

Abstract
Aims
A domain peptide (DP) matching the Gly2460–Pro2495 region of the cardiac type-2 ryanodine receptor (RyR2), DPc10, is known to mimic channel dysfunction associated with catecholaminergic polymorphic ventricular tachycardia (CPVT), owing to its interference in a normal interaction of the N-terminal (1–600) and central (2000–2500) domains (viz. domain unzipping). Using DPc10 and two other DPs harboring different mutation sites, we investigated the underlying mechanism of abnormal Ca2+ cycling in failing hearts.
Methods and results
Sarcoplasmic reticulum (SR) vesicles and cardiomyocytes were isolated from dog left ventricular muscles for Ca2+ leak and spark assays. The RyR2 moiety of the SR was fluorescently labelled with methylcoumarin acetate (MCA) using DPs corresponding to the 163–195 and 4090–4123 regions of RyR2 (DP163–195 and DP4090–4123, respectively) as site-directed carriers. Both DPs mediated a specific MCA fluorescence labelling of RyR2. Addition of either DP to the MCA-labelled SR induced domain unzipping, as evidenced by an increased accessibility of the bound MCA to a large-size fluorescence quencher. Both SR Ca2+ leak and Ca2+ spark frequency (SpF) were markedly increased in failing cardiomyocytes. Upon introduction of DP163–195 or DP4090–4123 into normal SR or cardiomyocytes, both Ca2+ leak and SpF increased to the levels comparable with those of failing myocytes. K201 (JTV519) suppressed all of the effects induced by DP163–195 (domain unzipping and increased Ca2+ leak and SpF) or those in failing cardiomyocytes, but did not suppress the effects induced by DP4090–4123.
Conclusion
Defective inter-domain interaction between N-terminal and central domains induces diastolic Ca2+ leak, leading to heart failure and lethal arrhythmia. Mutation at the C-terminal region seen in CPVT does not seem to communicate with the aforementioned N-terminal and central inter-domain interaction, although spontaneous Ca2+ leak is similarly induced.
Keywords: Calcium (cellular), Heart failure, Ion channels, SR (function)
1. Introduction
A Ca2+ release channel protein, the ryanodine receptor (RyR2) in sarcoplasmic reticulum (SR), plays a key role in a transient increase of the intracellular Ca2+ concentration from nmol/L to µmol/L during cardiac muscle contraction.1 A considerable amount of evidence has been accumulated that diastolic Ca2+ leak through the RyR2 is one of the problems in failing hearts.1 The Ca2+ leak reduces SR Ca2+ load, thereby decreasing the SR Ca2+ required for an efficient contraction, causing contractile dysfunction. Moreover, the diastolic Ca2+ leak triggers delayed afterdepolarization (DAD) caused by the entry of Na+ via Na+-Ca2+ exchanger, which occasionally results in lethal arrhythmia.2 It has been suggested that dissociation of RyR2-bound FKBP12.6 due to PKA-mediated hyper-phosphorylation may be the cause of diastolic Ca2+ leak seen in various types of heart failure.3 However, several reports suggest that PKA hyper-phosphorylation and subsequent FKBP12.6 dissociation may not necessarily be the primary cause of Ca2+ leak and heart failure.4,5
In our recent report we have shown that defective interactions between the N-terminal domain (1–600) and central domain (2000–2500) of RyR2, harboring many mutation sites of catecholaminergic polymorphic ventricular tachycardia (CPVT)6,7 or ARVC,8 cause Ca2+ leak in failing hearts and reduced SR Ca2+ load, leading to contractile dysfunction.9 The causative mechanism of such dysfunctions seems to be as follows. In normal hearts these domains are interacting with each other forming a zipped state, which stabilizes the closed state of the Ca2+ channel. However, in failing SR the interaction becomes loose (domain unzipping), which de-stabilizes the closed state making the channel leaky. Reversal of the unzipped state to a normal zipped state by K201 (JTV519) restores normal channel gating in otherwise leaky channels of failing SR.9
Almost 40% of the reported CPVT mutation sites are within C-terminal region of RyR2. Recently, Liu et al.10 reported that in the knock-in mouse model of human CPVT mutant of RyR2 (R4496C), sustained bi-directional ventricular tachycardia occurred, and that K201 was without effect on such lethal arrhythmia. These findings suggest that the mutation at C-terminal region may induce Ca2+ sparks or DAD, leading to the lethal arrhythmia, but the defectiveness of RyR2 due to the mutation within C-terminal domain may not be ascribable to the aforementioned defective inter-domain interaction between the N-terminal and central regions. George et al.11 made an attempt to reconstitute the channel gating function by co-expressing a truncated cytoplasmic N-terminal segment (segment P) and C-terminal segment containing channel domain (segment C) of RyR2, and found that in order to confer the agonist-regulated gating function to the channel domain, both Segments P and C must contain a common overlapping segment designated as I-domain that corresponds to residues 3722–4610. This suggests that the I-domain transduces cytoplasmic events to the Ca2+ pore-forming domain. Taken together, inter-domain interaction among N-terminal, central, I-domain, and transmembrane channel domain appears to play a pivotal regulatory role in Ca2+ channel function, although the precise mode of conformational alterations arising from mutations in distinct RyR2 domains may be different.
