

Abstract
Besides acting as potent free radical scavengers, tocopherols and tocotrienols have been known to have non-antioxidant properties such as the involvement of α-tocopherol (αT) in PKC pathway and the anti-cancer properties of γ-tocotrienol (γT3). This study aims to elucidate whether protective effects shown by αT and γT3 in H2O2-induced neuron cultures have anti-apoptotic or pro-apoptotic tendency toward the initiation of neuronal apoptosis. H2O2 is used to induce apoptosis in primary cerebellar neuron cultures which is attenuated by pretreatment of αT or γT3 at concentrations ≤10 μM. Similar to our previous work, γT3 was found to be neurotoxic at concentrations ≥100 μM, whereas αT showed no neurotoxicity. Cellular uptake of γT3 was higher than that of αT. Treating cells simultaneously with either γT3 or αT and with then H2O2 led to higher expression of Bax and Bcl-2 than in neurons exposed to H2O2 alone. Analysis of Bcl-2/Bax ratio as ‘survival index’ showed that both pretreatment of γT3 and αT followed by H2O2 increase the ‘survival index’ of Bcl-2/Bax ratio compared to H2O2-treated cells, while treatment of γT3 alone decrease the ratio compared to unchanged Bcl2/Bax ratio of similar treatment with αT alone. Similar treatment of γT3 decreased p53 expression and activates p38 MAPK phosphorylation, whereas αT did not alter its expression compared to H2O2-treated cells. Treating neurons with only γT3 or αT increased the expression of Bax, Bcl-2, p53, and p38 MAPK compared to control with γT3 exerting stronger expression for proteins involved than αT. In conclusion, low doses of γT3 and αT confer neuroprotection to H2O2-treated neurons via their antioxidant mechanism but γT3 has stronger pro-apoptosis tendency than αT by activating molecules involved in the neuronal apoptotic pathway in the absence of H2O2.
Keywords: Vitamin E, Oxidative stress, Antioxidant, Apoptosis, Cerebellar neurons
Introduction
Vitamin E has been touted as a potent free radical scavenger and is protective to neurons against oxidative stress. There has been considerable interest in the potential role of vitamin E in the treatment of age-related neurodegenerative diseases such as Alzheimer’s disease (AD) and Down’s syndrome (DS). In vitro studies using cell cultures suggest that vitamin E has neuroprotective effects; for example, vitamin E has been shown to prevent neurodegeneration in neuronal cultures from Down’s syndrome patients (Busciglio and Yankner 1995). The compounds, tocopherols and tocotrienols, which belong to the same family of vitamin E have been shown to act as neuroprotective agents in neuronal apoptosis induced by oxidative stress (Sen et al. 2000; Osakada et al. 2003, 2004). Epidemiological studies (Sano et al. 1997; Morris et al. 1998) and animal studies (Alvarado et al. 2006; Banudevi et al. 2006) have argued for the benefits of taking vitamin E and C and eating antioxidant-rich diets, respectively. But results from clinical studies in humans, however, have been controversial. Some studies have shown that dietary antioxidants (e.g., vitamin E and C) confer protection against neurodegenerative diseases (Golbe et al. 1998) and reduce the risk of AD (Engelhart et al. 2002); while a more recent study of a larger randomized controlled trial shown that high dose of vitamin E supplementation did not significantly slow down the progression of mild cognitive disorder to AD and showed no benefit on mild cognitive disorder (Petersen et al. 2005). Other studies have shown that dietary intake of antioxidant vitamins E and C confers no protection against the onset of amyotrophic lateral sclerosis (ALS) (Galbussera et al. 2006; Graf et al. 2005).
All tocopherol and tocotrienol derivatives come under one generic description that is vitamin E. Both tocopherols and tocotrienols have isomers, designated as α-, β-, γ-, δ-, which differ by the number and position of the methyl groups on the chromanol ring. Tocopherols have a phytyl chain, while tocotrienols have similar chain but with 3 double bonds at position 3′, 7′, and 11′ (Wang and Quinn 2000). The two classes of compound react differently with free radicals (Yoshida et al. 2003) and they show different potencies as neuroprotectants. They have been proposed to play important roles as signaling molecules (Azzi and Stocker 2000; Roy et al. 2002). Studies have shown that tocotrienols and tocopherols modulate the signal transduction activity of redox-sensitive proteins such as PKC-δ, PKB (Zingg 2007), as well as the initiation of apoptosis in cancer cells by tocotrienols (Agarwal et al. 2004; Srivastava and Gupta 2006). Tocotrienols have been shown to be pro-apoptotic in various cancer cell lines such as neoplastic mouse mammary tumor cells (Wali and Sylvester 2007) and prostate tumor cell (Kumar et al. 2006). A recent paper has reviewed that specific forms of vitamin E display potent apoptotic activity against a wide range of cancer cell types, while having little or no effect on normal cell function or viability. Experimental studies have also determined that the intracellular mechanisms mediating the apoptotic effects of specific vitamin E compounds display great diversity in different types of cancer cells and have been found to restore multidrug resistant tumor cells sensitivity to chemotherapeutic agents (Sylvester 2007).
Neuronal apoptosis appears to be either a cause or an important characteristic in most of the acute and chronic neurodegenerative diseases, such as Parkinson’s disease (Onyango 2007), amyotrophic lateral sclerosis (ALS) (Emerit et al. 2004), stroke, and Alzheimer’s disease (Moreira et al. 2005; Butterfield and Sultana 2007). Oxidative stress has been implicated either as a primary initiating event or as a secondary effect in the above neurodegenerative diseases by initiating neuronal apoptosis (Emerit et al. 2004; Mariani et al. 2005; Drögen and Schipper 2007). Reactive oxygen species (ROS) such as H2O2 are well-known triggers of apoptosis as H2O2 is the substrate for Fe2+-catalyzed generation of highly reactive hydroxyl radicals (Fenton reaction products) which rapidly oxidized and altered membrane lipids and proteins and also cleavage of DNA (Vervaart and Knight 1996; Wang et al. 2003; Krantic et al. 2005). This leads to gradual loss of specific sets of neurons and subsequent dysregulation of the central nervous system (CNS) function (Thompson 1995; Sastry and Rao 2000).
