

Abstract
Trehalose 6,6′-dimycolate (TDM) is the most abundant lipid extracted from Mycobacterium tuberculosis (MTB). TDM promotes MTB survival by decreasing phagosomal acidification and phagolysosomal fusion in macrophages. Delipidation of MTB using petroleum ether removes TDM and decreases MTB survival within host cells. TDM reconstituted onto MTB restores its virulent wild-type characteristics. We investigated the role of TDM in regulating surface marker expression in MTB-infected macrophages. Macrophages were infected with wild-type, delipidated, and TDM-reconstituted MTB for 24 hours and measured for changes in surface marker expression. TDM on MTB was found to specifically target MHCII, CD1d, CD40, CD80 and CD86. Both wild-type and TDM-reconstituted MTB suppressed or induced no change in expression of these surface markers, whereas delipidated MTB increased expression of the same markers. MTB-infected macrophages were also overlaid with MHCII-restricted T cell hybridomas which recognize Antigen 85B. Macrophages infected by wild-type and TDM-reconstituted MTB did not present antigen as well as delipidated MTB-infected macrophages. The evidence shown furthers supports the notion that TDM present on MTB promotes its survival and persistence in host macrophages.
Keywords: Mycobacterium tuberculosis, Trehalose dimycolate, Cord Factor, Macrophage, Surface Marker
1. Introduction
Trehalose 6,6′-dimycolate (TDM), also known as cord factor, is the most abundant extractable lipid on virulent Mycobacterium tuberculosis (MTB), constituting 90–95% of the extracted lipids from viable organisms [1]. Wild-type MTB reduces phagosomal acidification and inhibits phagosome-lysosome fusion in macrophages [2, 3]. Delipidated MTB (dMTB), where lipids including TDM are extracted off MTB, has been found to have a reduced ability to produce infection in vivo and survive in macrophages in vitro [4, 5], and promotes acidification and lysosomal fusion of the phagosome [6]. Delipidated MTB reconstituted with purified TDM (rMTB) restores the wild-type characteristics of MTB infection, implicating TDM as the cause. Our focus is to understand the role of TDM in promoting MTB survival when present in its native structural form on the surface of viable MTB.
We investigated how TDM modulates the activities of macrophages infected with virulent MTB by characterizing the expression of surface markers during infection. Macrophages respond to bacterial infection by expressing surface markers which regulate pathogen recognition and ingestion, cell adhesion and migration, antigen presentation, cell-cell interactions and costimulation of T cells [7]. Macrophage response to TDM was measured by the changes in expression of a selected panel of surface markers (CD1d, CD11a, CD11b, CD40, CD44, CD80, CD86, and MHCII). Our results demonstrate that the presence of TDM on the surface of MTB selectively suppresses expression of surface markers MHCII, CD1d, CD40, CD80 and CD86. In macrophages infected with TDM-delipidated MTB this suppression is reversed and surface marker expression increases significantly. No significant effect was seen in response to the presence or absence of TDM for CD11a, CD11b, or CD44. This suggests that TDM on MTB targets surface markers which are important for antigen presentation and T cell costimulation.
Macrophage antigen presentation was also found to be regulated by TDM on MTB. MTB-infected macrophages were overlaid with the MHCII-restricted BB7 T cell hybridoma which recognizes the mycobacterial Antigen 85B (Ag85B). TDM-intact MTB inhibited macrophages from effectively presenting Ag85B via MHCII to the hybridoma T cells. We propose that TDM on the surface of MTB contributes to the persistence of MTB in humans by inhibiting proper macrophage responses which are necessary for stimulation of the host immune response.
2. Materials and Methods
2.1 Macrophages
AMJ2-C8 (ATCC CRL-2455) and PMJ2-PC (ATCC CRL-2457) macrophages are derived from C57BL/6 murine alveolar and peritoneal macrophages, respectively. Both cell lines were immortalized by in vitro infection with the J2 retrovirus containing the oncogenes v-raf and v-myc, which enhances the cell’s ability to divide while preventing terminal differentiation. Cell types were found to closely resemble their parental cells and retained their macrophage-like characteristics [8, 9]. Cell lines were passaged every 3–4 days in Dulbecco’s Modified Essential Medium (Sigma-Aldrich) containing 0.15% NaHCO3, 0.35% glucose, 0.12% HEPES, 5% heat-killed fetal bovine serum (FBS), and 1% penicillin-gentamicin. Cells were adjusted to 1 × 106 cells ml−1 and maintained in 75 cm2 cell culture flasks at 37° C in 5% CO2 prior to infection. Primary bone marrow-derived macrophages (BMMs) were harvested from C57BL/6 mice and cultured in Iscove’s Modified Dulbecco’s Medium (Sigma-Aldrich) with 0.3% NaHCO3, 10% heat-killed FBS, 1% penicillin-gentamicin, and 10 ng ml−1 mouse GM-CSF for 7 days. Cells were adjusted to 1 × 106 cell ml−1 and maintained in 75 cm2 cell culture flasks or aliquoted into 24-well co-culture plates for antigen presentation assays.
