.x. [DOI] [PubMed] [Google Scholar]
- 95.Butler WT, Brunn JC, Qin C. Connect Tissue Res. 2003;44 1:171–8. [PubMed] [Google Scholar]
- 96.Gerstenfeld LC. J Bone Miner Res. 1999;14:850. doi: 10.1359/jbmr.1999.14.6.850. [DOI] [PubMed] [Google Scholar]
- 97.Rittling SR, Denhardt DT. Exp Nephrol. 1999;7:103. doi: 10.1159/000020591. [DOI] [PubMed] [Google Scholar]
- 98.Jono S. Nippon Rinsho. 2005;63 10:635. [PubMed] [Google Scholar]
- 99.Saeki Y. Nippon Rinsho. 2005;63 10:627. [Google Scholar]
- 100.Zaka R, Williams CJ. Curr Opin Rheumatol. 2006;18:181. doi: 10.1097/01.bor.0000209432.36355.6e. [DOI] [PubMed] [Google Scholar]
- 101.Aubin JE, Liu F, Malaval L, Gupta AK. Bone. 1995;17:77S. doi: 10.1016/8756-3282(95)00183-e. [DOI] [PubMed] [Google Scholar]
- 102.Butler WT. Ann N Y Acad Sci. 1995;760:6. doi: 10.1111/j.1749-6632.1995.tb44615.x. [DOI] [PubMed] [Google Scholar]
- 103.Chellaiah MA, Hruska KA. Drug News Perspect. 1998;11:350. doi: 10.1358/dnp.1998.11.6.863656. [DOI] [PubMed] [Google Scholar]
- 104.Denhardt DT, Guo X. FASEB J. 1993;7:1475. [PubMed] [Google Scholar]
- 105.Oldberg A, Franzen A, Heinegard D. Proc Natl Acad Sci U S A. 1986;83:8819. doi: 10.1073/pnas.83.23.8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Prince CW. Connect Tissue Res. 1989;21:15. doi: 10.3109/03008208909049991. [DOI] [PubMed] [Google Scholar]
- 107.Han M, Wen JK, Zheng B, Liu Z, Chen Y. Cardiovasc Pathol. 2007;16:283. doi: 10.1016/j.carpath.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 108.Ashkar S, Schaffer JL, Salih E, Gerstenfeld LC, Glimcher MJ. Ann N Y Acad Sci. 1995;760:296. doi: 10.1111/j.1749-6632.1995.tb44639.x. [DOI] [PubMed] [Google Scholar]
- 109.Salih E, Ashkar S, Gerstenfeld LC, Glimcher MJ. J Bone Miner Res. 1996;11:1461. doi: 10.1002/jbmr.5650111013. [DOI] [PubMed] [Google Scholar]
- 110.Salih E, Ashkar S, Gerstenfeld LC, Glimcher MJ. J Biol Chem. 1997;272:13966. doi: 10.1074/jbc.272.21.13966. [DOI] [PubMed] [Google Scholar]
- 111.Salih E, Ashkar S, Zhou HY, Gerstenfeld L, Glimcher MJ. Connect Tissue Res. 1996;35:207. doi: 10.3109/03008209609029193. [DOI] [PubMed] [Google Scholar]
- 112.Salih E, Huang JC, Strawich E, Gouverneur M, Glimcher MJ. Connect Tissue Res. 1998;38:225–35. doi: 10.3109/03008209809017041. discussion 241. [DOI] [PubMed] [Google Scholar]
- 113.Salih E, Zhou HY, Glimcher MJ. J Biol Chem. 1996;271:16897. doi: 10.1074/jbc.271.28.16897. [DOI] [PubMed] [Google Scholar]
- 114.McKee MD, Nanci A. Connect Tissue Res. 1996;35:197. doi: 10.3109/03008209609029192. [DOI] [PubMed] [Google Scholar]
- 115.McKee MD, Nanci A. Microsc Res Tech. 1996;33:141. doi: 10.1002/(SICI)1097-0029(19960201)33:2<141::AID-JEMT5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 116.Chen J, Singh K, Mukherjee BB, Sodek J. Matrix. 1993;13:113. doi: 10.1016/s0934-8832(11)80070-3. [DOI] [PubMed] [Google Scholar]
- 117.Ibaraki K, Termine JD, Whitson SW, Young MF. J Bone Miner Res. 1992;7:743. doi: 10.1002/jbmr.5650070704. [DOI] [PubMed] [Google Scholar]
- 118.Campbell TM, Wong WT, Mackie EJ. Calcif Tissue Int. 2003;73:49. doi: 10.1007/s00223-002-2120-4. [DOI] [PubMed] [Google Scholar]
- 119.MacNeil RL, Berry J, D'Errico J, Strayhorn C, Somerman MJ. Ann N Y Acad Sci. 1995;760:166. doi: 10.1111/j.1749-6632.1995.tb44628.x. [DOI] [PubMed] [Google Scholar]
- 120.Rodrigues LR, Teixeira JA, Schmitt FL, Paulsson M, Lindmark-Mansson H. Cancer Epidemiol Biomarkers Prev. 2007;16:1087. doi: 10.1158/1055-9965.EPI-06-1008. [DOI] [PubMed] [Google Scholar]