ROS production has been linked to the apoptosis signaling pathway in neurons which leads to caspase activation (Chong et al. 2005). Proteolysis of pro-caspases initiates a signaling cascade culminating in cell apoptosis with caspase-3 cascade triggered either through the extrinsic pathway involving caspase-8 activation (Peter and Krammer 2003) or through the intrinsic pathway involving caspase-9 activation (Zou et al. 1999). Accumulation of DNA damage caused by oxidative stress induced p53 production in neurons and in turn induced genes that encodes the pro-apoptotic proteins Bax and the BH3- only proteins PUMA and Noxa. At the same time, p53 may also promote apoptosis through transcriptional repression of survival factors such as Bcl-2 (Culmsee and Mattson 2005).
Our previous study has also shown vitamin E isomers γ-tocotrienol (γT3) to be cytotoxic to astrocytes (Mazlan et al. 2006) and this has raise concern of the safety level of vitamin E especially in the usage of high dose of vitamin E as treatment in diseases. It is generally known that both tocopherol and tocotrienol mainly exert their neuroprotection against oxidative stress via their antioxidant properties, but as tocotrienol has known anti-cancer properties by initiating apoptosis in cancer cells, the involvement of tocotrienol in the initiation of apoptosis pathway in neurons has yet to be investigated. Therefore, in the present study, we investigated the role of αT and γT3 in modulating neuronal apoptosis by comparing the effects of the two compounds on the expression of transduction proteins in the apoptotic pathway in the presence and absence of oxidative stress.
Materials and Methods
Antioxidant Compounds
The chemicals α-tocopherol (αT) and γ-tocotrienol (γT3), extracted from palm oil, were supplied by the Malaysia Palm Oil Board.
Preparation of Primary Neuron Cultures from Rat
This study protocol was approved by the Universiti Kebangsaan Malaysia (UKM) Animal Ethics Committee (UKMAEC).
Primary cultures of cerebellar granule neurons were prepared as follows. The cerebellar region was removed from postnatal 2nd day (P2) Wistar rat under a stereoscopic microscope. The tissue was placed in Ca2+- and Mg2+-free Dulbecco’s phosphate buffer saline (CMF-DPBS, pH 7.4) containing 3 g/l BSA (Sigma, USA), 1 g/l glucose (Sigma), and 50 μg/ml gentamicin (Gibco, USA). The meninges was removed from the cerebellar tissue, which was then placed on sterile Teflon board. A sterile scalpel was used to slice the sample using perpendicular strokes before it was transferred to a 50 ml collection tube. It was then briefly centrifuged, and the pellet was resuspended in 12 ml of a warm solution of 0.025% trypsin (Flowlab, Australia) and 0.04% DNAse (Sigma). The suspension was incubated in a shaking water bath for 10 min, followed by addition of 1 ml of fetal calf serum (FCS) (Flowlab) to inhibit trypsin activity and 1 ml of 1 mg/ml DNAse to decrease the clumping of genomic DNA from lysed cells. The suspension was then centrifuged at 800 rpm for 5 min. After removing the supernatant, the pellet was resuspended and slowly triturated with a fire-polished glass pipette in Dulbecco’s Modified Eagle’s Medium (DMEM, pH 7.4) (Flowlab) containing 19 mM NaHCO3, 26.2 mM KCl, 7 μM p-aminobenzoic acid (Sigma), 100 mU/l insulin (Sigma), 50 μg/ml gentamicin, and 10% fetal calf serum. The cells were plated at a density of 1.5 × 106 per ml in 96-well plates, 10 cm Petri dishes and 8-well chamber slides. All culture surfaces were pretreated with poly-l-lysine. Cultures were maintained in 5% CO2/95% air at 37°C. Experiments were carried out on the third to fifth day in vitro (DIV). Immunocytochemical analysis showed that 90% of cells were neurons (data not shown).
Cell Viability and Apoptosis Assay
Cerebellar granule neuron cultures were incubated with varying concentrations of H2O2 (1–500 μM), αT, and γT3 (1–750 μM) for 24 h at 37°C. Neurotoxicity was assessed by determining cell viability using MTS and LDH release assays as previously described (Mazlan et al. 2006). To study the neuroprotective effects of αT and γT3, IC50 of H2O2 in cerebellar granule neurons was first determined to be 50 μM. The primary neuronal cell cultures described above were divided into various treatments: control, H2O2-treated, treatment with only αT or γT3 and pretreatment with concentrations of αT and γT3 ranging from 1 to 100 μM for 1 h at 37°C and H2O2 was added later. The cells were then incubated for 24 h at 37°C after which cell viability and apoptosis were assessed. Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay and lactate dehydrogenase (LDH) release assay. The rate of apoptosis was assessed using single-stranded DNA (ssDNA) ELISA kits as described previously (Mazlan et al. 2006). Briefly, treated cells were fixed with 80% methanol in PBS and then treated with formamide. DNA in apoptotic cells were then denatured by heating to 75°C for 10 min, immediately followed by cooling to 4°C for 5 min. Positive controls provided by the kit were pre-fixed in a separate 96-well plate with 100 μl of positive controls added to each well and dried overnight according to protocol. The fixed positive control wells were rinsed with PBS before use and can be stored dry, covered until ready to use. The fixed positive controls were then concurrently processed together with the experimental plates according to subsequent steps of protocol. The absorbance reading of the positive control should be between 1.5 and 2.8 which indicates good assay sensitivity and will be denoted as 100% apoptosis. The apoptosis rate of all sample readings are calculated relative to the positive control readings. For negative controls, 100 Units/ml SI nuclease was added to prevent unspecific breakage of single-stranded DNA and was incubated at 37°C for 30 min. To block non-specific binding sites, 3% nonfat milk was added and incubated at 37°C for 1 h before incubating with antibody mixture for 30 min. ABTS solution was added after washing. The absorbance was then read at 405 nm using a microplate reader (VeraMax, Molecular Devices, USA).