2.2 TDM delipidation and reconstitution of live mycobacteria
H37Rv (ATCC 25618) was sub-cultured to the exponential phase at 37° C in 5% CO2 in 7H9 media enriched with 0.2% glycerol, 0.5% bovine serum albumin (BSA), 0.2% dextrose, and 0.05% Tween80. Delipidation of mycobacterial surface lipids was performed as described by Silva et al [10]. MTB was suspended in water:petroleum ether and vortexed vigorously for 2 min, allowed to settle for 5 min at room temperature, then centrifuged at 3000 rpm for 5 min. The petroleum ether phase which contained the mycobacterial lipids was removed. This process was repeated twice more, then the pelleted bacterium was resuspended in sterile saline.
TDM reconstitution was performed by adding 50 μg purified TDM (Sigma; 100% pure by TLC, > 98% as 6,6′-mycolate esters) dissolved in petroleum ether to 3 × 108 delipidated bacterium. The petroleum ether was evaporated, and the reconstituted strain was suspended in sterile saline. Both delipidated and TDM-reconstituted MTB maintained their viability, which was confirmed through CFU counts, but dMTB was found to be characteristic for its inability to form cords and was easier to disperse in culture. dMTB had a lower rate of survival in vivo and in vitro compared to wild-type and rMTB [5, 6]. All bacterium was suspended at a concentration of 3 × 108 ml−1 and sonicated prior to infection for even dispersal. The fbpA mutant strain was also reconstituted with pure TDM using the same methods and concentrations.
2.3 Thin-layer chromatography
The collected petroleum ether extracts were analyzed for mycolic acids using thin-layer chromatography (TLC) as adapted by Belisle et al (Fig. 1) [11]. The extracts were plated on a silica gel 60A plate (Whatman) against purified TDM standard (Sigma). The plate was run in a 90 chloroform: 10 methanol: 1 ammonium hydroxide solution and developed in an ethanol solution containing 5% sulfuric acid and 0.5% a-naphthol followed by charring for 10 min. The band produced from the petroleum ether extracts was found to be similar to the standard TDM band, confirming previously published results [5].
Fig. 1.

Thin layer chromatography of petroleum ether extracts from MTB strain H37Rv (#2) against commercial purified TDM standard (#1).
2.4 TDM-coated microspheres
Poly-D-L-lactide coglycolide (PLGA) (50:50; Resomer RG 502; Boehringer Ingelheim, Mannheim, Germany) microspheres were chosen for their ability to biodegrade, breaking down into lactide and coglycolide and eventually into carbon dioxide [12]. 5 μg of poly-D-L-lactide coglycolide beads were suspended in 50% methanol with or without 100 μg purified TDM (Sigma) and the solvent evaporated overnight. The beads were resuspended in phosphate-buffered saline (PBS) and counted for bead concentration. To confirm uptake of TDM, the beads were solubilized in chloroform and analyzed by TLC.
2.5 Mycobacterial infection and TDM microsphere administration to macrophages
Macrophages were infected with wild-type, delipidated, or TDM-reconstituted MTB at a multiplicity of infection (MOI) of 2 mycobacteria: 1 macrophage for 24 hours. 10 ng ml−1 of lipopolysaccharide (LPS) was included as a positive control for all infection experiments. Macrophage uptake of mycobacterium was confirmed through acid-fast staining and CFU counts, and macrophage viability confirmed through trypan blue staining. Bone marrow macrophages were infected with the PLGA beads at a MOI of 2 beads:1 macrophage for 24 hours.
2.6 Surface receptor staining and flow cytometry
Cells were collected and centrifuged at 1000 rpm for 5 min. The supernatant was removed and the pellet suspended at 2 × 105 cells/100 ul in staining buffer (1% heat-killed fetal bovine serum and 0.09% NaN3 in PBS) and anti-FcγRII (BD PharMingen, San Diego, CA) for 15 min at 4° C. Cells were washed with staining buffer and incubated with the following antibodies: anti-F4/80, anti-MHCII, anti-CD1d, anti-CD40, anti-CD80 and anti-CD86 (BD PharMingen, San Diego, CA) for 30 min at 4° C. Isotype antibodies were included to account for non-specific binding. Cells were washed with staining buffer once more, and then fixed in 2% paraformaldehyde. Fluorescence scanning was measured with a FACSCalibur flow cytometer using CellQuest software (BD Immunocytometry Systems, San Jose, CA). Cells positive for the macrophage-specific F4/80 antibody was gated on the analysis dot plot. A minimum of 10,000 cells within the gated cell population were counted for each sample and the percentage of cells positive for each antibody type was calculated. Mean fluorescence intensity (MFIs) for each sample population was also recorded. Statistical significance was determined through Student’s t-test and analysis of variance (ANOVA), where p-value < 0.05 is significant.