- 121.Ogbureke KU, Fisher LW. J Histochem Cytochem. 2007;55:403. doi: 10.1369/jhc.6A7075.2007. [DOI] [PubMed] [Google Scholar]
- 122.Lee JY, Choo JE, Choi YS, Park JB, Min DS, Lee SJ, Rhyu HK, Jo IH, Chung CP, Park YJ. Biomaterials. 2007;28:4257. doi: 10.1016/j.biomaterials.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 123.Chen NX, Moe SM. J Investig Med. 2006;54:380. doi: 10.2310/6650.2006.06017. [DOI] [PubMed] [Google Scholar]
- 124.Rosenthal AK, Gohr CM, Uzuki M, Masuda I. Matrix Biol. 2007;26:96. doi: 10.1016/j.matbio.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Razzouk S, Brunn JC, Qin C, Tye CE, Goldberg HA, Butler WT. Bone. 2002;30:40. doi: 10.1016/s8756-3282(01)00637-8. [DOI] [PubMed] [Google Scholar]
- 126.Asselman M, Verkoelen CF. Curr Opin Urol. 2002;12:271. doi: 10.1097/00042307-200207000-00002. [DOI] [PubMed] [Google Scholar]
- 127.Giachelli CM. Z Kardiol. 2001;90 3:31. doi: 10.1007/s003920170039. [DOI] [PubMed] [Google Scholar]
- 128.Miller SJ, Watson WC, Kerr KA, Labarrere CA, Chen NX, Deeg MA, Unthank JL. Am J Physiol Heart Circ Physiol. 2007;293:H2634. doi: 10.1152/ajpheart.00397.2007. [DOI] [PubMed] [Google Scholar]
- 129.Schlieper G, Westenfeld R, Brandenburg V, Ketteler M. Semin Dial. 2007;20:113. doi: 10.1111/j.1525-139X.2007.00257.x. [DOI] [PubMed] [Google Scholar]
- 130.Giachelli CM. Orthod Craniofac Res. 2005;8:229. doi: 10.1111/j.1601-6343.2005.00345.x. [DOI] [PubMed] [Google Scholar]
- 131.Speer MY, Chien YC, Quan M, Yang HY, Vali H, McKee MD, Giachelli CM. Cardiovasc Res. 2005;66:324. doi: 10.1016/j.cardiores.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 132.Trion A, van der Laarse A. Am Heart J. 2004;147:808. doi: 10.1016/j.ahj.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 133.Boskey AL. Ann N Y Acad Sci. 1995;760:249. doi: 10.1111/j.1749-6632.1995.tb44635.x. [DOI] [PubMed] [Google Scholar]
- 134.Hunter GK, Hauschka PV, Poole AR, Rosenberg LC, Goldberg HA. Biochem J. 1996;317(Pt 1):59. doi: 10.1042/bj3170059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hunter GK, Kyle CL, Goldberg HA. Biochem J. 1994;300(Pt 3):723. doi: 10.1042/bj3000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ito S, Saito T, Amano K. J Biomed Mater Res A. 2004;69:11. doi: 10.1002/jbm.a.20066. [DOI] [PubMed] [Google Scholar]
- 137.Pampena DA, Robertson KA, Litvinova O, Lajoie G, Goldberg HA, Hunter GK. Biochem J. 2004;378:1083. doi: 10.1042/BJ20031150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Christensen B, Kazanecki CC, Petersen TB, Rittling SR, Denhardt DT. J Biol Chem. 2007;282:19463. doi: 10.1074/jbc.M703055200. [DOI] [PubMed] [Google Scholar]
- 139.Saad FA, Salih E, Glimcher MJ. J Cell Biochem. 2007 doi: 10.1002/jcb.21453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Grohe B, O'Young J, Ionescu DA, Lajoie G, Rogers KA, Karttunen M, Goldberg HA, Hunter GK. J Am Chem Soc. 2007 doi: 10.1021/ja0745613. [DOI] [PubMed] [Google Scholar]
- 141.Keykhosravani M, Doherty-Kirby A, Zhang C, Brewer D, Goldberg HA, Hunter GK, Lajoie G. Biochemistry. 2005;44:6990. doi: 10.1021/bi050109p. [DOI] [PubMed] [Google Scholar]
- 142.Boskey AL, Maresca M, Ullrich W, Doty SB, Butler WT, Prince CW. Bone Miner. 1993;22:147. doi: 10.1016/s0169-6009(08)80225-5. [DOI] [PubMed] [Google Scholar]
- 143.Gilbert M, Shaw WJ, Long JR, Nelson K, Drobny GP, Giachelli CM, Stayton PS. J Biol Chem. 2000;275:16213. doi: 10.1074/jbc.M001773200. [DOI] [PubMed] [Google Scholar]