Morphological analyses of live and dead cells were done according to previously described method (Mazlan et al. 2006). Briefly, 30 μg/ml calcein-AM and 7.5 μg/ml propidium iodide were added concurrently to cultured granule neurons in chamber slides and incubated for 30 min. Thereafter, cultures were washed with PBS, fixed with fresh 2% paraformaldehyde, and then coverslips were mounted for microscopic examination.
Determination of Vitamin E Uptake by HPLC
Uptake of γT3 and αT was carried out using reverse-phase high performance liquid chromatography (HPLC) Fluorescent EM 330 nm, EX294 detector (Shimadzu, Japan) as describe previously (Mazlan et al. 2006). Briefly, neuron cultures treated with 10 μM of either γT3 or αT and untreated controls were harvested after 24 h and counted before adding 50 mg/dL BHT in 95% ethanol to stop auto-oxidation. After incubation for 10 min at room temperature, ice-cold 95% ethanol was added to precipitate the protein. To extract vitamin E, cells were sonicated for 40 s before addition of ice cold HPLC grade hexane. Cells were then centrifuged at 3000 g, 5 min before collecting the hexane layer of supernatant. Vitamin E extracts were then vacuum dried and stored at −70°C before analysis with HPLC. To prepare HPLC samples, 100 μl hexane was added before further dilution with hexane for HPLC analysis. Peaks of samples were compared with tocotrienol-rich fraction (TRF) standard and concentration of γT3 and αT uptake in cells were calculated in μM per million cells.
Activity of Caspase-3, -8, and -9
Activity of caspase-3, -8, and -9 was measured using the commercial kit available CPP32/Caspase-3 Fluorometric Protease Assay kit, FLICE/Caspase-8 Fluorometric Protease Assay kit and MCH6/Caspase-9 Fluorometric Protease Assay kit (Chemicon, USA), respectively. All three kits assay caspase activity by measuring the cleavage of a specific sequence recognized by the target caspase. For caspase-3, the recognition sequence is DEVD; for caspase-8, it is IETD; and for caspase-9, it is LEHD. In these three kits, the substrate for each specific sequence is conjugated to 7-amino-4-triflourometil coumarin (AFC), and the conjugate emits blue light at a wavelength of 400 nm. The cleavage of each recognition sequence releases AFC, which emits green light at 505 nm. The change in emission is quantified using a fluorescence microplate reader (Wallace, USA).
After the treatment of the cells as describe above, they were harvested. Aliquots of the cells (2 × 106) are resuspended in 50 μl of cold lysis buffer and allowed to sit on ice for 10 min. Next, 50 μl of reaction buffer and 5 μl of 1 mM DEVD/IETD/LEHD (leading to a final cleavage substrate concentration of 50 μM) were added to the samples and to the control and incubated at 37°C for 1 h. The samples were then quantified at 400 nm and 505 nm in the fluorescence microplate reader. The caspase activity levels in the samples were then compared to the corresponding control values.
SDS-PAGE and Western Blotting
The expression levels of proteins involved in apoptosis signaling were studied in the primary cultures of cerebellar granule neurons; these molecules included p53, Bax, Bcl-2, and p38 of the MAPK signaling pathway. Treated cells and untreated control cells were harvested into 250–300 μl complete RIPA lysis buffer, incubated on ice for 30 min, and centrifuged at 14,000g for 30 min. Supernatants were aliquoted and stored at −80°C. Total protein concentration was estimated by the Bradford assay. Aliquots containing 100 μg of total protein were loaded into 12% SDS-polyacrylamide gels. After electrophoresis, gels were transferred to a trans-blot machine, and proteins were blotted onto nitrocellulose membranes (GE Healthcare, USA) at 4°C. For detection of desired bands, membranes were blocked with 5% skim milk in PBS for 1 h, washed, and incubated with primary antibodies overnight at 4°C with gentle agitation. On the following day, membranes were washed with PBS-Tween and incubated for 1 h at room temperature with secondary antibodies; HRP-conjugated anti-mouse IgG for monoclonal antibodies (Bcl-2, Bax, p53 antibodies from Chemicon, USA, while PhosphoPlus p44/p42 MAP kinase kit from Cell Signaling Tech, USA) and HRP-conjugated anti-rabbit for polyclonal antibodies (Chemicon, USA). Finally, membranes were washed before substrate from Western Lightning Chemiluminescence Reagent Plus (Perkin-Elmer Life Science, USA) were added and bands were detected using X-ray film (Kodak, Japan). The intensity value of Bcl-2, Bax, p53, p38 MAPK and β-actin bands was obtained from scanned films using ImageMaster TotalLab software (version 1.11, GE Healthcare). The intensity values of Bcl-2, Bax, p53 bands were normalized to β-actin values, while phosphorylated p38 MAPK values were normalized to non-phosphorylated p38 MAPK in the same set of experiments.
Statistical Analysis
Each experiment of cultures in microplates was carried out in triplicate with at least 3 independent cultures. Experiments of cultures in petri dishes and chamber slides was carried out in 3 independent cultures. Data are reported as the mean ± SD from at least 3 independent experiments. Comparisons between groups were made using Student’s t-test and two-way ANOVA. A P-value less than 0.05 was considered statistically significant.
Results
Vitamin E toxicity and effects of vitamin E on H2O2-induced cell death
The neurotoxicity of γT3 was tested and found to be toxic for primary rat cerebellar neurons at concentrations higher than 100 μM, whereas αT was non-toxic up to 750 uM tested (Fig. 1a, b). αT and γT3 were able to protect neurons from H2O2-induced cell death at concentrations ≤10 μM as assessed by MTS assay (Fig. 1c) and also from ELISA-based ssDNA apoptosis assay (Fig. 1d), as well as from fluorescent staining (Fig. 2). Based on the calcien-AM and propidium iodide staining, 10 μM αT and γT3 were able to maintain the viability of neurons exposed to 50 μM H2O2 (Fig. 2c, d).
