

Abstract
G-protein-coupled receptors (GPCRs) form the largest superfamily of membrane proteins, and several GPCRs have been implicated in signaling between neurons and glia to protect neurons from pathological stresses. Here, we have used a screening strategy to investigate GPCRs that are involved in neuronal protection. The real-time PCR was performed using 274 primers targeting nonsensory GPCR mRNAs, which were listed on the database. The cDNAs from control and nerve-injured hypoglossal nuclei of mouse brain were used, and the alterations of PCR products were compared. This screen and the subsequent in situ hybridization screen exhibited six GPCR mRNAs which were prominently and convincingly induced in nerve-injured hypoglossal nuclei. Among these candidates, the chemokine receptor CCR5 was selected, based on the marked induction in CCR5 mRNA in microglia after nerve injury. The mRNA expression of ligands for CCR5, such as regulated on activation normal T-cell expressed and secreted (RANTES/CCL5), MIP-1α, and MIP-1β, were induced in injured motor neurons, indicating that CCR5 and its ligands were expressed in microglia and neurons, respectively, in response to nerve injury. In vitro, lipopolysaccharide (LPS)-induced expression of mRNAs for inflammatory cytokines (IL-1β, IL-6, and tumor necrosis factor-α) and inducible nitric oxide synthase (iNOS) in microglia were all suppressed by RANTES. Those suppressions were not observed in microglia from CCR5 null mice. In addition, nerve injury-induced motor neuron death seen in wild type C56BL/6J mice was accelerated in CCR5 knock-out C57BL/6J. These results may suggest that CCR5-mediated neuron–glia signaling functions to protect neurons by suppressing microglia toxicity.
Keywords: GPCR, chemokine, microglia, regeneration, neuroprotection, axotomy
Introduction
The bidirectional interactions between neuron and glia are crucial for maintaining normal neuronal activities such as signal transmission and plasticity (Araque et al., 1999; Perea and Araque, 2007). This bidirectional interaction is also important for neurons under pathological conditions to be protected from various deleterious stresses and to regenerate their axons (Floyd and Lyeth, 2007). However, the mediators between neuronal and glial cells and their functional consequences have not yet been fully revealed. To identify molecules that are associated with nerve regeneration, we have attempted transcriptomic and proteomic screenings, such as a differential display, expressed sequence tag (EST) approach, DNA microarray, and mass spectrometry analysis, using nerve-injured hypoglossal nuclei (Kiryu et al., 1995; Tanabe et al., 1999; Konishi et al., 2006). Those screenings did provide us with several nerve injury-associated molecules and the mechanisms involved (Kiryu-Seo et al., 2000; Nakagomi et al., 2003; Namikawa et al., 2006). However, most of those functioned in neurons, and molecules that mediated the neuron–glia signaling were less in number than expected. This prompted us to take an alternative approach focusing on G-protein-coupled receptor (GPCR) for the following reasons.
GPCRs are the largest family of membrane-bound receptors, and it has been estimated that more than half of all drugs target these receptors (Lagerstrom and Schioth, 2008). Based on the identification of many novel GPCR sequences emerging from ESTs and other DNA sequencing programs, it has become clear that ∼1000–1700 GPCRs belong to this family (Foord et al., 2005; Bjarnadottir et al., 2006). GPCRs are activated by a wide variety of ligand types, including ions, amino acids, light, lipids, nucleotides, peptides, and proteins (Lagerstrom and Schioth, 2008). Recent evidence has revealed that some of those were in fact used as signal mediators between neurons and glial cells. For instance, an ATP receptor such as P2Y12 (Haynes et al., 2006; Kettenmann, 2006) and a chemokine receptor such as CX3CR1 (Meucci et al., 2000) were expressed by microglia, and these signaling molecules functioned as a vital mediator between neurons and microglia in nerve regeneration and neuropathic pain sensation. It is therefore likely that additional GPCRs might be involved in the neuron–glia signaling pathway and may function in promoting the process of proper nerve regeneration. Although the number of putative GPCRs based on ESTs is ∼800 in human and 1700 in mouse (Foord et al., 2005; Bjarnadottir et al., 2006), the large population of GPCRs is assumed to be associated with olfaction, light, and taste sensations. The International Union of Pharmacology published a GPCR list containing 359 nonsensory receptors and 446 sensory GPCRs (7 opsin-like, 39 taste, and ∼400 olfactory receptors) in human (Foord et al., 2005), and Bjarnadóttir et al. (2006) estimated 326 nonsensory GPCRs and 1371 sensory GPCRs (1037 olfactory, 34 taste, and 300 rhodopsin-like receptors) in mouse. These nonsensory GPCRs include previously characterized receptors for peptides, monoamines, amino acids, and purines as well as several orphan GPCRs. We thus estimated that, by preparing specific primers, PCR-based screening of those ∼300 GPCRs was achievable and examined the gene expressions of those GPCRs in response to nerve injury using real-time PCR and tissues from the mouse hypoglossal nerve injury model. For this gene screen, we selected 274 primers, and those sequences were designed to target putative nonsensory GPCR mRNAs, which were listed on the database, including the orphan receptors but excluding sensory GPCRs. Using this screen, we have successfully identified candidate GPCRs, and we focused on a chemokine receptor CCR5 among the candidates and analyzed a functional significance of CCR5 as a signal mediator between neuron and microglia in nerve injury.
Materials and Methods
Animals.
Seven-week-old male C57BL/6 mice (n = 142) and CCR5-deficient mice (n = 5) were anesthetized with pentobarbital (45 mg/kg, i.p.) and positioned supine; their right hypoglossal nerve was then cut with scissors. CCR5-deficient mice used in this study were established by Murai et al. (2003). All mouse strains had an inbred C57BL/6 genetic background or had been backcrossed at least six times. This study was approved by the Animal Ethics Committee of Osaka City University, and all experiments were performed in compliance with institutional guidelines.
Histology.
C57BL/6 mice were decapitated 0, 1, 3, 5, 7, 14, 21, 28, 35, 42, 49, and 56 d after the operation (n = 3 in each time point except 35 d; 35 d, n = 8), and CCR5-deficient mice were decapitated 35 d after the operation (n = 5). Brains were quickly removed and frozen in powdered dry ice. Sections, 13 μm thick, were cut using a cryostat, thaw mounted onto 3-amino propyltriethoxysilane-coated slides, and stored at −80°C until use. The defined level of the hypoglossal nucleus was serially cut, and the serial sections were divided into six groups. Every sixth section was attached on the same slide. The adjacent section was placed on another slide from the next group, and accordingly six groups of slides from one animal were prepared.