In the present domain peptide (DP) probe study, we investigated the mode of inter-domain interactions among the three mutable domains (i.e. N-terminal, central, and I- domains) and their involvement in channel regulation. Here, we show that defective inter-domain interactions among these mutable domains indeed cause abnormal Ca2+ cycling in failing hearts.
2. Methods
2.1. Materials
FK506 and K201 were provided by Fujisawa Pharmaceutical Co. Ltd (Osaka, Japan) and Aetas Co. Ltd (Tokyo Japan), respectively.
2.2. Animal model
In beagle dogs weighing 10–13 kg, we induced heart failure by continuous application of rapid ventricular (RV) pacing at 250 b.p.m. using an externally programmed miniature pacemaker (Medtronic Inc., Minneapolis) for 28 days, as described elsewhere.12 This study conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85–23, revised 1996). The care of the animals and the protocols used were in accord with guidelines laid down by the Animal Ethics Committee of Yamaguchi University School of Medicine.
2.3. Preparation of sarcoplasmic reticulum vesicles
We prepared SR vesicles from dog LV muscle as described elsewhere.12 Left ventricles were homogenized in a solution containing 30 mM Tris–malate, 0.3 M sucrose, 5 mg/L leupeptin, and 0.1 mmol/L PMSF,pH 7.0 (solution I). The homogenate was centrifuged at 5500g for 10 min and the resultant supernatant was filtered through four layers of cheesecloth before centrifugation at 12 000g for 20 min. The supernatant was again filtered through cheesecloth and centrifuged at 143 000g for 30 min. The pellet was resuspended in a solution containing 0.6 mol/L KCl, 30 mmol/L Tris–malate, 0.3 mol/L sucrose, 5 mg/L leupeptin, 0.1 mmol/L PMSF, at pH 7.0 (solution II). This suspension was centrifuged at 143 000g for 45 min. The pellet was resuspended again in solution II, homogenized, and centrifuged at 143 000g as described earlier. The pellet was suspended in solution I and centrifuged at 143 000g. The resultant pellet represents the microsomal fraction that is enriched in SR vesicles, and it was homogenized in a solution containing 0.1 mol/L KCl, 20 mmol/L Tris–malate, 0.3 mol/L sucrose, 5 mg/L leupeptin, 0.1 mmol/L PMSF, at pH 7.0, to a final concentration of about 10–20 mg protein/mL. This fraction was rapidly frozen in liquid nitrogen and stored at –80°C. An aliquot was retained for determination of protein concentration by the method of Lowry et al.13
2.4. Peptides used and peptide synthesis
We used the following DPs: DPc10 (DP2460–2495), DPc10-mut, DP163–195, DP163–195-mut, DP4090–4123, and DP4090–4123-mut.
DPc 10 (DP2460–2495) 2460GFCPDHKAAMVLFLDRVYGIEVQDFLLHLLEVGFLP2495
DPc 10-mut (DP 2460–2495-mut R2474S) 2460GFCPDHKAAMVLFLDSVYGIEVQDFLLHLLEVGFLP2495
DP163–195 163HPASKQRSEGEKVRVGDDLILVSVSSERYLHLS195
DP163–195-mut (R176Q) 163HPASKQRSEGEKVQVGDDLILVSVSSERYLHLS195
DP4090–4123 4090PAKDIGFNVAVLLTNLSEHMPNDTRLQTFLELAE4123
DP4090–4123-mut (N4104K) 4090PAKDIGFNVAVLLTKLSEHMPNDTRLQTFLELAE4123
Peptides were synthesized on an Applied Biosystems model 431A synthesizer employing Fmoc (N-(9-fluorenyl)methoxycarbonyl) as the alpha-amino protecting group, as described previously.14 The peptides were cleaved and de-protected with 95% trifluoroacetic acid and purified by reversed-phase high-pressure liquid chromatography.
2.5. Ca2+-uptake and Ca2+-leak assays
Ca2+-uptake and the following Ca2+ leak assays were done as described previously.12 SR vesicles (0.2 mg/mL) were incubated in 0.5 mL of solution containing 0.15 mmol/L potassium gluconate, 1 mmol/L MgCl2, 0.2 mmol/L EGTA-calcium buffer (free [Ca2+] = 0.3 µmol/L), 10 mmol/L NaN3, and 20 mmol/L MOPS, pH 6.8. Ca2+ uptake was initiated by the addition of 0.5 mmol/L ATP into the reaction solution, and the time course of Ca2+ uptake was monitored spectrophotometrically with fluo 3 as a Ca2+ indicator (excitation at 480 nm, emission at 530 nm). After the Ca2+ uptake had reached a plateau, DP was added in the presence of 0.5 µmol/L thapsigargin to inhibit SR Ca2+-ATPase activity, and the resultant Ca2+ leak was measured. The Ca2+ leak was expressed as the ratio of the amount of Ca2+ leaked out from the SR at 60 s after the addition of thapsigargin to the amount of total Ca2+ uptake.