Fig. 1.

Cytotoxic effects of α-tocopherol (αT) and γ-tocotrienol (γT3) on cerebellar granule neurons as assessed by a in MTS assay and b in LDH release assays. Percent of MTS reduction corresponds to the viable cell number. Cerebellar neurons were incubated with increasing concentrations of either αT or γT3 for 24 h at 37°C. There was no significant reduction in viable cell number when incubated with αT. However, the viable cell number reduced significantly to 80% when cells were incubated with γT3 at 100 μM and cell viability were reduced further as concentration of γT3 increased. Similar results were obtained with the LDH release assay. *P < 0.05 compared to control. Data are presented as means ± SD, n = 9. Protective effects of α-tocopherol (αT) and γ-tocotrienol (γT3) against H2O2-induced cell loss in cerebellar granule neuron cultures, cell viability was determined using MTS assay (c) and apoptosis assay using ELISA kits for ssDNA (d). Neurons were pretreated with different concentrations of αT and γT3 for 1 h before exposure to 100 μM H2O2 for 24 h at 37°C. αT and γT3 increased cell viability and decreased apoptosis in H2O2-treated cells when used in the concentration range of 1 to 10 μM. At concentration above 10 μM, γT3 was found to increase apoptosis in H2O2-treated cells. *P < 0.05 compared to controls. Data are presented as mean ± SD, from 3 independent experiments of triplicate wells (n = 9)
Fig. 2.

Fluorescence cell death staining of neurons exposed to different treatments: a control, b 50 μM H2O2 treatment, c pre-incubation with γT3 (10 μM) for 1 h followed by H2O2 treatment, d pre-incubation with αT (10 μM) for 1 h followed by H2O2 treatment. Live cells were stained with 30 μM calcein-AM (green) and dead cells were stained with propidium iodide (PI, red). Neurons undergo early apoptosis as membrane permeability increases, allowing PI to stain cell nuclei. Nuclei show condensation (i) and formation of apoptotic bodies (ii) when exposed to 50 μM H2O2. When neurons were pre-incubated with either c γT3 or d αT, cells remained viable and membrane integrity was retained to a greater extent than in cells treated with H2O2 alone. Scale bar: 40 μm (refer online version for color figures)
Uptake of α-Tocopherol and γ-Tocotrienol into Neurons
HPLC analysis of hexane extract of neurons treated with αT and γT3 shows that there was a significant difference between the uptake of αT and γT3 in cerebellar granule neurons. Figure 3 shows that uptake of γT3 and αT was significantly higher than control and γT3 uptake was significantly higher compared to αT.
Fig. 3.

Uptake of α-tocopherol (αT) and γ-tocotrienol (γT3) in cerebellar granule neurons. Neuron cultures were incubated with 10 μM of either αT or γT3 for 24 hrs at 37°C. Cells were harvested and prepared for HPLC as described in text. Cellular uptake of γT3 and αT is significantly higher than control. Uptake of γT3 is significantly higher than αT uptake into neurons. Data are presented as mean ± SD, n = 3
Activation of Caspases
Figure 4 shows that exposure of neuronal cells to H2O2 significantly increased the activity of caspase-3 and caspase-8, but not of caspase-9. This suggests that under these experimental conditions, H2O2 induces apoptosis by activating the extrinsic pathway. Pretreatment with 10 μM γT3 significantly reduced the activities of caspase-3 and caspase-8, but not of caspase-9. Although αT at a concentration of 10 μM was able to reduce the activity of caspase-8, it was insufficient to reduce the activity of caspase-3.
Fig. 4.

Activity of caspases (caspase-3, caspase-8, and caspase-9) in H2O2-treated granule neurons pretreated with α-tocopherol (αT) or γ-tocotrienol (γT3) for 1 h. Exposure to H2O2 led to much higher activity of caspase-3 and caspase-8 than in control cultures of cerebellar neurons. In contrast, there were no significant differences in caspase-9 activity in cultures exposed to H2O2 alone, cultures pre-treated with γT3 or αT followed by H2O2, or control untreated cultures. Pre-incubation with γT3 lowered the activity of both caspase-3 and caspase-8 relative to cultures treated with H2O2 alone. Pre-incubation of αT reduced the activity of caspase-8 to the levels in control cultures, but it did not reduce the activity of caspase-3. *P < 0.05 compared to controls, and # P < 0.05 compared to cells treated only with H2O2. Data are presented as mean ± SD (n = 9)
Western Blot Analysis of Proteins
Exposing the primary neuronal cultures to H2O2 for a different periods of time (up to 24 h) increased the expression of the apoptosis-related proteins Bax, Bcl-2, and p53. Maximal expression was observed at 6 h for Bax in Fig. 5a (i) and Bcl-2 in Fig. 5a (ii), and at 1 h for p53 in Fig. 5a (iii) and 12 h for activation of p38 MAPK in Fig. 5a (iv). Relative band intensity of Bax and Bcl-2 is shown as a Bcl-2/Bax ratio in Fig. 5b (i). Various studies have used the ratio of Bax expression to Bcl-2 expression (Bax/Bcl-2) as indicator of apoptosis activity (Srinivas et al. 2000; Shabnam et al. 2004). This study uses the Bcl-2/Bax ratio to reflect survival index.
Fig. 5.