Cell counts were done blind for treatment condition, using a well established counting method that effectively eliminated the possibility of counting the same cell twice (Clarke and Oppenheim, 1995). According to the literature, the following criteria were used for counting valid cells: a large soma, a clear nucleus with an intact nuclear membrane, and at least one large clump of nucleolar material. For quantification of motor neuron survival after unilateral nerve transection, thionine-stained hypoglossal motor neurons in the right (operated side) and left (control side) hypoglossal nuclei were counted separately in every sixth section of each animal examined. Counts were performed using four independent groups of slides per animal. Data were presented as percentages of surviving neurons on operated and control sides. Statistical significance (p value) was evaluated using the two-tailed Student's t test.
Real-time quantitative reverse transcription-PCR.
Real-time quantitative reverse transcription (RT)-PCR using the SYBR Green-based method was performed as described previously (Aoki et al., 2002). Total RNA was isolated from the hypoglossal nuclei on either side from 100 mice 5 d after surgery (Tanabe et al., 1999). These RNAs (16 μg) were treated with DNase I and converted to cDNA with Superscript reverse transcriptase (Invitrogen) and random hexamer primers according to the instructions of the manufacturer. The efficiency of reverse transcription and the quality of cDNA were evaluated by PCR of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene. Primers were designed for 274 mouse GPCR genes (supplemental table, available at www.jneurosci.org as supplemental material) in the GPCR database (http://www.gpcr.org/7tm/) using Primer Express software (PerkinElmer Life and Analytical Sciences). SYBR Green-based real-time RT-PCR was performed in 12.5 μl reactions (ABI PRISM 7700 Sequence Detection System; PerkinElmer Life and Analytical Sciences). PCR products were analyzed by agarose gel electrophoresis (data not shown); we also verified products resulting from PCR with all primer sets. The primer sets that were not specific for target GPCR genes were redesigned and checked again by PCR, and these processes were repeated. Information on primer sequences can be obtained from S.A. (by e-mail: aokis@bio.kyutech.ac.jp). We further ensured that no bands resulted from PCR reactions containing primer sets and distilled water or non-reverse-transcribed RNAs (data not shown). The quantitative RT-PCR method (User Bulletin #2; Applied Biosystems) was modified to establish an expression level index for mRNA (Aoki et al., 2002), and the SYBR Green signal for GAPDH (GenBank accession number BC083149; forward primer, 5′-gaaggtggtgaagcaggcatc-3′; reverse primer, 5′-ccactcttccaccttcgatgc-3′) amplicon was used as a reference. Amplification efficiency was determined in a control PCR experiment using serial cDNA dilutions as template. The real-time RT-PCR products were analyzed using the Applied Biosystems sequence detection system software version 1.7.
In situ hybridization.
The following cDNA fragments were isolated by RT-PCR used as probes: CCR5, 1408–2368 of GenBank accession number D83648; CCL3 (MIP-1α), 81–359 of GenBank accession number NM_011337; CCL4 (MIP-1β), 78–356 of GenBank accession number NM_013652; and CCL5, 40–315 of GenBank accession number NM_013653. These plasmids were linearized, and 35S-labeled cDNA probes were prepared by in vitro transcription using SP6 or T7 RNA polymerase (Promega) and 35S-UTP (PerkinElmer Life and Analytical Sciences). In situ hybridization (ISH) was performed as described by Kiryu et al. (1995). Optical density of hybridization signal on film autoradiography was measured as an absolute value, using a computerized image analysis system (MCID; Imaging Research).
Semiquantitative RT-PCR.
Total RNAs were isolated from the hypoglossal nuclei of 20 mice and primary culture of microglial cells and reverse transcribed, respectively, using superscript II (Invitrogen) and oligo-dT. Aliquots from the RT reaction were used for PCR amplification using primer pairs ubiquitously expressing GAPDH as a control. The specific primers for the mRNA of mouse MIP-1α (GenBank accession number NM_011337; forward primer, 5′-atgaaggtctccaccactgcccttgc-3′; reverse primer, 5′-tcaggcattcagttccaggtcagtga-3′), MIP-1β (GenBank accession number NM_013652; forward primer, 5′-atgaagctctgcgtgtctgccctct-3′; reverse primer, 5′-tcagttcaactccaagtcactcatg-3′), and regulated on activation normal T-cell expressed and secreted (RANTES) (GenBank accession number NM_013653; forward primer, 5′-atgaagatctctgcagctgccctc-3′; reverse primer, 5′-ctagctcatctccaaatagttgatg-3′) were used and amplified by PCR. The reaction products were electrophoretically separated on a 1% agarose gel and visualized by staining with ethidium bromide.
Combined immunohistochemistry and in situ hybridization.
Immunohistochemistry was combined with ISH to identify the cellular source of CCR5 in the injured hypoglossal nucleus. Every sixth brain section was processed by the avidin–biotin method with peroxidase as a substrate, as described previously. Microglia were labeled with an antibody against iba1 (Wako Pure Chemicals). Briefly, sections were rinsed with potassium-PBS (K-PBS) and incubated with primary antibody. Sections were once more rinsed in K-PBS, incubated with biotinylated secondary antibodies (Vector Laboratories), and then rinsed again with K-PBS before a final incubation with an avidin–biotin–peroxidase complex (Vectastain ABC Elite kit; Vector Laboratories). After several washes in K-PBS, the brain slices were reacted in 0.05% diaminobenzidine and 0.003% hydrogen peroxide. Thereafter, sections were rinsed in K-PBS, mounted, desiccated, fixed in 4% paraformaldehyde, and digested by the proteinase K. ISH was performed according to the above description.
Microglial culture and anti-inflammatory assay.
Microglia were obtained from a primary mix culture according to a previous report (Nakajima et al., 1992). Briefly, cerebral cortices dissected from neonatal mice were dispersed with trypsin and DNase I. After 14–21 d in culture, detached microglia were collected and used for the following assay. Microglia were plated on culture dishes, serum starved for 5 h, and treated with or without 1 μg/ml lipopolysaccharide (LPS) for 6 h in the presence or absence of 0.5 μg/ml RANTES, and then cells were used for RNA extraction. Anti-inflammatory assay was performed by semiquantitative RT-PCR with the specific primers for the mRNA of mouse IL-1β (GenBank accession number NM_008361; forward primer, 5′-aagcaacgacaaaatacctgtggc-3′; reverse primer, 5′-agacacagattccatggtgaagtc-3′), IL-6 (GenBank accession number NM_031168; forward primer, 5′-gcagaaaacaatctgaaacttccag-3′; reverse primer, 5′-actccttctgtgactccagcttatc-3′), inducible nitric oxide synthase (iNOS) (GenBank accession number NM_010927; forward primer, 5′-gttccaggtgcacacaggctactc-3′; reverse primer, 5′-cttttttgccccataggaaaagac-3′), and tumor necrosis factor-α (TNF-α) (GenBank accession number NM_013693; forward primer, 5′-accttgtctactcccaggttctcttc-3′; reverse primer, 5′-agagcaatgactccaaagtagacc-3′).