2.6. Site-directed fluorescent labelling of the RyR
Specific fluorescent labelling of RyR2 in SR vesicles was performed using the cleavable hetero-bifunctional cross-linking reagent sulfosuccinimidyl 3-((2-(7-azido-4-methylcoumarin-3-acetamido) ethyl) dithio)propionate (SAED) from PIERCE (IL), with various DPs (DPc10, DP163–195, or DP4090–4123), as site-specific carriers. This method for the site-directed fluorescent labelling of the RyR2 was basically the same as the method used for the DP4-mediated MCA (methylcoumarin acetate)-labelling of RyR1.15 Briefly, peptide-SAED conjugate was formed by incubating 0.5 mmol/L peptide with 0.5 mmol/L SAED in a 20 mmol/L HEPES (pH 7.5) solution for 60 min at 22°C in the dark. The reaction was quenched by 20 mmol/L lysine. Unreacted SAED was removed using ion exchange column (GE HiTrap Q XL). The peptide-SAED conjugate (5 µmol/L in a final concentration) was mixed with 2 mg/mL SR protein in the sample solution containing a 1 mmol/L EGTA/calcium buffer (1.0 µmol/L free Ca2+) in the dark and photolyzed with UV light in a Pyrex tube at 4°C for 2 min. Beta-mercaptoethanol was added (100 mmol/L in a final concentration) to cleave the disulfide bond of SAED. After incubation on ice for 1 h, the mixture was centrifuged at 100 000g for 15 min, and the sedimented vesicles were resuspended in the sample solution to a final protein concentration of 10 mg/mL.
2.7. Fluorescence quenching assay of the MCA probe attached to the binding sites of DPc10, DP163–195, or DP4090–4123
The zipping/unzipping mode of regulatory domains within RyR2 was evaluated as described previously.9,16 To make the large-size quencher, QSY® 7 carboxylic acid (Mr = 791.32) was conjugated with BSA by incubating 5 mmol/L QSY® 7 carboxylic acid with 0.5 mmol/L bovine serum albumin (BSA) in 20 mmol/L HEPES (pH 7.5) for 60 min at 22°C in the dark. Unreacted QSY 7 carboxylic acid was removed by means of Sephadex G50 gel filtration. Fluorescence quenching by both QSY® 7 carboxylic acid–BSA conjugate (a macromolecular quencher) and unconjugated QSY® (a small-size quencher) was performed by measuring steady-state fluorescence of the labelled MCA (excitation at 368 nm, emission at 455 nm) in the presence or absence of various peptides and/or compounds. The data were analysed using the Stern–Volmer equation:15 Fo/F = 1 + KQ [Q] where F and Fo are fluorescence intensities in the presence and in the absence of added quencher; KQ, quenching constant, which is the measure of the accessibility of the protein-bound probe to the quencher; [Q], the concentration of QSY® 7 carboxylic acid–BSA conjugate.
The principle of the fluorescence quench assay of domain unzipping is that a large-size quencher QSY® 7 carboxylic acid–BSA conjugate is not accessible to the attached MCA in the zipped state, whereas it becomes accessible to the MCA in the unzipped state.
2.8. Immunoblot analysis
We performed immunoblot analyses of RyR2-bound FKBP12.6 as described previously.12 Co-immunoprecipitation of FKBP12.6 with RyR2 from the solubilized SR was performed using anti-RyR antibody (Oncogene Research Products), followed by immunoblotting with anti-FKBP12 (C-19) antibody (Santa Cruz Biotechnology).
2.9. Preparation of isolated cardiomyocytes
Cardiomyocytes were isolated from the LV free wall as described previously.9,17 In brief, a wedge of LV free wall, perfused by a branch of the left circumflex coronary artery, was dissected free from the heart and perfused with collagenase-containing buffer. LV myocardium was minced with scissors in fresh collagenase-containing buffer. Then, rod-shaped adult canine cardiomyocytes were prepared by retrograde perfusion of quickly excised hearts with 95%O2/5%CO2-bubbled Minimal Essential Medium (Sigma) supplemented with 50 µmol/L Ca2+, 0.5 mg/mL collagenase B, 0.5 mg/mL collagenase D, and 0.02 mg/mL protease type XIV. The concentration of Ca2+ was then gradually increased to a final concentration of 1 mmol/L by changing the incubation medium (50 µmol/L, 125 µmol/L, 300 µmol/L, and then 1 mmol/L). The isolated canine cardiomyocytes were transferred to laminin-coated glass culture dishes, and incubated for 12 h at 37°C in 5%CO2/95%O2 atmosphere.
2.10. Incorporation of domain peptides into cardiomyocytes
The DP was incorporated into the cells using a protein delivery reagent (Bioporter, Gene Therapy Systems, Inc., CA), as described previously.9 Successful incorporation of the DP into the cell was confirmed by detecting the fluorescence signal of the peptide pre-labelled with Alexa Fluor 488 (Molecular Probes, OR).