- 144.Rittling SR, Matsumoto HN, McKee MD, Nanci A, An XR, Novick KE, Kowalski AJ, Noda M, Denhardt DT. J Bone Miner Res. 1998;13:1101. doi: 10.1359/jbmr.1998.13.7.1101. [DOI] [PubMed] [Google Scholar]
- 145.Boskey AL, Spevak L, Paschalis E, Doty SB, McKee MD. Calcif Tissue Int. 2002;71:145. doi: 10.1007/s00223-001-1121-z. [DOI] [PubMed] [Google Scholar]
- 146.Kumura H, Minato N, Shimazaki K. J Dairy Res. 2006;73:449. doi: 10.1017/S0022029906001877. [DOI] [PubMed] [Google Scholar]
- 147.Gorski JP, Kremer E, Ruiz-Perez J, Wise GE, Artigues A. Ann N Y Acad Sci. 1995;760:12. doi: 10.1111/j.1749-6632.1995.tb44616.x. [DOI] [PubMed] [Google Scholar]
- 148.Shimabayashi S, Itoi K. Chem Pharm Bull (Tokyo) 1989;37:1437. doi: 10.1248/cpb.37.1437. [DOI] [PubMed] [Google Scholar]
- 149.Oldberg A, Franzen A, Heinegard D. J Biol Chem. 1988;263:19430. [PubMed] [Google Scholar]
- 150.Heinegard D, Oldberg A. FASEB J. 1989;3:2042. doi: 10.1096/fasebj.3.9.2663581. [DOI] [PubMed] [Google Scholar]
- 151.Fisher LW, McBride OW, Termine JD, Young MF. J Biol Chem. 1990;265:2347. [PubMed] [Google Scholar]
- 152.Kerr JM, Fisher LW, Termine JD, Wang MG, McBride OW, Young MF. Genomics. 1993;17(2):408. doi: 10.1006/geno.1993.1340. [DOI] [PubMed] [Google Scholar]
- 153.Kim RH, Shapiro HS, Li JJ, Wrana JL, Sodek J. Matrix Biol. 1994;14:31. doi: 10.1016/0945-053x(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 154.Shapiro HS, Chen J, Wrana JL, Zhang Q, Blum M, Sodek J. Matrix. 1993;13:431. doi: 10.1016/s0934-8832(11)80109-5. [DOI] [PubMed] [Google Scholar]
- 155.Ohnishi T, Nakamura O, Ozawa M, Arakaki N, Muramatsu T, Daikuhara Y. J Bone Miner Res. 1993;8:367. doi: 10.1002/jbmr.5650080314. [DOI] [PubMed] [Google Scholar]
- 156.Larsson E, Luning B, Heinegard D. Acta Chem Scand. 1993;47:565. doi: 10.3891/acta.chem.scand.47-0565. [DOI] [PubMed] [Google Scholar]
- 157.Wuttke M, Muller S, Nitsche DP, Paulsson M, Hanisch FG, Maurer P. J Biol Chem. 2001;276:36839. doi: 10.1074/jbc.M105689200. [DOI] [PubMed] [Google Scholar]
- 158.Chen J, McCulloch CA, Sodek J. Arch Oral Biol. 1993;38:241. doi: 10.1016/0003-9969(93)90034-j. [DOI] [PubMed] [Google Scholar]
- 159.Bianco P, Riminucci M, Silvestrini G, Bonucci E, Termine JD, Fisher LW, Robey PG. J Histochem Cytochem. 1993;41:193. doi: 10.1177/41.2.8419459. [DOI] [PubMed] [Google Scholar]
- 160.Chen J, Zhang Q, McCulloch CA, Sodek J. Histochem J. 1991;23:281–9. doi: 10.1007/BF01045047. [DOI] [PubMed] [Google Scholar]
- 161.Chen JK, Shapiro HS, Wrana JL, Reimers S, Heersche JN, Sodek J. Matrix. 1991;11:133. doi: 10.1016/s0934-8832(11)80217-9. [DOI] [PubMed] [Google Scholar]
- 162.Kasugai S, Todescan R, Jr, Nagata T, Yao KL, Butler WT, Sodek J. J Cell Physiol. 1991;147:111. doi: 10.1002/jcp.1041470115. [DOI] [PubMed] [Google Scholar]
- 163.Kasugai S, Zhang Q, Overall CM, Wrana JL, Butler WT, Sodek J. Bone Miner. 1991;13:235. doi: 10.1016/0169-6009(91)90071-7. [DOI] [PubMed] [Google Scholar]
- 164.Kasugai S, Nagata T, Sodek J. J Cell Physiol. 1992;152:467. doi: 10.1002/jcp.1041520305. [DOI] [PubMed] [Google Scholar]
- 165.Nefussi JR, Brami G, Modrowski D, Oboeuf M, Forest N. J Histochem Cytochem. 1997;45:493. doi: 10.1177/002215549704500402. [DOI] [PubMed] [Google Scholar]
- 166.Chen J, McKee MD, Nanci A, Sodek J. Histochem J. 1994;26:67. [PubMed] [Google Scholar]