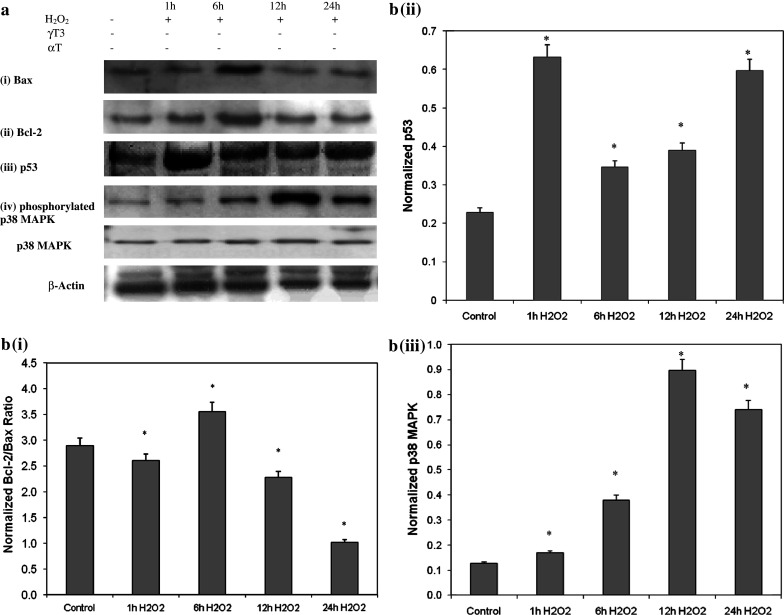
H2O2 activates apoptosis pathway proteins, Bax, Bcl-2, and p53 protein levels and phosphorylation of p38 MAPK in granule neurons. a Protein levels in primary neurons treated with H2O2 for different incubation periods (1, 6, 12, 24 h). During treatment with H2O2, the expression of both Bcl-2 and Bax increased and peaked at 6 h, then decreased at 12 and 24 h. H2O2 increased expression of p53, with a peak occurring at 1 h, above the levels in control cultures, while H2O2 activated phosphorylation of p38 MAPK at 6 h which peaked at 12 h. b Densitometric analysis of Bcl-2 and Bax as Bcl-2/Bax ratio in (i), p53 in (ii), and p38 MAPK (iii). Bcl-2 and Bax bands were quantified using Total Image software and the Bcl-2/Bax ratio was determined as ‘survival index.’ During exposure to H2O2, the ratio of Bcl-2/Bax increased at 6 h but progressively decreased thereafter while expression of p53 and activation of p38 MAPK increased. *P < 0.05 compared to control. Data are presented as mean ± SD (n = 3)
When primary cultures were treated with αT and γT3 prior to being exposed to H2O2, the expression of Bax, the expression of Bcl-2, and the ratio of Bcl-2 to Bax expression were higher than in cultures treated only with H2O2 in Fig. 6a (i), (ii), and b (i). Pretreatment of γT3 decreased p53 expression compared with cultures exposed only to H2O2, whereas αT did not in Fig. 6a (iii), b (ii). Pretreatment with γT3 did not reduce the level of phosphorylated p38 MAPK, whereas pretreatment with αT led to lower phosphorylation of p38 MAPK than in control cultures or cultures treated only with H2O2 in Fig. 6a (iv), b (iii).
Fig. 6.

Various treatment of cells in the following fashion: untreated control, incubation of neurons with H2O2 for 24 h (H2O2), incubation of neurons with γT3 for 24 h (γT3), 1 h of γT3 pretreatment in neurons followed by H2O2 incubation for 24 h (γT3 + H2O2), incubation of neurons with αT for 24 h (αT), and 1 h of αT pretreatment in neurons followed by H2O2 incubation for 24 h (αT + H2O2). a Protein levels in primary neurons with various treatments as mentioned above. Expression levels of Bax were higher in neurons pre-treated with γT3 or αT followed by H2O2 and also in neurons treated only with γT3 or αT than they were in untreated control neurons or neurons treated only with H2O2. Similarly, pre-incubation with γT3 or αT followed by H2O2 treatment and cultures treated with only γT3 or αT increased Bcl-2 levels compared to control cultures and cultures treated only with H2O2. Pre-incubation with γT3 or αT increased the Bcl-2/Bax ratio back to control levels. Incubation with γT3 only decreased the Bcl-2/Bax ratio relative to control cultures, whereas αT did not change the ratio. Pre-incubation of cells with γT3 or αT followed by H2O2 reduced p53 levels to below those in control cultures and cultures treated only with H2O2. Incubation with only γT3 and αT induced higher p53 expression compared to the control. Cultures treated with αT alone had significantly higher p53 levels than cultures treated with αT followed by H2O2. Pre-incubation with γT3 did not alter p38 MAPK activation, whereas pre-incubation with αT decreased phosphorylation of p38 MAPK. Cultures treated only with γT3 or αT showed increased activation of p38 compared to controls. Activation of p38 in cultures treated with γT3 alone was less than in cultures treated with γT3 followed by H2O2. In contrast, cultures treated with αT alone showed greater phosphorylation of p38 than did cultures treated with αT followed by H2O2. b Densitometric analysis of Bcl-2 and Bax as Bcl-2/Bax ratio in (i), p53 in (ii), and p38 MAPK in (iii). Cultures treated with γT3 alone had lower Bcl-2/Bax ratios than did cultures treated first with γT3 and then with H2O2. The opposite results were seen with αT. *P < 0.05 compared to control, # P < 0.05 compared to cells treated only with H2O2, and **P < 0.05 compared to cells pre-incubated with γT3 or αT followed by H2O2 treatment. Data are presented as mean ± SD (n = 3)
To investigate the involvement of αT and γT3 in the induction of the apoptosis signaling pathway, neurons were treated with these two vitamin E isomers without subsequent treatment with H2O2. Treatment with either compound resulted in increased expression of Bax and Bcl-2 as showed in Fig. 6a (i), (ii). However, calculating the Bcl-2/Bax ratio shows that γT3 decreases this ‘survival ratio,’ whereas αT did not alter it from Fig. 6b (i). In addition, treatment with either αT or γT3 increased the expression of p53 in Fig. 6a (iii), b (ii). Treatment with either γT3 or αT increased phosphorylation of p38 MAPK with γT3 treatment showing a marked increase of phosphorylated p38 MAPK compared to αT in Fig. 6a (iv), b (iii).