Results
GPCR screening
We designed 274 primers for mouse GPCR genes (supplemental table, available at www.jneurosci.org as supplemental material) in the GPCR database (http://www.gpcr.org/7tm/) using Primer Express software (PerkinElmer Life and Analytical Sciences). Among 274 genes, 29 genes were upregulated by more than twofold after hypoglossal axotomy (Table 1), whereas 15 genes were downregulated by less than one-quarter (Table 2). The upregulated gene group consisted of various types of receptors; receptors for monoamine (serotonin), neuropeptides [bradykinin, somatostatin, melanocortins, vasoactive intestinal peptide (VIP)/pituitary adenylate cyclase-activating polypeptide (PACAP)], purine (ATP/ADP), chemokine (RANTES, fractalkine), lysophospholipid (S1P), cannabinoid, prostanoid (prostaglandin E3), nicotinic acid, and trypsin-like protease, in addition to several orphan receptors (GPR84, EMR1, GPR34, CMKLR1, GPR35). Although no remarkable preference in terms of ligand species was seen, three purinergic receptors (P2Y12, P2Y6, and P2Y13) and four neuropeptide receptors represented larger populations of groups. The nonsensory GPCRs are assigned to one of the four distinct classes of GPCRs on the basis of shared sequence motifs with a prototype of each class: classes 1–3 and Frizzled family GPCRs (Vassilatis et al., 2003; Foord et al., 2005). Those 29 belong to either class 1 or class 2 GPCRs, and no receptors were assigned to class 3, in which receptors for amino acids such as glutamate and GABA were dominant, and Frizzled family. On the contrary, most of the downregulated receptors belonged to neuropeptide receptors; VIPR2 (VIP/PACAP), GALR3 (galanin), TRHR (TRH), SSTR3 (somatostatin), NMUR1 (neuromedin U), GALR2 (galanin), AGTR2 (angiotensin II), and OPRM1 (β-endorphin) in addition to six orphans. Those also belonged to either class 1 or class 2.
Table 1.
GPCR genes that were increased more than twofold after hypoglossal axotomy
| Gene name | Accession number | Ligand | Expression level of injured side (fold induction) |
|
|---|---|---|---|---|
| Relative to control | Relative to GAPDH | |||
| GPR84 | Q8CIM5 | Orphan | 47.16 | 0.0013 |
| GPR109 | Q9EP66 | Nicotinic acid | 25.23 | 0.00019 |
| F2RL1 | P55086 | Trypsin, trypsin-like enzyme | 25.07 | 0.00049 |
| P2RY13 | Q9BE53 | ADP | 19.44 | 0.000046 |
| GHRHR | P32082 | GRF | 17.08 | 0.0000059 |
| MC1R | Q01727 | MSH(α, β, γ), ACTH | 9.59 | 0.00011 |
| P2RY6 | Q9ERK9 | UTP, ADP, ATP | 9.53 | 0.00093 |
| CCR5 | P51682 | MIP-1α, β, RANTES, CCL8 | 8.63 | 0.0021 |
| ADORA3 | Q61618 | Adenosine | 8.15 | 0.00064 |
| CX3CR1 | Q9Z0D9 | Fractalkine | 6.88 | 0.00407 |
| BDKRB2 | P32299 | Bradykinin | 6.48 | 0.00037 |
| P2RY12 | Q9CPV9 | ATP, ADP | 4.83 | 0.019 |
| CNR2 | P47936 | Cannabinoid, 2-arachidonoyl glycerol, anandamide | 4.44 | 0.00012 |
| C5AR1 | P30993 | Anaphylatoxin C5a | 4.40 | 0.00067 |
| VIPR1 | P97751 | VIP | 4.39 | 0.000011 |
| EMR1 | Q61549 | Orphan | 4.32 | 0.0023 |
| GPR34 | Q9CTM7 | Orphan | 4.10 | 0.0077 |
| SSTR1 | P30873 | Somatostatin | 4.06 | 0.00039 |
| HTR2B | Q9QWS2 | Serotonin | 3.78 | 0.00011 |
| PTAFR | Q62035 | Platelet activating factor | 3.58 | 0.000068 |
| GPR65 | Q61038 | Glycosphingolipid psychosine | 3.41 | 0.00020 |
| EDG5 | P52592 | S1P | 3.27 | 0.0010 |
| CASR | Q9QY96 | Calium ions | 3.27 | 0.0000049 |
| CMKLR1 | P97468 | Orphan | 3.16 | 0.00062 |
| GPR35 | Q8CB97 | Orphan | 3.14 | 0.00011 |
| FPRL1 | O08790 | Lipoxin A4 | 2.58 | 0.000025 |
| PTGER3 | P30557 | Prostaglandin E2 | 2.44 | 0.00023 |
| TAAR1 | Q923Y8 | β-Phenylethylamine, p-tyramine | 2.33 | 0.000048 |
| MC4R | P56450 | MSH(α, β, γ), ACTH | 2.00 | 0.0082 |
Table 2.
GPCR genes that were decreased less than 0.25-fold after hypoglossal axotomy
| Gene name | Accession number | Ligand | Expression level of injured side (fold induction) |
|
|---|---|---|---|---|
| Relative to control | Relative to GAPDH | |||
| VIPR2 | P41588 | VIP, PACAP-38, PACAP-27 | 0.057 | 0.000071 |
| MRGPRA8 | Q91ZC4 | Orphan | 0.17 | 0.00000061 |
| GPRC5B | Q923Z0 | Orphan | 0.20 | 0.0010 |
| GALR3 | O88853 | Galanin | 0.21 | 0.000023 |
| AGTRL1 | Q9WV08 | Apelin | 0.21 | 0.000054 |
| TCP10C | Q9JLS1 | Orphan | 0.21 | 0.000059 |
| TRHR | P21761 | TRH | 0.22 | 0.00012 |
| SSTR3 | P30935 | Somatostatin-14, somatostatin-28 | 0.22 | 0.000061 |
| V2R7 | O35195 | Orphan | 0.23 | 0.000012 |
| NMUR1 | O55040 | Neuromedin U | 0.23 | 0.00080 |
| GALR2 | O88854 | Galanin, GALP | 0.23 | 0.000025 |
| RRH | Q9D1T9 | Orphan | 0.23 | 0.0000039 |
| AGTR2 | P35374 | Angiotensin II | 0.24 | 0.00044 |
| OPRM1 | Q9JIY1 | β -Endorphin | 0.24 | 0.00031 |
| GPR44 | Q9Z2J6 | Orphan | 0.24 | 0.000034 |
In situ hybridization screening
The above listed GPCRs (29 upregulated and 15 downregulated) were further examined by ISH, using sections from unilateral hypoglossal nerve-injured mouse brain. We preferred the radioactive probe rather than a nonradioactive probe, because the sensitivity of the radioactive probe was apparently higher than the other. This ISH screening eventually addressed six upregulated and three downregulated GPCRs, which demonstrated prominent alteration and cellular localization of mRNAs on the emulsion autoradiography base (Table 3, Fig. 1). The mRNAs for GPR34 (orphan), EMR1(orphan), CNR2 (cannabinoid), and ADORA3 (adenosine), which showed significant upregulation in ISH, were excluded from the final list, because the clear silver grain accumulations were not observed to determine cell species. Among the six upregulated GPCRs, F2RL1, which is a receptor for trypsin-like enzyme, and MC4R for melanocortin were expressed by nerve-injured motor neurons, because the silver grain accumulation was observed on large-sized cells (Fig. 1). Conversely, mRNA hybridization signals for four other receptors, CCR5, CX3CR1, P2RY12, and GPR84, were all observed on small cells surrounding neurons, indicating those mRNAs were expressed by microglia. The hybridization signals of downregulated receptors AGTR2 for angiotensin II, TRHR for TRH, and VIPR2 for VIP/PACAP were clearly identified on the motor neurons of the control side (left), whereas no hybridization signals were identified on the neurons of the nerve-injured side (Fig 1B, right), suggesting that those mRNAs were normally expressed by neurons and downregulated in response to nerve injury.