- 167.Laboux O, Ste-Marie LG, Glorieux FH, Nanci A. J Histochem Cytochem. 2003;51:61. doi: 10.1177/002215540305100108. [DOI] [PubMed] [Google Scholar]
- 168.Acebes C, de la Piedra C, Traba ML, Seibel MJ, Garcia Martin C, Armas J, Herrero-Beaumont G. Clin Chim Acta. 1999;289:99. doi: 10.1016/s0009-8981(99)00170-9. [DOI] [PubMed] [Google Scholar]
- 169.Riminucci M, Bradbeer JN, Corsi A, Gentili C, Descalzi F, Cancedda R, Bianco P. J Bone Miner Res. 1998;13:1852. doi: 10.1359/jbmr.1998.13.12.1852. [DOI] [PubMed] [Google Scholar]
- 170.Hultenby K, Reinholt FP, Norgard M, Oldberg A, Wendel M, Heinegard D. Eur J Cell Biol. 1994;63:230. [PubMed] [Google Scholar]
- 171.Zhu XL, Ganss B, Goldberg HA, Sodek J. Biochem Cell Biol. 2001;79:737. [PubMed] [Google Scholar]
- 172.Sorensen ES, Hojrup P, Petersen TE. Protein Sci. 1995;4:2040. doi: 10.1002/pro.5560041009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Salih E. Connect Tissue Res. 2003;44 1:223. [PubMed] [Google Scholar]
- 174.Salih E, Fluckiger R. J Biol Chem. 2004;279:19808. doi: 10.1074/jbc.M310299200. [DOI] [PubMed] [Google Scholar]
- 175.Suzuki Y, Kubota T, Koizumi T, Satoyoshi M, Teranaka T, Kawase T, Ikeda T, Yamaguchi A, Saito S, Mikuni-Takagaki Y. Connect Tissue Res. 1996;35:223. doi: 10.3109/03008209609029195. [DOI] [PubMed] [Google Scholar]
- 176.Fujisawa R, Nodasaka Y, Kuboki Y. Calcif Tissue Int. 1995;56:140. doi: 10.1007/BF00296346. [DOI] [PubMed] [Google Scholar]
- 177.Tye CE, Hunter GK, Goldberg HA. J Biol Chem. 2005;280:13487. doi: 10.1074/jbc.M408923200. [DOI] [PubMed] [Google Scholar]
- 178.Stubbs JT, 3rd, Mintz KP, Eanes ED, Torchia DA, Fisher LW. J Bone Miner Res. 1997;12:1210. doi: 10.1359/jbmr.1997.12.8.1210. [DOI] [PubMed] [Google Scholar]
- 179.Hunter GK, Goldberg HA. Proc Natl Acad Sci U S A. 1993;90:8562. doi: 10.1073/pnas.90.18.8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Stubbs JT., 3rd Connect Tissue Res. 1996;35:393. doi: 10.3109/03008209609029217. [DOI] [PubMed] [Google Scholar]
- 181.Hunter GK, Goldberg HA. Biochem J. 1994;302(Pt 1):175. doi: 10.1042/bj3020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wazen RM, Tye CE, Goldberg HA, Hunter GK, Smith CE, Nanci A. J Histochem Cytochem. 2007;55:35. doi: 10.1369/jhc.6A7046.2006. [DOI] [PubMed] [Google Scholar]
- 183.Tye CE, Rattray KR, Warner KJ, Gordon JA, Sodek J, Hunter GK, Goldberg HA. J Biol Chem. 2003;278:7949. doi: 10.1074/jbc.M211915200. [DOI] [PubMed] [Google Scholar]
- 184.Harris NL, Rattray KR, Tye CE, Underhill TM, Somerman MJ, D'Errico JA, Chambers AF, Hunter GK, Goldberg HA. Bone. 2000;27:795. doi: 10.1016/s8756-3282(00)00392-6. [DOI] [PubMed] [Google Scholar]
- 185.Ganss B, Kim RH, Sodek J. Crit Rev Oral Biol Med. 1999;10:79. doi: 10.1177/10454411990100010401. [DOI] [PubMed] [Google Scholar]
- 186.Goldberg HA, Warner KJ, Stillman MJ, Hunter GK. Connect Tissue Res. 1996;35:385. doi: 10.3109/03008209609029216. [DOI] [PubMed] [Google Scholar]
- 187.Petersen DN, Tkalcevic GT, Mansolf AL, Rivera-Gonzalez R, Brown TA. J Biol Chem. 2000;275:36172. doi: 10.1074/jbc.M003622200. [DOI] [PubMed] [Google Scholar]
- 188.Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, Oudet CL. Genomics. 2000;67:54. doi: 10.1006/geno.2000.6235. [DOI] [PubMed] [Google Scholar]
- 189.Gowen LC, Petersen DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT, Simmons HA, Crawford DT, Chidsey-Frink KL, Ke HZ, McNeish JD, Brown TA. J Biol Chem. 2003;278:1998. doi: 10.1074/jbc.M203250200. [DOI] [PubMed] [Google Scholar]