2.7 Antigen presentation assay
The Ag85B epitope-specific T cell hybridoma (BB7) was a gift from Drs. C. Harding and H. Boom (Case Western Reserve University, Cleveland, OH). The BB7 cells were used to measure Ag85B peptide complexed with MHC class II in macrophages [13–15]. Differentiated BMMs were plated at a concentration of 1 × 105 cells ml−1 in 24-well plates and used as naive cells or were activated for 24 hours with 50 ng ml−1 mouse recombinant IFN-γ (Cell Sciences) prior to infection. Both naïve and stimulated macrophages were infected with mycobacteria at a MOI of 2:1 for 4 hours at 37° C. Macrophages were washed 3 times with PBS and directly overlaid with BB7 T cells at a ratio of 5 T cells: 1 macrophage. Supernatants were collected 24 hours later, and measured for T hybridoma response using a sandwich ELISA immunoassay kit specific for IL-2 (BioLegend). Similar experiments were performed using naïve BMMs infected with the fbpA mutant strain.
2.8 SDS-PAGE and Western blot
BMMs were infected with wild-type, dMTB, and rMTB at an MOI of 2:1 for 4 hours. LPS (10 ng ml−1) and M. bovis BCG (Pasteur; MOI 2:1) were included as positive controls. Cells were washed 3 times with PBS, then gently scraped and pelleted. Samples were suspended in a protease inhibitor cocktail (Sigma) and lysed with SDS sample buffer (250 mM tris HCl, 500 mM DTT, 10% SDS, 50% glycerol, bromophenol blue). Samples were heated for 5 min at 100° C and quantitated for protein using BCA Protein Assay Kit (Pierce). 5 μg of protein for each sample were loaded onto a 10% Tris-HCL pre-made gel (Criterion) and ran at 150 volts for 90 minutes using tris-glycine-SDS buffer (TGS; Fisher). The gel was transferred to a PVDF membrane at 60 volts for 60 minutes in TGS buffer with 20% MeOH. The transferred membranes were blocked with 1% BSA in TTBS (tris-buffer saline with 1% Tween20) and probed with SOCS-1 (sc-9021), SOCS-3 (sc-9023), and actin (sc-1616) antibodies (Santa Cruz Biotechnology). Bands were visualized using HRP-tagged IgG antibodies (Santa Cruz Biotechnology) and ECL Western Blotting Detection kit (Amersham Biosciences).
3. Results
3.1 TDM on the surface of MTB inhibits surface marker expression in macrophages
The macrophage cell lines were chosen because of their similarity to primary alveolar and peritoneal murine macrophages [8, 9]. We began with the alveolar macrophage-like cells to determine whether the presence of TDM on MTB affected specific surface markers during infection. The cells were infected with wild-type MTB, delipidated (dMTB) or TDM-reconstituted MTB (rMTB) for 24 hours, and then counted for cells positive for expression of each surface marker within each cell population. To confirm that all cell types were responsive to a known stimulus, LPS was included as a positive control. Changes in expression were found to be most significant for MHCII, CD1d, and CD40 (Fig. 2a). The cells infected by wild-type MTB produced a 25–40% decrease in percent positive cells of these markers compared to uninfected controls. Infection by dMTB nearly doubled the expression of MHCII, CD1d, and CD40 compared to the control. rMTB infection produced similar results to wild-type, reducing surface expression for all three markers in alveolar macrophage-like cells.
Fig. 2. Effects of TDM on MTB on the expression of MHCII, CD1d, CD40, CD80 and CD86 by murine macrophages.