2.11. Analysis of Ca2+ sparks with laser scanning confocal microscopy
Ca2+ sparks were measured with a laser scanning confocal microscope (LSM-510, Carl Zeiss) equipped with an argon ion laser coupled to an inverted microscope (Axiovert 100, Carl Zeiss) with a Zeiss ×40 oil-immersion Plan-Neofluor objective (numerical aperture, 1.3; excitation at 488 nm; emission >505 nm), as previously described.18 Briefly, cardiomyocytes were loaded with fluo-4 AM (20 µmol/L; Molecular Probes) for 30 min at room temperature in the dark. To record Ca2+ sparks, line-scan mode was used, where a single cardiomyocyte was scanned repeatedly (325.7 Hz) along a line parallel to the longitudinal axis, avoiding nuclei. To monitor diastolic Ca2+ sparks, cardiomyocytes were stimulated until the Ca2+ transient reached steady state, then stimulation was stopped, and Ca2+ sparks recorded during the subsequent ∼10 s rest. Data were analysed with SparkMaster, an automated analysis program which allows rapid and reliable spark analysis.19 The analysis involved general image parameters (number of detected sparks, spark frequency) as well as individual spark parameters (Amplitude; FWHM, full width at half maximum; FDHM, full duration at half maximum).
2.12. Statistics
Paired or unpaired t-test was used for statistical comparison of the data corresponding to the normal and defective inter-domain interactions. We also used ANOVA with a post hoc Scheffe’s test for statistical comparison of concentration-dependent data. Data are expressed as means±SD. We accepted a P-value less than 0.05 as statistically significant.
3. Results
3.1. Both DP163–195 and DP4090–4123 induce Ca2+ leak without dissociating FKBP12.6 from RyR2
Addition of 0.5 µmol/L thapsigargin to normal SR vesicles at the steady-state of ATP-dependent Ca2+ uptake produced little Ca2+ leak, while addition of 3–100 µmol/L DP163–195, or DP4090–4123, together with 0.5 µmol/L thapsigargin produced a pronounced leak (Figure 1A). However, neither DP163–195-mut nor DP4090–4123-mut induced Ca2+ leak (data not shown). Figure 1B shows the effect of DP2460–2495 (DPc10), DP163–195, or DP4090–4123 on dissociation of FKBP12.6 from RyR2 in normal SR. None of these peptides dissociated FKBP12.6 from RyR2. The Ca2+ leak induced by DP163–195 was almost completely inhibited by 0.3 µmol/L K201, whereas the Ca2+ leak induced by DP4090–4123 was not inhibited by K201 (Figure 1C). In failing SR, spontaneous Ca2+ leak took place without addition of the peptides (DP163–195 and DP4090–4123), and the addition of these peptides produced little or no further increase in Ca2+ leak. The Ca2+ leak was inhibited by K201, although K201 was without effect in the presence of DP4090–4123 (Figure 1D).
Figure 1.

(A) Concentration-dependent effects of DP163–195 and DP4090–4123 on Ca2+ leak from normal sarcoplasmic reticulum. Representative time courses of Ca2+ leak following Ca2+ uptake (top figure) and summarized data (bottom figure). *P < 0.01 vs. baseline. (B) Effects of DPc10, DP163–195, and DP4090–4123 on dissociation of FKBP12.6 from RyR2. Before immunoprecipitation of RyR, sarcoplasmic reticulum vesicles were mixed with DP163–195 or DP4090–4123 for 30 min and then centrifuged, followed by western blotting. (C) Effect of K201 (0.3 µmol/L) on the DP163–195-induced or DP4090–4123-induced Ca2+ leak in normal sarcoplasmic reticulum. (D) Effect of K201 (0.3 µmol/L) on spontaneous Ca2+ leak seen in failing sarcoplasmic reticulum, in the presence of DP163–195 (30 µmol/L) or DP4090–4123 (30 µmol/L).
3.2. Specific fluorescent labelling of the peptide binding domains of RyR2
Figure 2 shows the fluorescence gel pictures of the MCA-labelled RyR2. Each of these peptides used as a site direction carrier (DPc10, DP163–195, or DP4090–4123) mediated a specific MCA fluorescence labelling of RyR2. However, each of the human ARVC or CPVT mutants of these peptides {Arg-2474-Ser mutation (DPc10mut), Arg-176-Gln mutation (DP163–195-mut), or Asn-4104-Lys mutation (DP4090-4123-mut)} did not mediate fluorescence labelling (Figure 2, left). An excess concentration of each unlabelled peptide (10 mmol/L) also prevented DP(DP163–195 or DP4090–4123)-mediated MCA labelling (Figure 2, right). In our previous report,9 an excess concentration of unlabelled DPc10 also prevented the DPc10-mediated MCA labelling.
Figure 2.

Site-directed fluorescence labelling of RyR2 with methylcoumarin acetate. Site-specific methylcoumarin acetate fluorescence labelling was performed using either DPc10, DP163–195, or DP4090–4123 as a site-directing carrier. No methylcoumarin acetate fluorescence was seen when corresponding DPmut was used (left figure). An excess concentration of each unlabelled peptide (10 mmol/L) also inhibited each domain peptide-mediated methylcoumarin acetate labelling (right figure).