Table 3.
GPCR genes that could be detected by in situ hybridization

Figure 1.

In situ hybridization screening. ISH screening demonstrated six upregulated (A) and three downregulated (B) gene expressions 5 d after nerve injury. The left columns (a–i) show emulsion autoradiography under dark-field illumination, and the right columns (j–r) show high-power bright-field photograph counterstained with thionine. The mRNA-positive signal (silver grain) for F2RL1 (a, j), MC4R (b, k), AGTR2 (g, p), TRHR (h, q), and VIPR2 (i, r) were observed on neurons. Those for CCR5 (c, l), CX3CR1 (d, m), GPR84 (e, n), and P2RY12 (f, o) are seen on microglia, which locate adjacent to the large-sized neurons and heavily stained with thionine. Scale bars: left, 50 μm; right, 25 μm.
Expression of CCR5 ligands
Previous papers have demonstrated localizations and putative functions for CX3CR1 and P2Y12 in microglia (Meucci et al., 2000; Haynes et al., 2006), and GPR84 is an orphan receptor. Thus, in this paper, we focused on CCR5 and addressed the functional relevance in expression of CCR5 in response to nerve injury. We first confirmed the expression of CCR5 in microglia by examining a simultaneous expression of CCR5 mRNA by ISH and a microglial marker Iba1 by immunohistochemistry. The hybridization signal for CCR5 mRNA was obviously accumulated on microglial cells, confirming that CCR5 was expressed by microglia (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Next question was on ligands for CCR5 in the nerve-injured hypoglossal nucleus. To determine the existences and expression responses of the CCR5 ligands in nerve-injured hypoglossal nucleus, the expression of mRNAs for MIP-1α, MIP-1β, and RANTES (CCL5) were examined by ISH. Film autoradiography demonstrated apparent upregulation of all three mRNAs in the injured side of the hypoglossal nucleus, and subsequent emulsion autoradiography showed clear accumulation of silver grain on neurons (Fig. 2). The increases of mRNAs were also proved by RT-PCR (Fig. 2). Semiquantification of the optical density unit of the bands derived by RT-PCR demonstrated significant increases of mRNAs for the all ligands (optical unit mean ± SD: RANTES, 100.195 ± 11.865 in injured side vs 6.514 ± 1.813 in control; MIP-1α, 72.133 ± 3.829 in injured vs 13.575 ± 2.186 in control; MIP-1β; 73.449 ± 9.921 in injured vs 23.302 ± 4.091 in control; t test, p < 0.001). The RT-PCR and ISH concluded that the RANTES mRNA induction was the most prominent among these ligands, and the mRNA expression was mainly observed in motor neurons. Furthermore, the expression profiles of mRNAs for CCR5 and ligands on film autoradiography (ISH) indicated that both RANTES and CCR5 mRNAs were upregulated substantially and in parallel after nerve injury (Fig. 3). Those suggest that the RANTES-mediated chemokine signaling would function between neuron and microglia, and the next question is whether the ligands are playing a positive (prosurvival/nerve regeneration) or negative (neuron death/axon degeneration) role.
Figure 2.

mRNAs for MIP-1α, MIP-1β, and RANTES are upregulated in nerve-injured hypoglossal motor neurons. A, Expression of mRNAs for CCR5 ligands 5 d after axotomy. The left columns show film autoradiography demonstrating that mRNA expressions are observed in injured side of hypoglossal nucleus (right side), and the right columns show emulsion autoradiography, indicating that those hybridization signals (silver grain) are mainly observed on neurons. Scale bars: left, 1 mm; right, 25 μm. B, RT-PCR analysis of mRNA expression for MIP-1α, MIP-1β, and RANTES in control and injured hypoglossal nuclei 5 d after axotomy.
Figure 3.

Expression profiles of mRNAs for CCR5 and its ligands (MIP-1α, MIP-1β, RANTES) in injured hypoglossal nucleus after axotomy. Semiquantification of mRNA expression levels from film autoradiography of hybridized tissue sections. Data represent the average intensity of the positive signals from film autoradiograms. Error bars indicate SE (statistically significant differences by paired t test, *p < 0.01).
RANTES suppresses inflammatory mediators through CCR5
Because the substantial and synchronized expressions of RANTES and CCR5 mRNAs in the early phase of the regeneration process suggested their functional implication in the neuron protection rather than the neurite elongation, we next examined the functional significance of the RANTES signaling in vitro using cultured microglia. To investigate a function of CCR5/RANTES (CCL5)-mediated signal on microglia, we first evaluated the effect of RANTES on the production of inflammatory mediators by microglia stimulated with LPS. Microglia from mouse brain was treated with RANTES when LPS was challenged to induce microglial activation. RANTES inhibited the production of mRNAs for the proinflammatory cytokines, such as IL-1β (by 20%), IL-6 (40%), and TNF-α (25%) (Fig. 4). In addition, iNOS mRNA expression was also suppressed by 50% by the RANTES application (Fig. 4). The suppression of mRNAs for inflammatory mediators by RANTES was also examined using microglia from CCR5−/− mice. CCR5-deficient microglia did not show significant suppression of those inflammatory mediators (Fig. 4), suggesting that the suppression of inflammatory mediators in microglia by RANTES was via CCR5.
Figure 4.