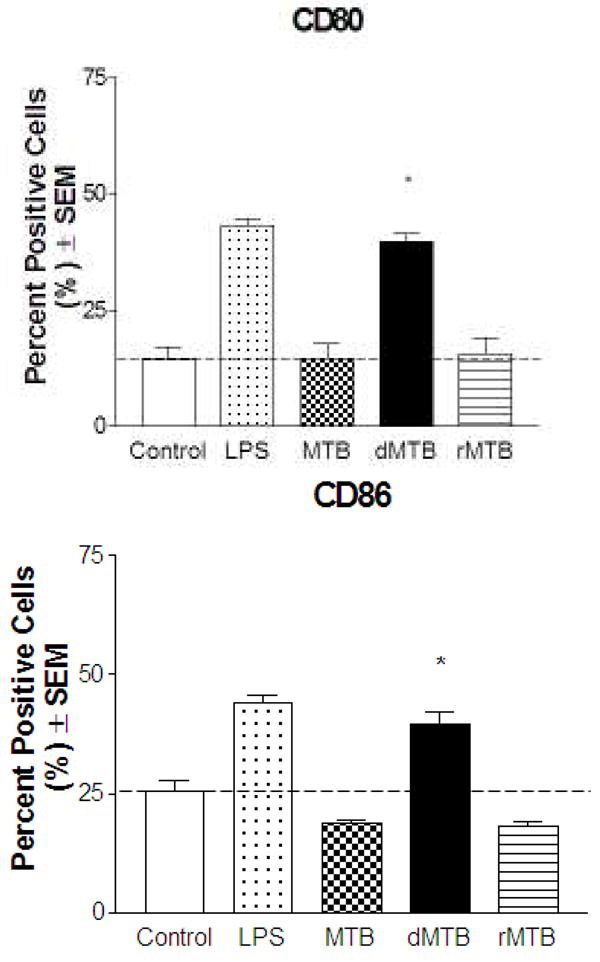

Macrophages were infected with wild-type, delipidated (dMTB), or TDM-reconstituted (rMTB) MTB at a MOI of 2:1 for 24 hours, and examined for surface expression of MHCII, CD1d, and CD40 using fluorescent antibodies and flow cytometry. a) Data shows percent cells positive for MHCII, CD1d, and CD40 in alveolar-like, peritoneal-like, and bone marrow macrophages. b) Percent positive cells for CD80 and CD86 in BMMs. Data collected from triplicate experiments (SEM; * p < 0.05 vs. uninfected macrophage controls). Horizontal dotted line represents fold change of percent positive cells compared to uninfected controls. c) Histograms of BMM experiments show mean fluorescence intensity (MFI) against number of cells for MHCII, CD1d, CD40, CD80 and CD86. Grey solid line is isotype control, black solid line is uninfected control, and dotted line is infection with MTB, dMTB, or rMTB.
The peritoneal macrophage-like cells were infected in the same manner as before. Cells infected by wild-type MTB demonstrated small increases in expression of MHCII and CD1d and a small decrease in expression of CD40, but were not all significant to the uninfected controls. In contrast, dMTB infection induced an increase in expression for all three markers, more than twofold for MHCII and CD1d, and a one-third increase for CD40. rMTB infection once again reduced the expression of MHCII, CD1d, and CD40 to similar responses as seen by wild-type MTB infection.
To ensure that this trend was not an artifact of the cell lines, the same experiments were conducted using C57BL/6 murine bone marrow-derived macrophages (BMMs). The basal levels of MHCII, CD1d, and CD40 were found to be much lower than the cell lines, and infection with wild-type MTB did not significantly increase marker expression. Once TDM was removed from MTB, expression of MHCII, CD1d, and CD40 increased to at least 40–50%, and the fold-increase exceeded that of the previous two cell lines. As expected, infection with rMTB restored the characteristics seen by wild-type MTB infection. Interestingly, the patterned response to the presence of TDM on MTB was also seen for CD80 and CD86 in our BMM infections (Fig. 2b), but no changes in expression were seen by any of the macrophage types for CD11a, CD11b, and CD44. Histograms of the BMMs comparing mean fluorescence intensity (MFIs) of the infected cell populations were found to correlate with the percent positive cell data (Fig. 2c).
BMMs were also infected with TDM-coated biodegradable beads for 24 hours. The TDM-coated beads acted in a similar manner to wild-type MTB, showing no significant increase in surface marker expression for MHCII, CD1d, CD40, and CD80 and CD86 (Fig. 3). Plain uncoated beads appeared to have a slight increase in surface marker expression, but were found to be statistically insignificant.
Fig. 3. Effects of TDM-coated beads on expression of surface markers in bone marrow macrophages.

BMMs were treated for 24 hours with TDM-coated or plain biodegradable beads and assayed for surface marker expression. Histograms show mean fluorescence intensity (MFI) against number of cells for MHCII, CD1d, CD40, CD80 and CD86. Solid black line is untreated control BMMs, grey solid line is plain bead treatment, and black dotted line is TDM-coated bead treatment.
3.2 TDM-induced suppression of macrophage Ag85B presentation
We investigated the effects of TDM on mycobacterial antigen presentation by MTB-infected bone marrow macrophages using the BB7 T cell hybridoma to detect Ag85B:MHCII complexes [13, 15]. BMMs infected with wild-type MTB induced the hybridoma T cells to produce about 200 pg ml−1 of IL-2 (Fig. 4a). BMMs infected in the same manner with dMTB increased IL-2 concentrations by more than 500 pg ml−1. TDM-restored rMTB reduced IL-2 production to the same levels as wild-type MTB infection. The cells were also activated with IFN-γ 24 hours prior to infection with MTB. BMMs activated with IFN-γ induced an increase in IL-2 concentrations for all infections, but wild-type and TDM-reconstituted MTB did not meet or exceed the levels seen by dMTB infection.