- 190.Six N, Septier D, Chaussain-Miller C, Blacher R, DenBesten P, Goldberg M. J Dent Res. 2007;86:780. doi: 10.1177/154405910708600818. [DOI] [PubMed] [Google Scholar]
- 191.Goldberg M, Lacerda-Pinheiro S, Jegat N, Six N, Septier D, Priam F, Bonnefoix M, Tompkins K, Chardin H, Denbesten P, Veis A, Poliard A. Dent Clin North Am. 2006;50:277. doi: 10.1016/j.cden.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 192.Liu H, Li W, Gao C, Kumagai Y, Blacher RW, DenBesten PK. J Dent Res. 2004;83:496. doi: 10.1177/154405910408300612. [DOI] [PubMed] [Google Scholar]
- 193.Rowe PS, Kumagai Y, Gutierrez G, Garrett IR, Blacher R, Rosen D, Cundy J, Navvab S, Chen D, Drezner MK, Quarles LD, Mundy GR. Bone. 2004;34:303. doi: 10.1016/j.bone.2003.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wei X, Ling J, Wu L, Liu L, Xiao Y. J Endod. 2007;33:703. doi: 10.1016/j.joen.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 195.Gluhak-Heinrich J, Pavlin D, Yang W, MacDougall M, Harris SE. Arch Oral Biol. 2007;52:684. doi: 10.1016/j.archoralbio.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Nampei A, Hashimoto J, Hayashida K, Tsuboi H, Shi K, Tsuji I, Miyashita H, Yamada T, Matsukawa N, Matsumoto M, Morimoto S, Ogihara T, Ochi T, Yoshikawa H. J Bone Miner Metab. 2004;22:176. doi: 10.1007/s00774-003-0468-9. [DOI] [PubMed] [Google Scholar]
- 197.Ogbureke KU, Fisher LW. J Dent Res. 2004;83:664. doi: 10.1177/154405910408300902. [DOI] [PubMed] [Google Scholar]
- 198.Siggelkow H, Schmidt E, Hennies B, Hufner M. Bone. 2004;35:570. doi: 10.1016/j.bone.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 199.Rowe PS, Garrett IR, Schwarz PM, Carnes DL, Lafer EM, Mundy GR, Gutierrez GE. Bone. 2005;36:33. doi: 10.1016/j.bone.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Quarles LD. Am J Physiol Endocrinol Metab. 2003;285:E1. doi: 10.1152/ajpendo.00016.2003. [DOI] [PubMed] [Google Scholar]
- 201.Wu L, Zhu F, Wu Y, Lin Y, Nie X, Jing W, Qiao J, Liu L, Tang W, Zheng X, Tian W. Cells Tissues Organs. 2008;187:103. doi: 10.1159/000110079. [DOI] [PubMed] [Google Scholar]
- 202.Rowe PS, Matsumoto N, Jo OD, Shih RN, Oconnor J, Roudier MP, Bain S, Liu S, Harrison J, Yanagawa N. Bone. 2006;39:773. doi: 10.1016/j.bone.2006.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bresler D, Bruder J, Mohnike K, Fraser WD, Rowe PS. J Endocrinol. 2004;183:R1–9. doi: 10.1677/joe.1.05989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.George A, Gui J, Jenkins NA, Gilbert DJ, Copeland NG, Veis A. J Histochem Cytochem. 1994;42:1527. doi: 10.1177/42.12.7983353. [DOI] [PubMed] [Google Scholar]
- 205.MacDougall M, DuPont BR, Simmons D, Leach RJ. Cytogenet Cell Genet. 1996;74:189. doi: 10.1159/000134410. [DOI] [PubMed] [Google Scholar]
- 206.Butler WT. Connect Tissue Res. 1995;33:59. doi: 10.3109/03008209509016983. [DOI] [PubMed] [Google Scholar]
- 207.Kulkarni GV, Chen B, Malone JP, Narayanan AS, George A. Arch Oral Biol. 2000;45:475. doi: 10.1016/s0003-9969(00)00010-8. [DOI] [PubMed] [Google Scholar]
- 208.Qin C, Brunn JC, Cook RG, Orkiszewski RS, Malone JP, Veis A, Butler WT. J Biol Chem. 2003;278:34700. doi: 10.1074/jbc.M305315200. [DOI] [PubMed] [Google Scholar]
- 209.Feng JQ, Huang H, Lu Y, Ye L, Xie Y, Tsutsui TW, Kunieda T, Castranio T, Scott G, Bonewald LB, Mishina Y. J Dent Res. 2003;82:776. doi: 10.1177/154405910308201003. [DOI] [PubMed] [Google Scholar]