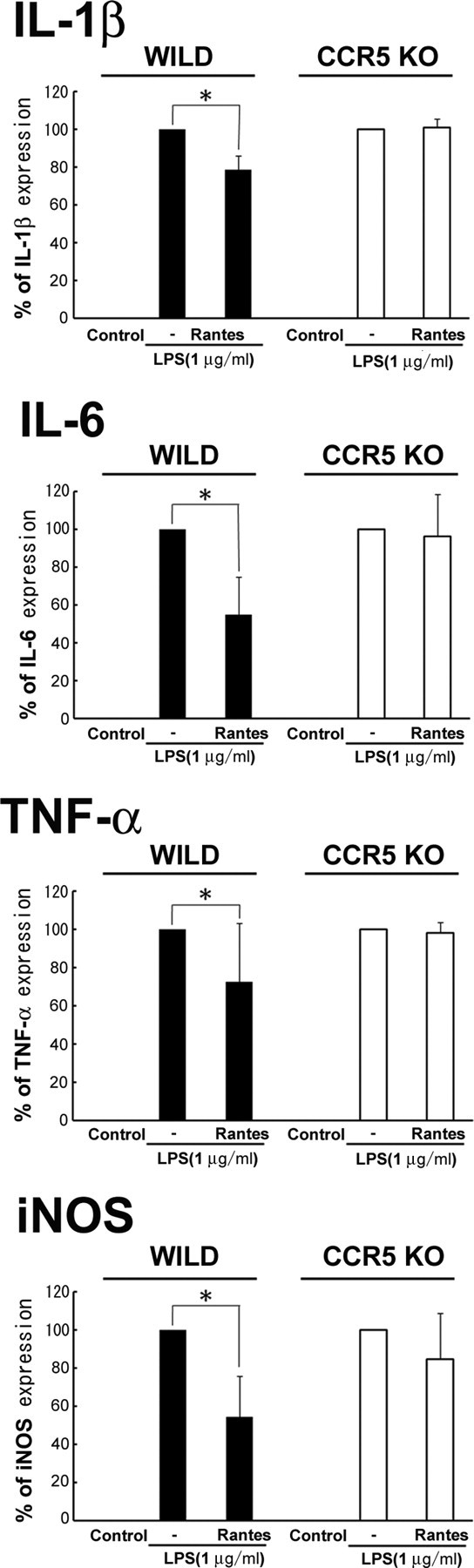
RANTES suppressed LPS-induced mRNA expression of inflammatory cytokines in microglia. Microglia from wild-type (WILD) and CCR5−/− (CCR5 KO) mice were cultured and treated for 6 h with 1 μg/ml LPS with and without 0.5 μg/ml RANTES. mRNA expression for IL-1β, IL-6, iNOS, and TNF-α was examined by RT-PCR. Error bars indicate SD (statistically significant differences by paired t test, *p < 0.001). Each bar represents results of five separate experiments.
Nerve injury-induced motor neuron death is accelerated in CCR5 knock-out mice
Next, to evaluate the significance of CCR5-mediated anti-toxic function in animal, we examined the motor neuron survival after nerve injury using CCR5−/− mouse. Motor neurons in the C57BL/6J mice strain are relatively susceptible to nerve injury, whereas rats and some other strains of mice were resistant to the insult (Kiryu-Seo et al., 2005, 2006). We thus hypothesized that the microglial toxicity by inflammatory mediators might be controlled by RANTES as is done by CX3CR1-mediated signal in microglia. Given the anti-toxic action by CCR5-mediated signaling, the microglial toxicity elicited by expression of inflammatory mediators may be exaggerated by the ablation of CCR5, leading to exacerbation of motor neuron survival after nerve injury. We then compared the survival of motor neurons between wild-type and CCR5−/− mice after nerve injury (Fig. 5). The nerve injury-induced motor neuron death progresses gradually, taking a few months, and finally >50% of motor neurons die. Thus, we took a time point at 35 d after axotomy, because >60% of neurons were still surviving at this time point. In CCR5−/− mouse, 50.7 ± 8.6% of motor neurons survived in the hypoglossal nucleus, whereas 64.4 ± 11.8% of motor neurons survived in wild type (p < 0.01, t test). This suggests that, although the difference of surviving motor neurons between wild-type and CCR5−/− was not dramatic, the acceleration of cell death in CCR5−/− is apparently significant.
Figure 5.
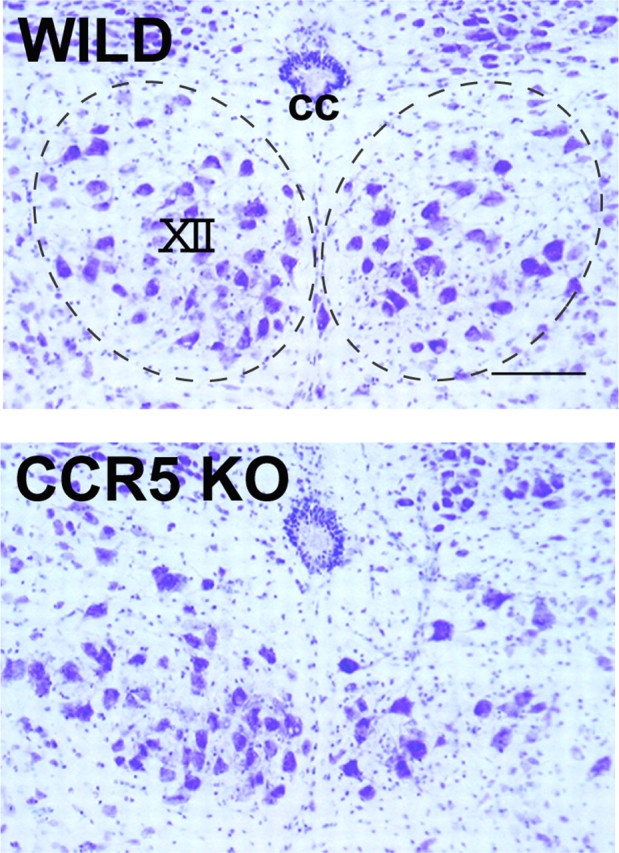
Motor neuron death is accelerated in CCR5-deficient mice. The representative thionine-stained sections of hypoglossal nucleus from wild-type (WILD) and CCR−/− (CCR5 KO) mice. The hypoglossal nucleus is shown by the dashed line. The right side hypoglossal nerve was transected. Note that the number of motor neurons survived in the nerve-injured side of CCR5 null mouse is significantly less than those in wild type. cc, Central canal; XII, hypoglossal nucleus. Scale bar, 50 μm.
Discussion
The present study demonstrated that CCR5/RANTES could be one of the signal mediators between neurons and microglia in response to nerve injury and that RANTES was capable of suppressing the expression of the inflammatory cytokines and iNOS, which are assumed to be toxic to neurons.