Fig. 4. Removal of TDM from MTB increases Ag85B presentation to BB7 T cells.

a) BMMs with or without IFN-γ stimulation were infected with MTB, dMTB, or rMTB for 4 hours, then washed and overlaid with BB7 T cells. Supernatants were collected at 24 hours and measured for IL-2. Graph displays pg ml−1 of IL-2 concentration of triplicate experiments (SEM; * p < 0.05 vs wild-type MTB infection). b) BMMs infected with fbpA mutant or TDM-reconstituted fbpA mutant, then overlaid with BB7 T cells. Data collected from triplicate experiments (SEM; * p < 0.05 vs. uninfected macrophage controls).
We included experiments using the fbpA mutant MTB strain to study the effects of TDM on MTB. The fbpA gene encodes for Ag85A, part of the antigen 85 complex, and has been found to have mycolyl transferase activity [16]. This gene is disrupted in the mutant strain; therefore TDM cannot be produced and transported to the extracellular surface. Similar to delipidated MTB, this mutant strain has been found to have a lower survival rate in macrophages compared to wild-type MTB [16]. BMMs were infected with the fbpA mutant or with TDM-reconstituted fbpA mutant, and overlaid with BB7 T cells (Fig. 4b). IL-2 concentrations for macrophages infected with the fbpA mutant were found to be similar to the responses seen for dMTB, but upon reconstitution with pure TDM, presentation of Ag85B decreased by more than one-half.
3.3 TDM-induced SOCS expression
Suppressor of cytokine signaling (SOCS) has been implicated in suppressing certain immune signaling pathways by mycobacteria in murine macrophages [17]. SOCS negatively regulates the JAK-STAT pathway, which is important in many IFN-γ-mediated responses in macrophages, including the upregulation of MHCII. We investigated whether the presence or absence of TDM affected the production of SOCS in MTB-infected macrophages. BMMs were infected with wild-type, dMTB, or rMTB for 4 hours and the whole cell lysates were probed for SOCS-1 and SOCS-3 (Fig. 5). LPS and BCG are known inducers of SOCS and were included as positive controls in our experiments. In the presence of TDM, both SOCS-1 and SOCS-3 were produced, whereas dMTB induced little to no SOCS.
Fig 5. TDM promotes SOCS production in bone marrow macrophages.

BMMs were infected with LPS, BCG, MTB, dMTB, or rMTB for 4 hours. Whole cell lysates were immunoblotted for SOCS-1, SOCS-3, and actin.
4. Discussion
TDM is a unique molecule because its biologic activity changes depending on its environment. On the surface of MTB, TDM protects the organisms from killing within macrophages via mechanisms that include inhibition of phagosome-lysosome fusion and decreased acidification of phagosomes [6, 18]. When TDM is isolated and becomes associated with a hydrophobic substance such as oil or lipids, it becomes highly toxic in macrophages and is a potent T cell immunogen [19–21]. Pure TDM suspended in saline induces no toxicity in mice in quantities as high as 50 milligrams, whereas as little as 10 micrograms of TDM in an oil emulsion is highly toxic [22]. In immunized mice, TDM associated with lipid can induce allergic or caseating granulomas [19, 21]. Once dismissed as “unphysiologic”, the ability of TDM to exhibit two distinct sets of biologic activity has now been recognized as central to the pathogenesis of tuberculosis during primary infection and reactivation [21].
TDM is found on other mycobacterial species, pathogenic and non-pathogenic, and even non-mycobacterial species such as corynebacterium [23]. Interestingly, the structural arrangement and size of the mycolic acids on TDM tends to differ when isolated from these different species. One study has attempted to classify TDM by mycolic acid subclasses and found that the different compositions of TDM can affect how TDM aggregates and induces toxicity in mice [24]. Rao et al emphasizes the importance of TDM structure through their studies on mutant strains with altered TDM structures. The loss of trans-cyclopropanation of mycolic acids in their mutant strains produced a hypervirulent strain which was found to enhance inflammatory responses by macrophages [25]. Therefore the varying structures of TDM have been shown to play an important role in MTB pathogenicity. However, our study is focused on the biologic properties of TDM in its native structural form on MTB, specifically the laboratory strain H37Rv, and its effect on macrophage activity when TDM is absent from MTB.