- 210.D'Souza RN, Cavender A, Sunavala G, Alvarez J, Ohshima T, Kulkarni AB, MacDougall M. J Bone Miner Res. 1997;12:2040. doi: 10.1359/jbmr.1997.12.12.2040. [DOI] [PubMed] [Google Scholar]
- 211.Kalajzic I, Braut A, Guo D, Jiang X, Kronenberg MS, Mina M, Harris MA, Harris SE, Rowe DW. Bone. 2004;35:74. doi: 10.1016/j.bone.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 212.Gluhak-Heinrich J, Ye L, Bonewald LF, Feng JQ, MacDougall M, Harris SE, Pavlin D. J Bone Miner Res. 2003;18:807. doi: 10.1359/jbmr.2003.18.5.807. [DOI] [PubMed] [Google Scholar]
- 213.Hao J, Zou B, Narayanan K, George A. Bone. 2004;34:921. doi: 10.1016/j.bone.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 214.Narayanan K, Ramachandran A, Hao J, George A. Connect Tissue Res. 2002;43:365. doi: 10.1080/03008200290000592. [DOI] [PubMed] [Google Scholar]
- 215.He G, Dahl T, Veis A, George A. Connect Tissue Res. 2003;44 1:240. [PubMed] [Google Scholar]
- 216.He G, Dahl T, Veis A, George A. Nat Mater. 2003;2:552. doi: 10.1038/nmat945. [DOI] [PubMed] [Google Scholar]
- 217.He G, Gajjeraman S, Schultz D, Cookson D, Qin C, Butler WT, Hao J, George A. Biochemistry. 2005;44:16140. doi: 10.1021/bi051045l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.He G, George A. J Biol Chem. 2004;279:11649. doi: 10.1074/jbc.M309296200. [DOI] [PubMed] [Google Scholar]
- 219.Tartaix PH, Doulaverakis M, George A, Fisher LW, Butler WT, Qin C, Salih E, Tan M, Fujimoto Y, Spevak L, Boskey AL. J Biol Chem. 2004;279:18115. doi: 10.1074/jbc.M314114200. [DOI] [PubMed] [Google Scholar]
- 220.Gajjeraman S, Narayanan K, Hao J, Qin C, George A. J Biol Chem. 2007;282:1193. doi: 10.1074/jbc.M604732200. [DOI] [PubMed] [Google Scholar]
- 221.Ling Y, Rios HF, Myers ER, Lu Y, Feng JQ, Boskey AL. J Bone Miner Res. 2005;20:2169. doi: 10.1359/JBMR.050815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Dimuzio MT, Veis A. J Biol Chem. 1978;253:6845. [PubMed] [Google Scholar]
- 223.Dimuzio MT, Veis A. Calcif Tiss Res. 1978;25:169. doi: 10.1007/BF02010765. [DOI] [PubMed] [Google Scholar]
- 224.Curley-Joseph J, Veis A. J Dent Res. 1979;58:1625. doi: 10.1177/00220345790580061201. [DOI] [PubMed] [Google Scholar]
- 225.Butler WT, Ritchie HH, Bronckers AL. Ciba Found Symp. 1997;205:107–15. doi: 10.1002/9780470515303.ch8. discussion 115. [DOI] [PubMed] [Google Scholar]
- 226.Chang SR, Chiego D, Jr, Clarkson BH. Calcif Tissue Int. 1996;59:149. doi: 10.1007/s002239900101. [DOI] [PubMed] [Google Scholar]
- 227.Clarkson BH, Chang SR, Holland GR. Caries Res. 1998;32:357. doi: 10.1159/000016472. [DOI] [PubMed] [Google Scholar]
- 228.Dahlin S, Angstrom J, Linde A. Eur J Oral Sci. 1998;106 1:239. doi: 10.1111/j.1600-0722.1998.tb02182.x. [DOI] [PubMed] [Google Scholar]
- 229.DiMuzio MT, Bhown M, Butler WT. Calcif Tissue Int. 1985;37:242. doi: 10.1007/BF02554870. [DOI] [PubMed] [Google Scholar]
- 230.Feng JQ, Luan X, Wallace J, Jing D, Ohshima T, Kulkarni AB, D'Souza RN, Kozak CA, MacDougall M. J Biol Chem. 1998;273:9457. doi: 10.1074/jbc.273.16.9457. [DOI] [PubMed] [Google Scholar]
- 231.Ritchie HH, Wang LH. J Biol Chem. 1996;271:21695. doi: 10.1074/jbc.271.36.21695. [DOI] [PubMed] [Google Scholar]
- 232.Ritchie HH, Wang LH, Knudtson K. Biochim Biophys Acta. 2001;1520:212. doi: 10.1016/s0167-4781(01)00274-3. [DOI] [PubMed] [Google Scholar]