GPCR screen
One of the intriguing results in this study is that, among the six prominently upregulated GPCRs, four were expressed by microglia. We have taken some transcriptomic and proteomic approaches to identify nerve injury-associated molecules using the tissue from nerve-injured hypoglossal nuclei, and, using this approach, we have identified several molecules (Kiryu et al., 1995; Tanabe et al., 1999; Konishi et al., 2006). In those previous screens, most of the molecules identified were expressed in neurons rather than in glial cells. This suggests that the population of GPCRs may be more abundant in microglia than in neurons. This is likely because monocytes and macrophages express an extensive repertoire of GPCRs that regulate inflammation and immunity (Lattin et al., 2007). Although we eventually focused on six GPCRs after ISH screening, additional GPCRs such as the adenosine receptor ADRA3, cannabinoid receptor CB2, and orphan receptors EMR1 and GRP34, whose mRNA expressions were significantly upregulated in the real-time PCR base, were likely to be expressed by glial cells. The silver grains indicated those mRNA localizations that were intense and relatively even in the injured hypoglossal nucleus, and this pattern was of typical non-neuronal pattern, presumably astrocyte or microglia (Tanabe et al., 1999), although the emulsion autoradiograms of those were not clear enough to determine exact cellular localizations of those mRNAs. It may be concluded that the GPCR screen performed here was of practical use in identifying functional receptors on microglia.
Among four GPCRs expressed by microglia, the localizations and putative functions of CX3CR1 and P2RY12 were reported previously. CX3CR1 is characterized as fractalkine (CX3CL1) receptor, and the expression in microglia and its anti-inflammatory function have been reported (discuss later). P2RY12 is an ATP/ADP receptor and is suggested to function in neuropathic pain in which the P2RY12 is expressed by microglia in the dorsal horn of spinal cord in response to sciatic nerve injury and plays a crucial role in the generation of neuropathic pain (Haynes et al., 2006; Kobayashi et al., 2008; Tozaki-Saitoh et al., 2008). As for the orphan GPR84, the function is still obscure, although localization in microglia has been shown (Bouchard et al., 2007). GRP84 was strikingly restricted to bone marrow-derived macrophages and microglia in an unstimulated state and was strongly upregulated by LPS in both bone marrow-derived macrophages and activated peritoneal macrophages (Lattin et al., 2008). The similar induction of GPR84 in macrophage was consistently observed in an endotoxin shock model as well as in the experimental autoimmune encephalomyelitis model of multiple sclerosis, suggesting that GPR84 has a role in neuroinflammatory events (Bouchard et al., 2007).
CCR5/CCL5 localization
Several studies have suggested that CCR5 is expressed by a variety of cells such as microglia, astrocytes, neurons, and endotherial cells (Rottman et al., 1997; Meucci et al., 2000; Kaul et al., 2007). Our real-time PCR study apparently demonstrated the marked increase of mRNAs for CCR5 in nerve-injured hypoglossal nuclei. ISH demonstrated an increase and accumulation of silver grains on microglia, which localized adjacent to large motor neurons in response to nerve injury. This was also confirmed by simultaneous labeling with CCR5 hybridization signal and Iba1, a microglial marker of immunoreactivity. In contrast to the obvious increase of CCR5 mRNA signal on microglia, the silver grain accumulation on neurons and astrocytes was not clear in our emulsion autoradiography. Concomitantly, several reports addressed the expression of CCR5 in microglia and CCR5-mediated functions on microglia (Hanisch, 2002; Garden and Moller, 2006). For instance, in human control and Alzheimer's disease (AD) patient brains, CCR5 were present on microglia, with increased expression on some reactive microglia in AD (Xia et al., 1998). These studies together with the present observations suggested that CCR5 is highly expressed by microglia in response to nerve injury, although CCR5 might also be expressed on other types of cells with lower levels of protein.
Ligands for CCR5, RANTES, MIP-1α and MIP-1β have been identified (Cocchi et al., 1995), and their localization in CNS has also been widely reported in the literature. For instance, immunohistochemistry revealed the presence of MIP-1β predominantly in a subpopulation of reactive astrocytes, which were more widespread in AD than control brains, and MIP-1α predominantly in neurons and weakly in some microglia in both AD and controls (Xia et al., 1998). Another double-labeling study showed that MIP-1β, but not RANTES, was expressed by reactive astrocytes near the lesion site (Ghirnikar et al., 1996). RANTES immunoreactivity was observed in epineurial macrophage, and a weak positive signal was seen in perineurium, Schwann cell, and axon (Taskinen and Roytta, 2000). RANTES protein was detected immunohistochemically in embryonic day 12.5 dorsal root ganglion and the cutaneous layers of the developing hindlimb (Bolin et al., 1998). Thus, the great controversies on the localization of the ligands exist, and this is presumably attributable to the specificities of antibodies used, less amounts of those chemokines in CNS, and difference of experimental preparations such as in vivo and in vitro conditions. In terms of expression response to nerve injury, the increase of both CCR5 and RANTES mRNAs were shown in nerve-injured DRG neurons, although no mRNA expression was detectable in normal DRG neurons (Bhangoo et al., 2007). Flügel et al. (2001) found RANTES mRNA expressed in motor neurons, but the level of expression and cellular distribution of RANTES was not altered by nerve injury (Flügel et al., 2001). On the contrary, the present data demonstrated that hypoglossal motor neurons expressed mRNAs for RANTES in response to nerve injury, and this increase was confirmed by ISH and PCR. This discrepancy seen in the gene expression response may be attributable to the difference between motor and sensory neurons, a difference of nerve injury operations, or a difference of probes to detect mRNA. A slight but not significant increase of RANTES mRNA in nerve-injured motor neurons was actually seen in the study by Flügel et al. (2001), although the quantitative analysis was not done. Immunohistochemical demonstration of RANTES was attempted using several commercially available antibodies, but we could not demonstrate clear localization of RANTES immunoreactivity. In any event, RANTES expression in nerve-injured motor neurons appears to be beneficial and sound.