3.3. Spectroscopic monitoring of the changes in the mode of inter-domain interactions induced by domain peptide (DP163–195 or DP4090–4123)
In order to monitor the mode of the inter-domain interactions, we adopted the fluorescence quenching technique.16
The MCA probe that has been attached to the critical domain is expected to be inaccessible to a bulky fluorescence quencher (QSY-BSA conjugate) in a zipped configuration (because in this case the probe is occluded between the interacting domains), while it will become accessible to the quencher upon unzipping (see Methods). The accessibility of the protein-bound MCA to the QSY-BSA quencher, i.e. the extent of domain unzipping, can be quantitatively determined as the slope (=the quenching constant, KQ) of the Stern–Volmer plot (Fo/F = 1+KQ [Q]).
The data of fluorescence quenching assay are shown in Figure 3. When MCA labelling was mediated by DP163–195 (Figure 3A), DP163–195 increased the slope (KQ) in a concentration-dependent manner, indicating that this peptide interfered with the inter-domain interaction between the N-terminal and central domains, causing domain unzipping (Figure 5, a). In this MCA-labelled RyR2 using DP163–165 as a carrier, DP4090–4123 had no effect on the KQ, indicating that DP4090–4123 has no effect on the inter-domain interaction between N-terminal and central domains (Figure 5, b). K201 inhibited the increase in KQ induced by DP163–195 (cf: Scheme 1-f), but DP163–195mut had no effect. The data with the RyR2, MCA-labelled with DP4090–4123, are shown in Figure 3B. DP4090–4123 increased KQ, indicating that the peptide produced domain unzipping between I-domain and the putative partner domain of I-domain (IP domain) (Figure 5, b). DP4090-4123-mut had no effect. Interestingly in this case when MCA was introduced via DP4090–4123, DP163–195 also increased the KQ (Figure 5, a). K201 inhibited the increase of KQ induced by DP163–195 (Figure 5, f), but not that by DP4090–4123 (Figure 5, c). These results suggest that domain unzipping between N-terminal and central domain causes another domain unzipping between IP-domain and I-domain in a coupled manner; hence inhibition of the former by K201 results in the inhibition of the latter as well. In contrast, domain unzipping between the IP-domain and the I-domain does not cause domain unzipping between the central domain and the N-terminal domain.
Figure 3.


Fluorescence quenching analysis of domain unzipping (upper figures). A fluorescent probe methylcoumarin acetate was attached to the RyR2 of normal and failing sarcoplasmic reticulum in a site-specific manner using either DP163–195 (A) or DP4090–4123 (B) as a carrier. Then, the accessibility of the RyR2-bound MCA to a macromolecular fluorescence quencher BSA-QSY conjugate, the Stern–Volmer fluorescence quenching constant (i.e. the slope of the Fo/F vs. [BSA-QSY] plot), was determined as a measure of the degree of domain unzipping. Statistical comparison of the slope of each plot, which is equivalent to the Stern–Volmer quenching constant (KQ) (bottom figures). DP163–195 increased the accessibility of the protein-bound MCA (carrier: DP163–195) to the quencher, as shown by an increase of the slope of the plot. K201 reversed the effect of DP163–195 in normal SR and decreased an elevated level of quencher accessibility in failing SR. DP4090–4123 also increased the accessibility of the bound MCA (carrier: DP4090–4123) to the quencher in normal SR, but in this case K201 was without effect.
Figure 5.
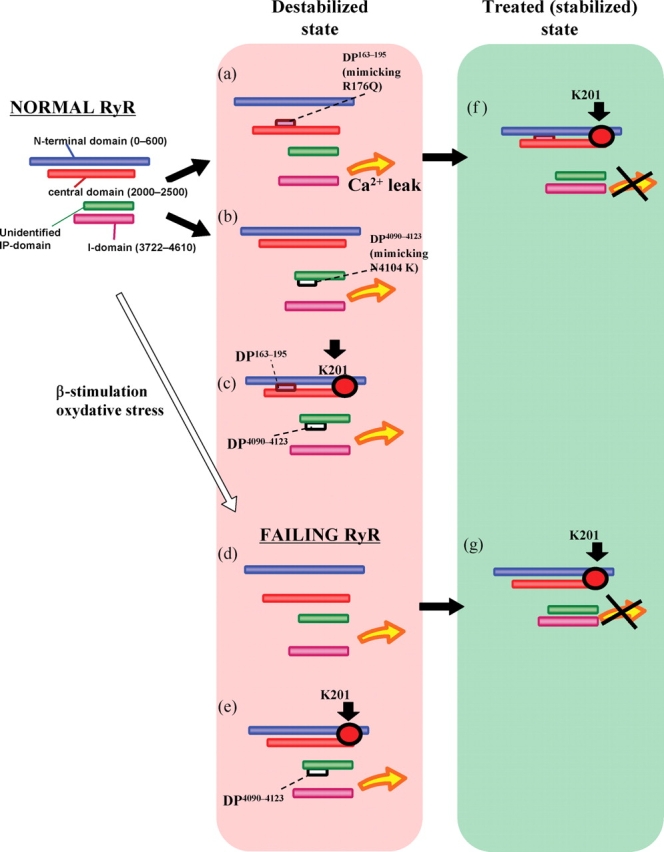
Model illustrating how defects in the inter-domain interactions within RyR2 cause channel dysfunctions (diastolic Ca2+ leak) in heart failure and ARVC2/CPVT. N, N-terminal domain (1–600); C, central domain (2000–2500); I, I-domain; IP, IP-domain (putative partner domain of I-domain; unidentified).