Discussion
Neuroprotective Effects of α-Tocopherol and γ-Tocotrienol
Numerous in vitro studies have demonstrated the ability of αT to protect against cellular damage induced by oxidative stress in striatal neurons (Osakada et al. 2003), and damages due to Fe2+-mediated oxidation in hippocampal neurons (Ferreira et al. 2005; Crouzin et al. 2007). Recent in vitro and in vivo studies by Zhang et al. (2004) showed that tocopherol reduces brain infarction and brain damage in cerebral ischemia by activating hypoxia-inducible factor-1 (HIF-1), which protects the brain from the effects of ischemia. Similar results have been obtained in studies involving long-term administration of αT to OXYS rats (an animal model of aging and oxidative stress) for up to nine months which reduces protein carbonyl production and lipid peroxidation, while simultaneously enhancing cognitive function (Kolosova et al. 2006). Finally, Ferreira et al. (2005) reported that pre-treating hippocampal neurons with αT prior to exposing them to Fe2+ protected the cells from oxidative damage, and that this neuroprotection was due to the induction of gene expression and the synthesis of new protein.
Our previous work has shown similar neuroprotective effects of γT3 in astrocytes (Mazlan et al. 2006). In both that report and the present study, we found high concentrations of γT3 (≥100 μM) to be detrimental to neuronal survival. In contrast, we found that concentrations below 10 μM were insufficient to be cytotoxic to neurons, yet sufficient to confer neuroprotection (Fig. 1a, b) from cell viability assay and apoptosis assay. In addition, cellular uptake of γT3 is higher than αT in neurons under identical conditions (Fig. 3). This is in contrast to our previous study showing no significant differences between uptake of αT and γT3 by astrocytes (Mazlan et al. 2006). A recent study also showed that α-tocotrienol is localized at the same place as α-tocopherol which is near the membrane surface and tocotrienol has higher membrane permeability than tocopherol which strongly suggested that transport of γT3 across the cell membrane of cerebellar granule neurons is more efficient than transport of αT (Yoshida et al. 2007). These data may explain why γT3 is neuroprotective at low concentrations but toxic at higher concentrations as the recent study above also implies. However, the mechanism for γT3 cellular transport from liver is still unclear. Osakada et al. (2004) have demonstrated neuroprotection by α-, γ-, and δ-tocotrienol in striatal neurons exposed to various pro-oxidants, including H2O2. Similar to our results in cerebellar neurons, their work suggests that γT3 protects granule neurons at low concentration (≤10 μM) through its antioxidant properties. Sen et al. (2000) showed that α-tocotrienol at nanomolar concentrations conferred neuroprotection against glutamate-induced oxidative stress in the HT4 neuronal cell line; in this case, however, the protection was not due to the compound’s antioxidant properties but through its effects on the c-Src signaling pathway. Furthermore, recent reports from their group suggest that α-tocotrienol also interacts with 12-lipogenase (12-LOX) to prevent glutamate-induced cell death (Khanna et al. 2006).
Apoptosis Signaling Pathway
In the apoptosis signaling pathway, caspases, Bcl-2, Bax, p53, and other molecules play an important role in the initiation of apoptosis. In the present study, we observed H2O2 to activate both the intrinsic pathway via caspase-9 and the extrinsic pathway via caspase-8. As a result, caspase-3 was activated (Fig. 4). In contrast to the activity of H2O2, γT3 and αT were found to act through the extrinsic pathway but not through the intrinsic one, since pre-treating cultures with either γT3 or αT did not prevent the activation of caspase-9.
Expression of Bcl-2 and Bax is essential for caspase activation in the apoptosis signaling pathway. The Bcl-2 family appears to function by conformation-induced insertion into the outer mitochondrial membrane, which leads to the formation of channels or pores through which cytochrome c can be released. Cytochrome c then activates caspase-3. Bcl-2 and Bax are able to form homodimers (Bcl-2:Bcl-2 and Bax:Bax) and heterodimers (Bcl-2:Bax) with one another. When Bax molecules are present in excess, the anti-apoptosis activity of Bcl-2 is antagonized. On the other hand, Bax heterodimerization with Bcl-2 prevents apoptosis by blocking the release of cytochrome c (Antonsson et al. 1997) or by modulating the mitochondrial membrane potential and volume homeostasis (Vander Heiden et al. 1997). This study looks into the Bcl-2/Bax ratio from densitometric analysis rather than individually assessed expression of Bcl-2 and Bax to reflex the interaction of the heterodimerization with Bcl-2 as a measure of anti-apoptosis. After incubating primary neuronal cultures for 24 h with H2O2, the Bcl-2/Bax ratio fell significantly (Fig. 5b). However, it is interesting to note that the initial cellular response was a rise in Bcl-2/Bax ratio, with a peak at 6 h. This may represent an attempt by the cells to defend themselves against H2O2-induced oxidative damage. Pretreatment of cells with either αT or γT3 before exposure to H2O2 increased the survival index. However, γT3 treatment without exposure to H2O2 decreased the index, a result that was not observed in any of the αT experiments (Fig. 6b). Numakawa et al. (2006) showed that αT induced up-regulation of Bcl-2 after 24 h treatment, but Bad levels remain unchanged whereby this study also show increased expression of Bcl-2 as well as increased Bax level.
Oxidative damage to nuclear DNA activates p53 protein, which then activates the downstream effectors Bax and Apaf1. Bax is directly regulated by p53 (Morrison et al. 2003). The results of this study show that treatment with either αT or γT3 without exposure to H2O2 increases p53 expression. Thus the present data suggest that the ability of αT or γT3 to prevent H2O2-induced apoptosis in neurons is due primarily to their antioxidant properties. However, in the absence of any pro-oxidant, αT and γT3 seems to initiate the apoptosis pathway in the activation of Bax and p53.
p38 MAPK is preferentially activated by oxidative stress with increased activation of p38 MAPK under conditions of oxidative stress has been implicated in the accumulation of tau proteins in the brain, which is linked to Alzheimer’s disease (Ferrer et al. 2005). Most studies in neurons concentrate on the role of p38 MAPK in the apoptotic pathway; in this pathway, reactive oxygen species (ROS), including H2O2 trigger phosphorylation of p38 MAPK (Chen et al. 2005; Wang et al. 2003). In fact, H2O2 acts in pancreatic cells as a secondary signaling molecule to activate activator protein-1 and the ERK 1/2, JNK, and p38 MAPK pathways (Kikuta et al. 2006). In the present study, we found that in the absence of ROS, γT3 acts as a pro-oxidant in that it induces a marked increase in the phosphorylation of p38 MAPK, while with αT it is very minimal from Fig. 6a (iv), b (iii). Other studies have also shown that αT was able to reduce p38 activation in malonate-treated cerebellar granule neurons which induced oxidative stress in cells (Gomez-Lazaro et al. 2007). Thus, it appears that in this study αT is more protective toward neuron as it does not activate p38 MAPK under non-stressed condition.