The responses seen in the different macrophage types revealed a trend: TDM on the surface of MTB in wild-type and rMTB resulted in a characteristic expression of macrophage surface markers which were distinct from TDM-removed dMTB infection. Wild-type MTB induced no change or reduced expression of MHCII, CD1d, and CD40. This response was also seen for CD80 and CD86 in BMMs. Surface marker expression increased when lipids were extracted from MTB while still maintaining its viability. TDM was implicated as the cause because the wild-type phenotype was restored by reconstituting delipidated MTB with pure TDM. We found this to be unique for only these receptors because no differences were seen for the surface markers CD11a, CD11b, and CD44.
The significance of these surface markers is their importance in antigen presentation (MHCII and CD1d) and T cell co-stimulation (CD40, CD80 and CD86). MHCII is a major antigen presenting molecule, and its expression is impaired upon infection by MTB [26]. CD1d participates in development of cell-mediated immunity and is important for the T cell response to TDM, which has been recognized as a major MTB antigen [19, 27]. A decrease in CD40 expression has been correlated with a decrease in antigen-stimulated IFN-γ secretion from T cells [28]. CD80 and CD86 play a vital role as costimulatory molecules for MHCII during cross-stimulation with the T cell receptor. High levels of expression of MHCII along with CD40, CD80, and CD86 contribute to the efficiency of antigen presenting cells for stimulating T cells [7]. TDM on the surface of MTB may reduce MHCII binding efficiency by blocking expression of secondary signals necessary for antigen presentation. dMTB significantly increases surface marker expression, suggesting that TDM prevents macrophages from stimulating an effective immune response by targeting specific surface markers, therefore facilitating the persistence of MTB within macrophages.
A consequence of decreased surface marker expression is an impairment of cell-cell interactions such as antigen presentation to T cells. Our data shows a link between decreasing surface marker expression of MHCII and presentation of Ag85B to T cell hybridomas. Macrophages infected with wild-type or rMTB were found to be less efficient at presenting Ag85B to T hybridomas compared to dMTB. Macrophages treated with IFN-γ prior to infection exhibited enhanced antigen presentation [15], but TDM- intact MTB still reduced antigen presentation in IFN-γ-stimulated macrophages compared to delipidated MTB. This model implies that TDM may be involved in halting the antigen processing and presentation pathway in macrophages, or the TDM-induced decrease in expression of MHCII may lower the chances of a successful interaction with a T cell receptor. Publications have shown that dMTB has a lower survival rate in macrophages and phagosome-lysosome fusion occurs more readily when TDM is absent [5, 6].
The mechanistic properties of TDM on MTB are still unclear, but we hypothesize that there are three possible theories to explore. First, TDM could be produced to cover up other proteins or ligands on the surface of MTB. Extensive studies have analyzed the components of lipid extractions from TDM, but little is known about what is left behind on viable MTB after the extraction process. If TDM and other lipids are removed from MTB, there is a likely possibility that other proteins will become exposed to the cellular environment. These exposed proteins may be highly antigenic, explaining why delipidated MTB induces such strong surface marker responses in macrophages. It is also possible that the exposed proteins on delipidated MTB could be recognized by different pattern recognition receptors and taken up through an alternative phagosomal pathway, leading to more effective bacterial killing than TDM-intact MTB [29, 30].
Secondly, TDM could be acting as passive protection, inducing no response in macrophages as a means of avoiding phagosomal degradation. The TDM-coated beads were found to induce little to no response in the BMMs, and the surface marker expression characteristics were not significantly different with plain beads or the uninfected controls. TDM is produced in excess by MTB, forming a thick, hydrophobic “lipid capsule” that is characteristic of its growth patterns in subculture. The lipid capsule which surrounds MTB may be a means of avoiding detection within the macrophage, or the highly hydrophobic properties of this capsule may also inhibit lysosomal fusion, as evidenced by Spargo et al, who showed that phospholipid vesicles failed to fuse when coated with TDM [18].
Lastly, TDM itself could be inducing active inhibition in the macrophage immune response. We show evidence of active inhibition involving the TDM-induced production of SOCS. SOCS negatively targets IFN-γ-mediated responses via the JAK-STAT pathway [17]. Macrophage response to IFN-γ is necessary for upregulation of MHCII expression [31]. The TDM-induced overproduction of SOCS may inhibit the signal transduction pathways leading to MHCII expression, therefore decreasing antigen presentation efficiency. TDM may also target expression of other surface markers by blocking its assembly or transport to the cell surface, but the mechanisms are unknown.
MTB has a unique ability to evade host defenses, survive in human tissues, and eventually escape to infect new people. The immunomodifying effects of TDM demonstrated in our study reveal the complexities of TDM and how it protects MTB within the host cell. When TDM is present, macrophages are unable to produce optimal expression of surface markers important for antigen presentation and T cell costimulation. It is clear that TDM has a significant role in protecting and promoting MTB survival, and deserves more attention by the scientific community in understanding the virulence mechanisms of tuberculosis.