Putative functions of CCR5/RANTES signaling
CCR5 regulates both trafficking and effector functions of several immune cells such as Th1 cells, macrophages, NK cells, and immature dendritic cells (Balistreri et al., 2007). Therefore, these two regulatory functions are plausible in nervous system as well. Microglia are capable of changing their morphology quickly in response to injury stimuli and migrating toward injured brain regions. In addition, previous studies demonstrated that microglia synthesize various inflammatory cytokines, nitric oxide, and trophic factors in response to physiological and pathological stimuli (Hanisch, 2002; Garden and Moller, 2006). Those complicated features make their functional consequence obscure. The function of microglia is particularly unclear during motor neuron regeneration. In response to motor nerve injury, microglia located near the injured-motor neurons are activated and migrate toward the cell bodies of nerve-injured motor neurons. Those migrated microglial cells are then attached to the cell bodies of motor neurons and subsequently modify the surfaces by changing their morphology. As for the migration activity of microglia, CCR5 has been shown to be important in CNS using culture and slice preparations. In those experiments, a highly specific CCR5 antagonist, TAK-779, was used and demonstrated a significant reduction of the migration rate of microglia in CNS injury models (Marella and Chabry, 2004; Carbonell et al., 2005). These studies highlighted the trafficking function of CCR5. However, in the motor nerve injury model, a series of microglial behavioral effects in response to nerve injury seemed normal even in CCR5 null mice, suggesting that CCR5-mediated signaling may not be a major chemotaxis factor for microglia, at least in the present experimental paradigm, although we could not demonstrate the time-lapse-based change of microglial morphology in vivo. This was also suggested by another in vivo study using a hippocampal denervated model in which the infiltration of both T cells and macrophages was observed in CCR5-deficient hippocampi, showing that CCR5 ligands (including RANTES/CCL5) are not critical for the infiltration response (Babcock et al., 2003). Perhaps other signal mediators such as ATP/ADP and MCP-1 would be major chemoattractants for microglia to respond to nerve injury because membrane ruffling and chemotaxis of microglia were induced by ATP or ADP via Gi/o-coupled P2Y receptors (Honda et al., 2001), and the receptor antagonists suppressed the migration of microglia (Tsuda et al., 2003). Also, neither T cells nor macrophages infiltrated the denervated hippocampus of MCP-1 receptor (CCR2)-deficient mice, arguing for a critical role for the CCR2 ligand MCP-1/CCL2 in leukocyte migration (Babcock et al., 2003). Those observations suggested the trafficking regulation function of CCR5 may not be crucial under the motor nerve injury condition, and an alternative principal function of CCR5 such as an effector function of microglia may be the case.
One of the plausible effector functions was inspired by an intriguing function of CX3CR1, which was demonstrated recently. CX3CR1 was shown to be upregulated in perineuronal microglia in response to nerve transection, and its ligand, CX3CL1 (flactalkine), was found to be constitutively expressed by neurons, although axotomy of peripheral nerve led to downregulation of CX3CL1 mRNA in motor neurons (Harrison et al., 1998). This CX3CL1–CX3CR1 system was proved to be one of the major signaling pathways from neuron to microglia in response to nerve injury. CX3CL1 attenuates the secretion of IL-6 and TNF-α, upregulation of iNOS in LPS-activated microglia, and production of IL-1β (Cardona et al., 2006). Intriguingly, the present study revealed that RANTES has a similar function in which productions of IL-6, IL-1β, TNF-α, and iNOS were suppressed in LPS-activated microglia in vitro, although the precise mechanism underlying the suppression was unknown. Both RANTES and CX3CL1 are therefore advantageous to protect neurons from inflammation and oxidative stress originating from microglia. The simultaneous upregulation of both receptors, CCR5 and CX3CR1, in microglia together with the upregulation of RANTES in the injured neurons could suppress the toxicity of nerve injury-activated microglia. With these dual suppression mechanisms, nerve-injured neurons might be affected by relatively less microglial toxicity. This may lead to an accelerated death of nerve injury-induced motor neuron death in CCR5-deficient mice.
In conclusion, microglia assume both neurotoxic and neurotrophic phenotypes in response to nerve injury (Garden and Moller, 2006). However, under certain conditions, such as peripheral nerve injury, some signal mediators from nerve-injured motor neurons to adjacent microglia seem to play an important role in the suppression of harmful side effects of the microglia, such as inflammatory cytokine release and the nitric oxide production. This suppression makes the protective function of microglia predominant, and thereby the motor neurons are able to survive longer after injury. The present mechanism underlying the suppression of microglial toxicity may provide a therapeutic potential for the progressive neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS), on the basis of the suggestion made by Boillee et al. (2006) that microglia have a central role in neuronal damage by secreting potentially neurotoxic molecules in ALS. Therefore, a manipulation of CCL5 as well as CX3CL1 signaling in those diseases may prove to be therapeutic by minimizing microglial hostile effects and enhancing their beneficial effects.
Footnotes
This work was supported by grants from Ministry of Health, Labor, and Welfare of Japan, and Ministry of Education, Culture, Sports, Sciences, and Technology of Japan. We thank Dr. Marguerite Prior (Cleveland Clinic) for reading and correcting this manuscript. We are grateful to C. Kadono for her excellent technical assistance.
References
- Aoki S, Su Q, Li H, Nishikawa K, Ayukawa K, Hara Y, Namikawa K, Kiryu-Seo S, Kiyama H, Wada K. Identification of an axotomy-induced glycosylated protein, AIGP1, possibly involved in cell death triggered by endoplasmic reticulum-Golgi stress. J Neurosci. 2002;22:10751–10760. doi: 10.1523/JNEUROSCI.22-24-10751.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23:7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balistreri CR, Caruso C, Grimaldi MP, Listì F, Vasto S, Orlando V, Campagna AM, Lio D, Candore G. CCR5 receptor: biologic and genetic implications in age-related diseases. Ann NY Acad Sci. 2007;1100:162–172. doi: 10.1196/annals.1395.014. [DOI] [PubMed] [Google Scholar]
- Bhangoo S, Ren D, Miller RJ, Henry KJ, Lineswala J, Hamdouchi C, Li B, Monahan PE, Chan DM, Ripsch MS, White FA. Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: a mechanism for the development of chronic sensitization of peripheral nociceptors. Mol Pain. 2007;3:38. doi: 10.1186/1744-8069-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnadóttir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schiöth HB. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics. 2006;88:263–273. doi: 10.1016/j.ygeno.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Bolin LM, Murray R, Lukacs NW, Strieter RM, Kunkel SL, Schall TJ, Bacon KB. Primary sensory neurons migrate in response to the chemokine RANTES. J Neuroimmunol. 1998;81:49–57. doi: 10.1016/s0165-5728(97)00158-6. [DOI] [PubMed] [Google Scholar]