Figure 3 (right side) depicts the fluorescence quenching data obtained with the RyR2 of failing SR MCA-labelled with either DP163–195 (Figure 3A) or DP4090–4123 (Figure 3B) as well. The KQ in the RyR2 of failing SR was significantly larger compared with the KQ in the normal RyR2 without added peptide (Figure 5, d), but was comparable with the value of normal RyR2 that was reached after the addition of DP (4090–4123 or 163–195) (Figure 5, a or b). Addition of these DP to the failing RyR2 produced no further increase in the KQ value, indicating that similar domain unzipping has taken place in the failing RyR2 even without addition of DPs. K201 decreased the KQ of the failing RyR2 to a normal level (Figure 5, g).
3.4. Effect of domain peptides on Ca2+ sparks in normal and failing cardiomyocytes
In order to investigate how the domain-unzipping effect of DP163–195 or DP4090–4123 is reflected upon the cardiomyocyte function, we incorporated DP163–195 or DP4090–4123 into the normal and failing cardiomyocytes by mediation of BioPorter (see Methods). Successful incorporation of DP163–195 or DP4090–4123 into the cell was confirmed by detecting the intra-cellular fluorescence signal of Alexa-labelled DPs (Figure 4A).
Figure 4.


(A) Delivery of DP163–195 or DP4090–4123, fluorescently labelled with Alexa Fluor 488 (Molecular Probes, OR), into the isolated cardiomyocytes. Confocal microscopy clearly detects the fluorescence signal (shown as green) of DP163–195 or DP4090–4123 in the cardiomyocytes. Cell surface membrane was fluorescently labelled as red by wheat germ agglutinin-Alexa Fluor 633 conjugate (Molecular Probes, OR). (B) Spontaneous Ca2+ sparks in DP163–195- or DP4090–4123-incorporated normal and failing cardiomyocytes at either 2 or 5 mmol/L extra-cellular [Ca2+]. (C) Effect of K201 on spontaneous Ca2+ sparks in DP163–195- or DP4090–4123-incorporated normal and failing cardiomyocytes at 5 mmol/L extra-cellular [Ca2+]. Summarized data are shown at the right side. FWHM, full width at half maximum; FDHM, full duration at half maximum.
We monitored spontaneous Ca2+ sparks in cardiomyocytes at two different extracellular [Ca2+]s: 2 and 5 mmol/L. As shown in Figure 4B, the occurrence of Ca2+ sparks was less frequent in normal cardiomyocytes. However, incorporation of DP163–195 or DP4090–4123 into normal cardiomyocytes resulted in a significant increase in the Ca2+ spark frequency particularly at [Ca2+]=5 mmol/L. Failing cardiomyocytes showed high Ca2+ spark frequency similar to the peptide-treated normal myocytes, even without introduction of the peptides. By incorporation of the peptides, both FWHM and FDHM changed in the same direction as spark frequency, whereas only spark amplitude changed in an opposite direction. This indicates that as Ca2+ spark frequency increases, Ca2+ spark amplitude decreases, and a Ca2+ spark spreads more in time and spatial direction. Incorporation of either DP163–195-mut (ARVC Arg-176-Gln mutation) or DP4090–4123-mut (CPVT Asn-4104-Lys mutation) into normal cardiomycytes produced no appreciable effect on the Ca2+ spark frequency (data not shown). Protein delivery reagent (Bioporter) alone also had no effect on the Ca2+ spark frequency (data not shown). In normal cardiomyocytes, K201 inhibited the DP163–195-induced Ca2+ sparks, but not those induced by DP4090–4123 (Figure 4C). Addition of K201 inhibited the Ca2+ spark in failing cardiomyocytes, even in the presence of DP163–195. However, when DP4090–4123 was incorporated into the failing cardiomyocytes, K201 produced virtually no effect on the Ca2+ spark frequency. In response to K201, both FWHM and FDHM again changed in the same direction as Ca2+ spark frequency, whereas only Ca2+ spark amplitude changed in an opposite direction.
4. Discussion
We have previously reported that in failing hearts the inter-domain interaction between N-terminal (1–600) and central domain (2000–2500) of RyR2 becomes defective, resulting in abnormal intracellular Ca2+ handling, such as Ca2+ leak from RyR2 and prolonged Ca2+ transient.9 The present study has uncovered several new mechanisms of domain-mediated RyR2 channel regulation and its pathology. Thus, it was found that defective inter-domain interactions not only in the N-terminal domain/central domain but also in IP-domain (putative partner domain of I-domain, which remains to be identified)/I-domain causes various pro-arrhythmia states, such as increased frequency of spontaneous Ca2+ sparks and appearance of DAD.