Acknowledgments
We wish to thank Dr. Lisa Armitige (Dept. of Pathology, University of Texas- Houston Health Science Center) for providing the fbpA mutant strain. We also wish to thank Dr. Clifford V. Harding and Dr. Henry Boom (Case Western Reserve University) for providing the BB7 T cell hybridoma.
Supported by NIH R01 HL068537
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bloch H, Sorkin E, Erlenmeyer H. A toxic lipid component of the tubercle bacillus (cord factor). I. Isolation from petroleum ether extracts of young bacterial cultures. Am Rev Tuberc. 1953;67:629–643. doi: 10.1164/art.1953.67.5.629. [DOI] [PubMed] [Google Scholar]
- 2.Flynn JL, Chan J. Immune evasion by Mycobacterium tuberculosis: living with the enemy. Curr Opin Immunol. 2003;15:450–455. doi: 10.1016/s0952-7915(03)00075-x. [DOI] [PubMed] [Google Scholar]
- 3.Hestvik AL, Hmama Z, Av-Gay Y. Mycobacterial manipulation of the host cell. FEMS Microbiol Rev. 2005;29:1041–1050. doi: 10.1016/j.femsre.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Bloch H. Studies on the virulence of tubercle bacilli; isolation and biological properties of a constituent of virulent organisms. J Exp Med. 1950;91:197–218, pl. doi: 10.1084/jem.91.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Indrigo J, Hunter RL, Jr, Actor JK. Influence of trehalose 6,6′-dimycolate TDM during mycobacterial infection of bone marrow macrophages. Microbiology. 2002;148:1991–1998. doi: 10.1099/00221287-148-7-1991. [DOI] [PubMed] [Google Scholar]
- 6.Indrigo J, Hunter RL, Jr, Actor JK. Cord factor trehalose 6,6′-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology. 2003;149:2049–2059. doi: 10.1099/mic.0.26226-0. [DOI] [PubMed] [Google Scholar]
- 7.Hope JC, Thom ML, McCormick PA, Howard CJ. Interaction of antigen presenting cells with mycobacteria. Vet Immunol Immunopathol. 2004;100:187–195. doi: 10.1016/j.vetimm.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Adami C, Brunda MJ, Palleroni AV. In vivo immortalization of murine peritoneal macrophages: a new rapid and efficient method for obtaining macrophage cell lines. J Leukoc Biol. 1993;53:475–478. doi: 10.1002/jlb.53.4.475. [DOI] [PubMed] [Google Scholar]
- 9.Palleroni AV, Varesio L, Wright RB, Brunda MJ. Tumoricidal alveolar macrophage and tumor infiltrating macrophage cell lines. Int J Cancer. 1991;49:296–302. doi: 10.1002/ijc.2910490226. [DOI] [PubMed] [Google Scholar]
- 10.Silva CL, Ekizlerian SM, Fazioli RA. Role of cord factor in the modulation of infection caused by mycobacteria. Am J Pathol. 1985;118:238–247. [PMC free article] [PubMed] [Google Scholar]
- 11.Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science. 1997;276:1420–1422. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- 12.Lima KM, Rodrigues JM., Junior Poly-DL-lactide-co-glycolide microspheres as a controlled release antigen delivery system. Braz J Med Biol Res. 1999;32:171–180. doi: 10.1590/s0100-879x1999000200005. [DOI] [PubMed] [Google Scholar]
- 13.Noss EH, Harding CV, Boom WH. Mycobacterium tuberculosis inhibits MHC class II antigen processing in murine bone marrow macrophages. Cell Immunol. 2000;201:63–74. doi: 10.1006/cimm.2000.1633. [DOI] [PubMed] [Google Scholar]
- 14.Noss EH, Pai RK, Sellati TJ, Radolf JD, Belisle J, Golenbock DT, Boom WH, Harding CV. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. J Immunol. 2001;167:910–918. doi: 10.4049/jimmunol.167.2.910. [DOI] [PubMed] [Google Scholar]
- 15.Singh CR, Moulton RA, Armitige LY, Bidani A, Snuggs M, Dhandayuthapani S, Hunter RL, Jagannath C. Processing and presentation of a mycobacterial antigen 85B epitope by murine macrophages is dependent on the phagosomal acquisition of vacuolar proton ATPase and in situ activation of cathepsin D. J Immunol. 2006;177:3250–3259. doi: 10.4049/jimmunol.177.5.3250. [DOI] [PubMed] [Google Scholar]