- Bouchard C, Pagé J, Bédard A, Tremblay P, Vallières L. G protein-coupled receptor 84, a microglia-associated protein expressed in neuroinflammatory conditions. Glia. 2007;55:790–800. doi: 10.1002/glia.20506. [DOI] [PubMed] [Google Scholar]
- Carbonell WS, Murase S, Horwitz AF, Mandell JW. Migration of perilesional microglia after focal brain injury and modulation by CC chemokine receptor 5: an in situ time-lapse confocal imaging study. J Neurosci. 2005;25:7040–7047. doi: 10.1523/JNEUROSCI.5171-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Clarke PG, Oppenheim RW. Neuron death in vertebrate development: in vitro methods. Methods Cell Biol. 1995;46:277–321. [PubMed] [Google Scholar]
- Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- Floyd CL, Lyeth BG. Astroglia: important mediators of traumatic brain injury. Prog Brain Res. 2007;161:61–79. doi: 10.1016/S0079-6123(06)61005-4. [DOI] [PubMed] [Google Scholar]
- Flügel A, Hager G, Horvat A, Spitzer C, Singer GM, Graeber MB, Kreutzberg GW, Schwaiger FW. Neuronal MCP-1 expression in response to remote nerve injury. J Cereb Blood Flow Metab. 2001;21:69–76. doi: 10.1097/00004647-200101000-00009. [DOI] [PubMed] [Google Scholar]
- Foord SM, Bonner TI, Neubig RR, Rosser EM, Pin JP, Davenport AP, Spedding M, Harmar AJ. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol Rev. 2005;57:279–288. doi: 10.1124/pr.57.2.5. [DOI] [PubMed] [Google Scholar]
- Garden GA, Möller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- Ghirnikar RS, Lee YL, He TR, Eng LF. Chemokine expression in rat stab wound brain injury. J Neurosci Res. 1996;46:727–733. doi: 10.1002/(SICI)1097-4547(19961215)46:6<727::AID-JNR9>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci. 2001;21:1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Ma Q, Medders KE, Desai MK, Lipton SA. HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ. 2007;14:296–305. doi: 10.1038/sj.cdd.4402006. [DOI] [PubMed] [Google Scholar]
- Kettenmann H. Triggering the brain's pathology sensor. Nat Neurosci. 2006;9:1463–1464. doi: 10.1038/nn1206-1463. [DOI] [PubMed] [Google Scholar]
- Kiryu S, Yao GL, Morita N, Kato H, Kiyama H. Nerve injury enhances rat neuronal glutamate transporter expression: identification by differential display PCR. J Neurosci. 1995;15:7872–7878. doi: 10.1523/JNEUROSCI.15-12-07872.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Hirayama T, Kato R, Kiyama H. Noxa is a critical mediator of p53-dependent motor neuron death after nerve injury in adult mouse. J Neurosci. 2005;25:1442–1447. doi: 10.1523/JNEUROSCI.4041-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Gamo K, Tachibana T, Tanaka K, Kiyama H. Unique anti-apoptotic activity of EAAC1 in injured motor neurons. EMBO J. 2006;25:3411–3421. doi: 10.1038/sj.emboj.7601225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Sasaki M, Yokohama H, Nakagomi S, Hirayama T, Aoki S, Wada K, Kiyama H. Damage-induced neuronal endopeptidase (DINE) is a unique metallopeptidase expressed in response to neuronal damage and activates superoxide scavengers. Proc Natl Acad Sci USA. 2000;97:4345–4350. doi: 10.1073/pnas.070509897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci. 2008;28:2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H, Namikawa K, Kiyama H. Annexin III implicated in the microglial response to motor nerve injury. Glia. 2006;53:723–732. doi: 10.1002/glia.20327. [DOI] [PubMed] [Google Scholar]
- Lagerström MC, Schiöth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- Lattin J, Zidar DA, Schroder K, Kellie S, Hume DA, Sweet MJ. G-protein-coupled receptor expression, function, and signaling in macrophages. J Leukoc Biol. 2007;82:16–32. doi: 10.1189/jlb.0107051. [DOI] [PubMed] [Google Scholar]
- Lattin JE, Schroder K, Su AI, Walker JR, Zhang J, Wiltshire T, Saijo K, Glass CK, Hume DA, Kellie S, Sweet MJ. Expression analysis of G Protein-Coupled Receptors in mouse macrophages. Immunome Res. 2008;4:5. doi: 10.1186/1745-7580-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella M, Chabry J. Neurons and astrocytes respond to prion infection by inducing microglia recruitment. J Neurosci. 2004;24:620–627. doi: 10.1523/JNEUROSCI.4303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci USA. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai M, Yoneyama H, Ezaki T, Suematsu M, Terashima Y, Harada A, Hamada H, Asakura H, Ishikawa H, Matsushima K. Peyer's patch is the essential site in initiating murine acute and lethal graft-versus-host reaction. Nat Immunol. 2003;4:154–160. doi: 10.1038/ni879. [DOI] [PubMed] [Google Scholar]
- Nakagomi S, Suzuki Y, Namikawa K, Kiryu-Seo S, Kiyama H. Expression of the activating transcription factor 3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J Neurosci. 2003;23:5187–5196. doi: 10.1523/JNEUROSCI.23-12-05187.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Shimojo M, Hamanoue M, Ishiura S, Sugita H, Kohsaka S. Identification of elastase as a secretory protease from cultured rat microglia. J Neurochem. 1992;58:1401–1408. doi: 10.1111/j.1471-4159.1992.tb11356.x. [DOI] [PubMed] [Google Scholar]
- Namikawa K, Okamoto T, Suzuki A, Konishi H, Kiyama H. Pancreatitis-associated protein-III is a novel macrophage chemoattractant implicated in nerve regeneration. J Neurosci. 2006;26:7460–7467. doi: 10.1523/JNEUROSCI.0023-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science. 2007;317:1083–1086. doi: 10.1126/science.1144640. [DOI] [PubMed] [Google Scholar]
- Rottman JB, Ganley KP, Williams K, Wu L, Mackay CR, Ringler DJ. Cellular localization of the chemokine receptor CCR5. Correlation to cellular targets of HIV-1 infection. Am J Pathol. 1997;151:1341–1351. [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Nakagomi S, Kiryu-Seo S, Namikawa K, Imai Y, Ochi T, Tohyama M, Kiyama H. Expressed-sequence-tag approach to identify differentially expressed genes following peripheral nerve axotomy. Brain Res Mol Brain Res. 1999;64:34–40. doi: 10.1016/s0169-328x(98)00302-7. [DOI] [PubMed] [Google Scholar]
- Taskinen HS, Röyttä M. Increased expression of chemokines (MCP-1, MIP-1alpha, RANTES) after peripheral nerve transection. J Peripher Nerv Syst. 2000;5:75–81. doi: 10.1046/j.1529-8027.2000.00009.x. [DOI] [PubMed] [Google Scholar]
- Tozaki-Saitoh H, Tsuda M, Miyata H, Ueda K, Kohsaka S, Inoue K. P2Y12 receptors in spinal microglia are required for neuropathic pain after peripheral nerve injury. J Neurosci. 2008;28:4949–4956. doi: 10.1523/JNEUROSCI.0323-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE, Mortrud MT, Brown A, Rodriguez SS, Weller JR, Wright AC, Bergmann JE, Gaitanaris GA. The G protein-coupled receptor repertoires of human and mouse. Proc Natl Acad Sci USA. 2003;100:4903–4908. doi: 10.1073/pnas.0230374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia MQ, Qin SX, Wu LJ, Mackay CR, Hyman BT. Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer's disease brains. Am J Pathol. 1998;153:31–37. doi: 10.1016/s0002-9440(10)65542-3. [DOI] [PMC free article] [PubMed] [Google Scholar]