There are several reports supporting the view that the I-domain (3722–4610) is involved in the regulation of channel gating. George et al.11,20 have shown that functional coupling between the cytoplasmic and transmembrane domains of RyR2 is mediated by the I-domain. Thus, sudden cardiac death (SCD)-linked mutations occurring in the I-domain (N4104K and R4496C) cause RyR2 channel instability via defective inter-domain interaction, resulting in Ca2+ release dysfunction. Other investigators also empathized the important role of several sub-domains located within the I-domain in the regulation of channel gating. Xiong et al.21 found that the 4064–4210 residue segment of RyR1 shows a CaM-like sequence (CaMLD), and suggested that this region may be involved in the mechanism of Ca2+-dependent channel activation. The fact that CPVT mutations are not randomly scattered over the I-domain, but rather clustered in a rather restricted region; namely in the 3778–4200 region, especially in the 4090–4123 region, suggests that the sub-domain corresponding to DP4090–4123 plays a critical role on normal channel gating.
The main findings of the present study and the postulated model deduced from these findings are schematically illustrated in (Figure 5). Normal RyR: In normal (stabilized) RyR2 at a non-activated or resting state, domain–domain interaction between N-terminal and central domain is coupled with another domain–domain interaction between IP-domain and I-domain, each of which forms a ‘zipped’ complex. Destabilized state, top: Addition of (DPc10 or) DP163–195 unzips the N-terminal domain/central domain. This induces, in a coupled manner, unzipping of IP-domain/I-domain, causing destabilized channel and diastolic Ca2+ leak (state-a). Presumably, CPVT mutation in either N-terminal domain or central domain, beta-adrenergic stimulation9 or oxidative stress,22 produces diastolic Ca2+ leak following the same domain unzipping mechanism. Destabilized state, middle: As shown in the fluorescence quenching experiment, DP4090–4123 unzips IP-domain/I-domain, but not the N-terminal domain/central domain (state-b). Effect of K201: Addition of K201 to the destabilized RyR2 (with both regions unzipped) restores a zipped configuration as in the ‘normal RyR’ [see Treated (stabilized) state-f]. However, the drug is ineffective to the RyR2 with only IP-domain/I-domain unzipped (Destabilized state, bottom: c). Failing RyR: The most likely configuration of failing RyR2 would be as seen in the ‘top configuration of destabilized state’ of this diagram (state-d). Addition of K201 to this configuration will induce zipping first in the N-terminal domain/central domain, then in IP-domain/I-domain (state-g). Addition of DP4090–4123 to this re-stabilized failing RyR2 will destabilize the channel again, because of the induced unzipping in IP-domain/I-domain (state-e).
We recently identified the K201-binding site as domain2114–2149 of the ryanodine receptor (RyR2), and the binding of K201 to this domain corrects the defective inter-domain interaction between N-terminal (1–600) and central regions (2000–2500) of RyR2 in pacing-induced failing hearts.23 The effect of K201 on the mode of inter-domain interactions described here has provided a further, new insight into the underlying mechanism for diastolic spontaneous Ca2+ sparks and DAD seen in failing cardiomyocytes. As shown, the primary action of K201 on the channel function of RyR2 is the correction of unwanted domain unzipping between the N-terminal and central domains seen in failing SR or in DPc10-introduced normal SR.9 The increase in Ca2+ spark frequency seen in failing cardiomyocytes was inhibited by K201, but when DP4090–4123 was incorporated into the failing cardiomyocytes, Ca2+ sparks remained abnormal even in the presence of K201. These findings suggest that in the tachycardia-induced failing hearts Ca2+ sparks are indeed induced by defective inter-domain interaction between N-terminal and central domains. The fact that K201 is less effective in correcting the defective IP-domain/I-domain interaction suggests that K201 may also be less effective in correcting the problem produced by CPVT mutations within the I-domain. The fact that K201 was not effective in the R4496C RyR2 mutant in knock-in mice, which showed bi-directional VT,10 seems to be consistent with the present data that neither Ca2+ leak nor Ca2+ sparks, induced by DP4090–4123, was inhibited by K201. The point mutation, such as R4496C or N4104K at I-domain, seen in CPVT may directly induce domain unzipping between IP-domain and I domain, independent of the mode of inter-domain interaction between N-terminal and central domains.
While elevation of SR Ca2+ content obviously increases the amount of releasable Ca2+, it may become as a causative factor of spontaneous diastolic SR Ca2+ release.24 In failing hearts, in which SR Ca2+ content is decreased,9 Ca2+ spark frequency was quite high even in baseline conditions, and increasing [Ca2+] further increased the Ca2+ spark frequency. This suggests that the threshold of SR Ca2+ load for the induction of spontaneous Ca2+ spark is made much lower in failing hearts than in normal hearts. With regard to this, Jiang et al.25,26 recently proposed that in mutation-linked RyR2 disorder the threshold of SR Ca2+ content for spontaneous Ca2+ release, referred as store-overload-induced Ca2+ release (SOICR), was much lower than normal RyR2. The SOICR may also be involved in the defective channel gating in failing hearts, which may also be caused by defective inter-domain interaction in failing RyR2.