- 16.Armitige LY, Jagannath C, Wanger AR, Norris SJ. Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: effect on growth in culture and in macrophages. Infect Immun. 2000;68:767–778. doi: 10.1128/iai.68.2.767-778.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imai K, Kurita-Ochiai T, Ochiai K. Mycobacterium bovis bacillus Calmette-Guerin infection promotes SOCS induction and inhibits IFN-gamma-stimulated JAK/STAT signaling in J774 macrophages. FEMS Immunol Med Microbiol. 2003;39:173–180. doi: 10.1016/S0928-8244(03)00231-1. [DOI] [PubMed] [Google Scholar]
- 18.Spargo BJ, Crowe LM, Ioneda T, Beaman BL, Crowe JH. Cord factor (alpha, alpha-trehalose 6,6′-dimycolate) inhibits fusion between phospholipid vesicles. Proc Natl Acad Sci U S A. 1991;88:737–740. doi: 10.1073/pnas.88.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guidry TV, Olsen M, Kil KS, Hunter RL, Jr, Geng YJ, Actor JK. Failure of CD1D−/− mice to elicit hypersensitive granulomas to mycobacterial cord factor trehalose 6,6′-dimycolate. J Interferon Cytokine Res. 2004;24:362–371. doi: 10.1089/107999004323142222. [DOI] [PubMed] [Google Scholar]
- 20.Hunter RL, Olsen M, Jagannath C, Actor JK. Trehalose 6,6′-dimycolate and lipid in the pathogenesis of caseating granulomas of tuberculosis in mice. Am J Pathol. 2006;168:1249–1261. doi: 10.2353/ajpath.2006.050848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamagami H, Matsumoto T, Fujiwara N, Arakawa T, Kaneda K, Yano I, Kobayashi K. Trehalose 6,6′-dimycolate (cord factor) of Mycobacterium tuberculosis induces foreign-body- and hypersensitivity-type granulomas in mice. Infect Immun. 2001;69:810–815. doi: 10.1128/IAI.69.2.810-815.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunter RL, Olsen MR, Jagannath C, Actor JK. Multiple roles of cord factor in the pathogenesis of primary, secondary, and cavitary tuberculosis, including a revised description of the pathology of secondary disease. Ann Clin Lab Sci. 2006;36:371–386. [PubMed] [Google Scholar]
- 23.Lederer E. Cord factor and related trehalose esters. Chem Phys Lipids. 1976;16:91–106. doi: 10.1016/0009-3084(76)90001-3. [DOI] [PubMed] [Google Scholar]
- 24.Fujita Y, Okamoto Y, Uenishi Y, Sunagawa M, Uchiyama T, Yano I. Molecular and supra-molecular structure related differences in toxicity and granulomatogenic activity of mycobacterial cord factor in mice. Microb Pathog. 2007;43:10–21. doi: 10.1016/j.micpath.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Rao V, Gao F, Chen B, Jacobs WR, Jr, Glickman MS. Trans-cyclopropanation of mycolic acids on trehalose dimycolate suppresses Mycobacterium tuberculosis -induced inflammation and virulence. J Clin Invest. 2006;116:1660–1667. doi: 10.1172/JCI27335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hmama Z, Gabathuler R, Jefferies WA, de Jong G, Reiner NE. Attenuation of HLA-DR expression by mononuclear phagocytes infected with Mycobacterium tuberculosis is related to intracellular sequestration of immature class II heterodimers. J Immunol. 1998;161:4882–4893. [PubMed] [Google Scholar]
- 27.Guidry TV, Hunter RL, Jr, Actor JK. CD3+ cells transfer the hypersensitive granulomatous response to mycobacterial glycolipid trehalose 6,6′-dimycolate in mice. Microbiology. 2006;152:3765–3775. doi: 10.1099/mic.0.29290-0. [DOI] [PubMed] [Google Scholar]
- 28.Samten B, Thomas EK, Gong J, Barnes PF. Depressed CD40 ligand expression contributes to reduced gamma interferon production in human tuberculosis. Infect Immun. 2000;68:3002–3006. doi: 10.1128/iai.68.5.3002-3006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinberg BE, Grinstein S. Pathogen destruction versus intracellular survival: the role of lipids as phagosomal fate determinants. J Clin Invest. 2008;118:2002–2011. doi: 10.1172/JCI35433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Kooyk Y, Geijtenbeek TB. DC-SIGN: escape mechanism for pathogens. Nat Rev Immunol. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- 31.Hussain S, Zwilling BS, Lafuse WP. Mycobacterium avium infection of mouse macrophages inhibits IFN-gamma Janus kinase-STAT signaling and gene induction by down-regulation of the IFN-gamma receptor. J Immunol. 1999;163:2041–2048. [PubMed] [Google Scholar]