- 233.Sabsay B, Stetler-Stevenson WG, Lechner JH, Veis A. Biochem J. 1991;276(Pt 3):699. doi: 10.1042/bj2760699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.George A, Srinivasan R, Thotakura S, Veis A. Eur J Oral Sci. 1998;106 1:221. doi: 10.1111/j.1600-0722.1998.tb02179.x. [DOI] [PubMed] [Google Scholar]
- 235.Wu CB, Veis A. Biomaterials. 1990;11:35. [PubMed] [Google Scholar]
- 236.Wu CB, Pelech SL, Veis A. J Biol Chem. 1992;267:16588. [PubMed] [Google Scholar]
- 237.Veis A, Wei K, Sfeir C, George A, Malone J. Eur J Oral Sci. 1998;106 1:234. doi: 10.1111/j.1600-0722.1998.tb02181.x. [DOI] [PubMed] [Google Scholar]
- 238.Sfeir S, Veis A. J Bone Min Res. 1995;10:607. doi: 10.1002/jbmr.5650100414. [DOI] [PubMed] [Google Scholar]
- 239.Sfeir C, Veis A. Connect Tissue Res. 1996;35:215. doi: 10.3109/03008209609029194. [DOI] [PubMed] [Google Scholar]
- 240.Veis A, Wu CB, Sfeir C. Critical Rev Oral Biol Med. 1997;8:360. doi: 10.1177/10454411970080040101. [DOI] [PubMed] [Google Scholar]
- 241.Mikuni-Takagaki Y, Glimcher MJ. Biochem J. 1990;268(3):585. doi: 10.1042/bj2680585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Mikini-Takagaki Y, Glimcher MJ. Biochem J. 1990;268(3):593. doi: 10.1042/bj2680593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Satoyoshi M, Koizumi T, Teranaka T, Iwamoto T, Takita H, Kuboki Y, Saito S, Mikuni-Takagaki Y. Calcif Tissue Int. 1995;57(3):237. doi: 10.1007/BF00310265. [DOI] [PubMed] [Google Scholar]
- 244.Evans JS, Chan SI. Biopolymers. 1994;34:507. doi: 10.1002/bip.360340407. [DOI] [PubMed] [Google Scholar]
- 245.Evans JS, Chiu T, Chan SI. Biopolymers. 1994;34:1359. doi: 10.1002/bip.360341008. [DOI] [PubMed] [Google Scholar]
- 246.Rabie AM, Veis A. Connect Tissue Res. 1995;31:197. doi: 10.3109/03008209509010811. [DOI] [PubMed] [Google Scholar]
- 247.Laboux O, Menanteau J, Pelhate S, Aubry J, Blottiere HM, Pouezat D, Jean A, Pouezat JA. Hybridoma. 1994;13:143. doi: 10.1089/hyb.1994.13.143. [DOI] [PubMed] [Google Scholar]
- 248.Takagi Y, Fujisawa R, Sasaki S. Connect Tissue Res. 1986;14:279. doi: 10.3109/03008208609017471. [DOI] [PubMed] [Google Scholar]
- 249.Takagi Y, Sasaki S. J Oral Pathol. 1986;15:463. doi: 10.1111/j.1600-0714.1986.tb00659.x. [DOI] [PubMed] [Google Scholar]
- 250.Hao J, He G, Narayanan K, Zou B, Lin L, Muni T, Ramachandran A, George A. Bone. 2005;37:578. doi: 10.1016/j.bone.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 251.Alvares K, Kanyar YS, Veis A. Dev Dynam. 2006;235:2980. doi: 10.1002/dvdy.20935. [DOI] [PubMed] [Google Scholar]
- 252.Dahl T, Sabsay B, Veis A. J Struct Biol. 1998;123:162. doi: 10.1006/jsbi.1998.4025. [DOI] [PubMed] [Google Scholar]
- 253.Traub W, Jodaikin A, Arad T, Veis A, Sabsay B. Matrix. 1992;12:197. doi: 10.1016/s0934-8832(11)80062-4. [DOI] [PubMed] [Google Scholar]
- 254.He G, Ramachandran A, Dahl T, George S, Schultz D, Cookson D, Veis A, George A. J Biol Chem. 2005;280:33109. doi: 10.1074/jbc.M500159200. [DOI] [PubMed] [Google Scholar]
- 255.George A, Hao J. Cells Tissues Organs. 2005;181:232. doi: 10.1159/000091384. [DOI] [PubMed] [Google Scholar]
- 256.Veis A, Sfeir C, Wu CB. Crit Rev Oral Biol Med. 1997;8:360. doi: 10.1177/10454411970080040101. [DOI] [PubMed] [Google Scholar]
- 257.Lee SL, Veis A. Int J Pept Protein Res. 1980;16:231. doi: 10.1111/j.1399-3011.1980.tb02957.x. [DOI] [PubMed] [Google Scholar]
- 258.Burke EM, Guo Y, Colon L, Rahima M, Veis A, Nancollas GH. Colloids & Surfaces, B. 2000;17:49. [Google Scholar]