Although we have not identified IP-domain yet, the project for identification of IP-domain using LC/MS spectroscopy is now on-going.
In conclusion, three hot regions within RyR2 (N-terminal, central, and I- domains), harbouring many ARVC2 or CPVT mutations, may play a critical role in regulating channel function. Defective inter-domain interactions among these domains may lead to serious problems of channel gating, as seen in abnormal Ca2+ spark events and DAD, leading to heart failure and lethal arrhythmia.
Conflict of interest: none declared.
Funding
This work was supported by grants-in-aid for scientific research from The Ministry of Education in Japan (grant nos. 18390234, 20390226 to M.Y., 18590777, 20590868 to T.Y., 18591706, 20591805 to S.K., 19209030 to M.M.), and grant from the National Heart, Lung and Blood Institutes (HL072841 to N.I.).
References
- 1.Yano M, Yamamoto T, Ikemoto N, Matsuzaki M. Abnormal Ryanodine Receptor Function in Heart Failure. (Review) Pharmacol Therapeut. 2005;107:377–391. doi: 10.1016/j.pharmthera.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 3.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 4.Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–1022. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- 5.Xiao B, Sutherland C, Walsh MP, Chen SR. Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6) Circ Res. 2004;94:487–495. doi: 10.1161/01.RES.0000115945.89741.22. [DOI] [PubMed] [Google Scholar]
- 6.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 7.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–490. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 8.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum Mol Genet. 2001;10:189–194. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 9.Oda T, Yano M, Yamamoto T, Tokuhisa T, Okuda S, Doi M, et al. Defective regulation of inter-domain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. 2005;111:3244–3254. doi: 10.1161/CIRCULATIONAHA.104.507921. [DOI] [PubMed] [Google Scholar]
- 10.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia:insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 11.George CH, Jundi H, Thomas NL, Scoote M, Walters N, Williams AJ, et al. Ryanodine receptor regulation by intramolecular interaction between cytoplasmic and transmembrane domains. Mol Biol Cell. 2004;15:2627–2638. doi: 10.1091/mbc.E03-09-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, et al. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–2136. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 13.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 14.Yamamoto T, Ikemoto N. Peptide probe study of the critical regulatory domain of the cardiac ryanodine receptor. Biochem Biophys Res Commun. 2002;291:1102–1108. doi: 10.1006/bbrc.2002.6569. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto T, Ikemoto N. Spectroscopic monitoring of local conformational changes during the intramolecular domain-domain interaction of the ryanodine receptor. Biochemistry. 2002;41:1492–1501. doi: 10.1021/bi015581z. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto T, El-Hayek R, Ikemoto N. Postulated role of interdomain interaction within the ryanodine receptor in Ca(2+) channel regulation. J Biol Chem. 2000;275:11618–11625. doi: 10.1074/jbc.275.16.11618. [DOI] [PubMed] [Google Scholar]
- 17.Igarashi-Saito K, Tsutsui H, Yamamoto S, Takahashi M, Kinugawa S, Tagawa H, et al. Role of SR Ca -ATPase in contractile dysfunction of myocytes in tachycardia-induced heart failure. Am J Physiol. 1998;274:H31–H40. doi: 10.1152/ajpheart.1998.275.1.H31. [DOI] [PubMed] [Google Scholar]
- 18.Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Gyorke I, et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res. 2006;98:1151–1158. doi: 10.1161/01.RES.0000220647.93982.08. [DOI] [PubMed] [Google Scholar]
- 19.Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster - automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol. 2007;293:C1073–C1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- 20.George CH, Jundi H, Walters N, Thomas NL, West RR, Lai FA. Arrhythmogenic mutation-linked defects in ryanodine receptor autoregulation reveal a novel mechanism of Ca2+ release channel dysfunction. Circ Res. 2006;98:88–97. doi: 10.1161/01.RES.0000199296.70534.7c. [DOI] [PubMed] [Google Scholar]
- 21.Xiong L, Zhang JZ, He R, Hamilton SL. A Ca2+-binding domain in RyR1 that interacts with the calmodulin binding site and modulates channel activity. Biophys J. 2006;90:173–182. doi: 10.1529/biophysj.105.066092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yano M, Okuda S, Oda T, Tokuhisa T, Tateishi H, Mochizuki M, et al. Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation. 2005;112:3633–3643. doi: 10.1161/CIRCULATIONAHA.105.555623. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto T, Yano M, Xu X, Uchinoumi H, Tateishi H, Mochizuki M, et al. Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation. 2008;117:762–772. doi: 10.1161/CIRCULATIONAHA.107.718957. [DOI] [PubMed] [Google Scholar]
- 24.Bers DM. Sarcoplasmic reticulum Ca release in intact ventricular myocytes. Front Biosci. 2002;7:d1697–d1711. doi: 10.2741/A873. [DOI] [PubMed] [Google Scholar]
- 25.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci USA. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–1181. doi: 10.1161/01.RES.0000192146.85173.4b. [DOI] [PubMed] [Google Scholar]