- 259.Furedi-Milhofer H, Moradian-Oldak J, Weiner S, Veis A, Mintz KP, Addadi L. Connect Tissue Res. 1994;30:251. doi: 10.3109/03008209409015041. [DOI] [PubMed] [Google Scholar]
- 260.Milan AM, Sugars RV, Embery G, Waddington RJ. Eur J Oral Sci. 2006;114:223. doi: 10.1111/j.1600-0722.2006.00347.x. [DOI] [PubMed] [Google Scholar]
- 261.Verdelis K, Lukashova L, Yamauchi M, Atsawasuwan P, Wright JT, Peterson MG, Jha D, Boskey AL. Eur J Oral Sci. 2007;115:296. doi: 10.1111/j.1600-0722.2007.00463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ye L, Mishina Y, Chen D, Huang H, Dallas SL, Dallas MR, Sivakumar P, Kunieda T, Tsutsui TW, Boskey A, Bonewald LF, Feng JQ. J Biol Chem. 2005;280:6197. doi: 10.1074/jbc.M412911200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Ye L, MacDougall M, Zhang S, Xie Y, Zhang J, Li Z, Lu Y, Mishina Y, Feng JQ. J Biol Chem. 2004;279:19141. doi: 10.1074/jbc.M400490200. [DOI] [PubMed] [Google Scholar]
- 264.Sreenath T, Thyagarajan T, Hall B, Longenecker G, D'Souza R, Hong S, Wright JT, MacDougall M, Sauk J, Kulkarni AB. J Biol Chem. 2003;278:24874. doi: 10.1074/jbc.M303908200. [DOI] [PubMed] [Google Scholar]
- 265.Malaval L, Wade-Gueye NM, Boudiffa M, Fei J, Zirngibl R, Chen F, Laroche N, Roux JP, Burt-Pichat B, Duboeuf F, Boivin G, Jurdic P, Lafage-Proust MH, Amedee J, Vico L, Rossant J, Aubin JE. J Exp Med. 2008;205:1145. doi: 10.1084/jem.20071294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Gorski JP, Shimizu K. J Biol Chem. 1988;263:15938. [PubMed] [Google Scholar]
- 267.Gorski JP, Wang A, Lovitch D, Law D, Powell K, Midura RJ. J Biol Chem. 2004;279:25455. doi: 10.1074/jbc.M312408200. [DOI] [PubMed] [Google Scholar]
- 268.Midura RJ, Wang A, Lovitch D, Law D, Powell K, Gorski JP. J Biol Chem. 2004;279:25464. doi: 10.1074/jbc.M312409200. [DOI] [PubMed] [Google Scholar]
- 269.Gorski JP, Griffin D, Dudley G, Stanford C, Lai E, Karr, Solursh M. J Biol Chem. 1990;265:14956. [PubMed] [Google Scholar]
- 270.Gorski JP, Kremer EA, Chen Y, Ryan S, Fullenkamp C, Delvisico J, Jensen K, McKee MD. J Cell Biochem. 1997;64:547. [PubMed] [Google Scholar]
- 271.Midura RJ, Vasanji A, Su X, Wang A, Midura SB, Gorski JP. Bone. 2007 In Press, July 2007. [Google Scholar]
- 272.Huffman NT, Keightley JA, Chaoying C, Midura RJ, Lovitch D, Veno PA, Dallas SL, Gorski JP. J Biol Chem. 2007;282:26002. doi: 10.1074/jbc.M701332200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Irving JT. Fed Proc. 1976;35:109. [PubMed] [Google Scholar]
- 274.Fincham AG, Moradian-Oldak J, Sarte PE. Calcif Tissue Int. 1994;55:398. doi: 10.1007/BF00299322. [DOI] [PubMed] [Google Scholar]
- 275.Du C, Falini G, Fermani S, Abbott C, Moradian-Oldak J. Science. 2005;307:1450. doi: 10.1126/science.1105675. [DOI] [PubMed] [Google Scholar]
- 276.Moradian-Oldak J, Goldberg M. Cells Tissues Organs. 2005;18:202. doi: 10.1159/000091382. [DOI] [PubMed] [Google Scholar]
- 277.Bartlett JD, Ganss B, Goldberg M, Moradian-Oldak J, Paine ML, Snead ML, Wen X, White SN, Zhou YL. Curr Top Dev Biol. 2006;74:57. doi: 10.1016/S0070-2153(06)74003-0. [DOI] [PubMed] [Google Scholar]
- 278.Salih E, Huang JC, Strawich E, Gouverneur M, Glimcher MJ. Connect Tissue Res. 1998;38:225. doi: 10.3109/03008209809017041. [DOI] [PubMed] [Google Scholar]
- 279.Shaw WJ, Campbell AA, Paine ML, Snead ML. J Biol Chem. 2004;279:40263. doi: 10.1074/jbc.C400322200. [DOI] [PubMed] [Google Scholar]
- 280.Veis A. Reviews in Mineralogy and Geochemistry. 2003;54:249. [Google Scholar]