The present study also shows that treating neurons with γT3 or αT under non-stress condition causes early signs of apoptosis as reported by Srivastava and Gupta (2006) and by Kumar et al. (2006) in their work on cancer. Thus, the present study indicates that H2O2 acts not only as a pro-oxidant but also as a signaling molecule in apoptosis activation. Pretreatment of cells with αT or γT3 antagonized the effects of H2O2 on these signaling proteins; however, in the absence of H2O2, γT3 increased the expression of apoptotic proteins of p53 and phosphorylated p38 MAPK while decreased Bcl-2/Bax ratio indicating its strong pro-apoptotic tendency. αT on the other hand showed up-regulation of p53 expression but did not highly activate p38 MAPK and Bcl-2/Bax remained unchanged indicating αT is not as strongly involved in neuronal apoptosis signaling as γT3. In this way, our results suggest that at the concentrations used in this study, γT3 plays a stronger role than αT in modulating initiation of apoptosis in neurons.
Acknowledgments
This study was funded by the Ministry of Science, Technology, and Innovation of Malaysia under Intensified Research Prioritized Area (IRPA) grant 06-02-02-0022/PR0008/09-07. We thank Dr. Coral Sanfeliu from the Institute of Biomedical Research of Barcelona (IIBB) in Spain for her invaluable assistance in setting of the neuronal culture system in our laboratory.
References
- Agarwal MK, Agarwal ML, Athar M, Gupta S (2004) Tocotrienol-rich fraction of palm oil activates p53, modulates Bax/Bcl-2 ratio and induces apoptosis independent of cell cycle association. Cell Cycle 3:1–7 [DOI] [PubMed] [Google Scholar]
- Alvarado C, Alvarez P, Puerto M, Gausserès N, Jiménez L, De la Fuente M (2006) Dietary supplementation with antioxidants improves functions and decreases oxidative stress of leukocytes from prematurely aging mice. Nutrition 22:767–777. doi:10.1016/j.nut.2006.05.007 [DOI] [PubMed] [Google Scholar]
- Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC (1997) Inhibition of Bax channel-forming activity by Bcl-2. Science 277:370–372. doi:10.1126/science.277.5324.370 [DOI] [PubMed] [Google Scholar]
- Azzi A, Stocker A (2000) Vitamin E: non-antioxidant roles. Prog Lipid Res 39:231–255. doi:10.1016/S0163-7827(00)00006-0 [DOI] [PubMed] [Google Scholar]
- Banudevi S, Krishnamoorthy G, Venkataraman P, Vignesh C, Aruldhas MM, Arunakaran J (2006) Role of alpha-tocopherol on antioxidant status in liver, lung and kidney of PCB exposed male albino rats. Food Chem Toxicol 44:2040–2046. doi:10.1016/j.fct.2006.07.017 [DOI] [PubMed] [Google Scholar]
- Busciglio J, Yankner BA (1995) Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature 378:776–779. doi:10.1038/378776a0 [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Sultana R (2007) Redox Proteomics identification of oxidatively modified brain proteins in Alzheimer’s disease and mild cognitive impairment: insights into the progression of this dementing disorder. J Alzheimers Dis 12:61–72 [DOI] [PubMed] [Google Scholar]
- Chen J, Errico ST, Freed WJ (2005) Reactive oxygen species and p38 phosphorylation regulate the protective effects of Δ9-tetrahydrocannabinol in the apoptotic response to NMDA. Neurosci Lett 389:99–103. doi:10.1016/j.neulet.2005.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li FQ, Maeise K (2005) Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol 75:207–246. doi:10.1016/j.pneurobio.2005.02.004 [DOI] [PubMed] [Google Scholar]
- Crouzin N, Ferreira MC, Cohen-Solal C, Aimar RF, Vignes M, Guiramand J (2007) α-Tocopherol-mediated long-lasting protection against oxidative damage involves an attenuation of calcium entry through TRP-like channels in cultured hippocampal neurons. Free Radic Biol Med 42:1326–1337. doi:10.1016/j.freeradbiomed.2007.01.032 [DOI] [PubMed] [Google Scholar]
- Culmsee C, Mattson MP (2005) p53 in neuronal apoptosis. Biochem Biophy Res Comm 331:761–777 [DOI] [PubMed] [Google Scholar]
- Drögen W, Schipper HM (2007) Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell 6:361–370. doi:10.1111/j.1474-9726.2007.00294.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerit J, Edeas M, Bricaire F (2004) Neurodegenerative diseases and oxidative stress. Biomed Pharmacother 58:39–46. doi:10.1016/j.biopha.2003.11.004 [DOI] [PubMed] [Google Scholar]
- Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Wittenman JCM, Breteler MMB (2002) Dietary intake of antioxidant and risk of Alzheimer’s disease. JAMA 287:3223–3229. doi:10.1001/jama.287.24.3223 [DOI] [PubMed] [Google Scholar]
- Ferreira MC, Crouzin N, Barbanel G, Cohen-Solal C, Récasens M, Vignes M, Guiramand J (2005) A transient treatment of hippocampal neurons with α-tocopherol induces a long-lasting protection against oxidative damage via a genomic action. Free Radic Biol Med 39:1009–1020. doi:10.1016/j.freeradbiomed.2005.05.021 [DOI] [PubMed] [Google Scholar]
- Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribé E, Dalfó E, Avila J (2005) Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr Alzheimer Res 21:3–18. doi:10.2174/1567205052772713 [DOI] [PubMed] [Google Scholar]
- Galbussera A, Tremolizzo L, Brighina L, Testa D, Lovati R, Ferrarese C, Cavaletti G, Filippini G (2006) Vitamin E intake and the quality of life in amyotrophic lateral sclerosis patients: a follow-up case series study. Neurol Sci 27:190–193. doi:10.1007/s10072-006-0668-x [DOI] [PubMed] [Google Scholar]
- Gomez-Lazaro M, Galindo MF, Melero-Fernandez de Mera RM, Fernandez-Gómez FJ, Concannon CG, Segura MF, Comella JX, Prehn JHM, Jordan J (2007) Reactive oxygen species and p38 mitogen-activated protein kinase activate Bax to induce mitochondrial cytochrome c release and apoptosis in response to malonate. Mol Pharmacol 71:736–743. doi:10.1124/mol.106.030718 [DOI] [PubMed] [Google Scholar]
- Golbe LI, Farrell TM, Davis PH (1998) Case-control study of early life dietary factors in Parkinson’s disease. Arch Neurol 45:1350–1353 [DOI] [PubMed] [Google Scholar]
- Graf M, Ecker D, Horowski R, Kramer B, Riederer P, Gerlach M, Hager C, Ludolph AC, Becker G, Osterhage J, Jost WH, Schrank B, Stein C, Kostopulos P, Lubik S, Wekwerth K, Dengler R, Troeger M, Wuerz A, Hoge A, Schrader C, Schimke N, Krampfl K, Petri S, Zierz S, Eger K, Neudecker S, Traufeller K, Sievert M, Neundorfer B, Hecht M (2005) High-dose vitamin E therapy in amyotrophic lateral sclerosis as add-on therapy to riluzole: a result of placebo controlled double blind study. J Neural Transm 112(5):649–660. doi:10.1007/s00702-004-0220-1 [DOI] [PubMed] [Google Scholar]
- Khanna S, Roy S, Parinandi NL, Maurer M, Sen CK (2006) Characterization of the potent neuroprotective properties of the natural vitamin E a-tocotrienol. J Neurochem 98:1474–1486. doi:10.1111/j.1471-4159.2006.04000.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuta K, Masamune A, Satoh M, Suzuki N, Satoh K, Shimosegawa T (2006) Hydrogen peroxide activates activator protein-1 and mitogen-activated protein kinases in pancreatic stellate cells. Mol Cell Biochem 291:11–20. doi:10.1007/s11010-006-9189-4 [DOI] [PubMed] [Google Scholar]
- Kolosova NG, Shcheglova TV, Sergeeva SV, Loskutova LV (2006) Long-term antioxidant supplementation attenuates oxidative stress markers and cognitive deficits in senescent-accelerated OXYS rats. Neurobiol Aging 27:1289–1297. doi:10.1016/j.neurobiolaging.2005.07.022 [DOI] [PubMed] [Google Scholar]
- Krantic S, Mechawar N, Reix S, Quirion R (2005) Molecular basis of programmed cell death involved in neurodegeneration. Trends Neurosci 28:670–676 [DOI] [PubMed] [Google Scholar]
- Kumar KS, Raghavan M, Hieber K, Ege C, Mog S, Parra N, Hildabrand A, Singh V, Srinivasan V, Toles R, Karikari P, Petrovics G, Seed T, Srivastava S, Papas A (2006) Prefencial radiation sensitization of prostate cancer in nude mice by nutraceutical antioxidant γ-tocotrienol. Life Sci 78:2099–2104. doi:10.1016/j.lfs.2005.12.005 [DOI] [PubMed] [Google Scholar]
- Mariani E, Polidori MC, Cherubini A, Mecocci P (2005) Oxidative stress in brain aging, neurodegenerative and vascular disease: An overview. J Chromatogr B Analyt Technol Biomed Life Sci 827:65–75. doi:10.1016/j.jchromb.2005.04.023 [DOI] [PubMed] [Google Scholar]
- Mazlan M, Then SM, Gapor MT, Wan Zurinah WN (2006) Comparative effects of α-tocopherol and γ-tocotrienol against hydrogen peroxide induced apoptosis on primary-cultured astrocytes. J Neurol Sci 243:5–12. doi:10.1016/j.jns.2005.10.006 [DOI] [PubMed] [Google Scholar]
- Moreira PI, Siedlak SL, Aliev G, Zhu X, Cash AD, Smith MA, Perry G (2005) Oxidative stress mechanism and potential therapeutics in Alzheimer disease. J Neural Transm 112:921–932. doi:10.1007/s00702-004-0242-8 [DOI] [PubMed] [Google Scholar]
- Morris MC, Beckeet LA, Scherr PA, Herbert LE, Bennett DA, Field TS, Evans DA (1998) Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis Assoc Disord 12:121–126. doi:10.1097/00002093-199809000-00001 [DOI] [PubMed] [Google Scholar]
- Morrison RS, Kinoshita Y, Johnson MD, Guo W, Garden GA (2003) p53-dependent cell death signaling in neurons. Neurochem Res 28:15–27. doi:10.1023/A:1021687810103 [DOI] [PubMed] [Google Scholar]
- Numakawa Y, Numakawa T, Matsumoto T, Yagasaki Y, Kumamaru E, Kunugi H, Taguchi T, Niki E (2006) Vitamin E protected cultured cortical neurons from oxidative stressed-induced cell death through the activation of mitogen-activated protein kinase and phosphatidylinositol 3 kinase. J Neurochem 97:1191–1202. doi:10.1111/j.1471-4159.2006.03827.x [DOI] [PubMed] [Google Scholar]
- Onyango IG (2007) Mitochondrial and oxidative stress in Parkinson’s Disease. Neurochem Res 33:589–597. doi:10.1007/s11064-007-9482-y [DOI] [PubMed] [Google Scholar]
- Osakada F, Hashino A, Kume T, Katsuki H, Kaneko S, Akaike A (2003) Neuroprotective effects of α-tocopherol on oxidative stress in rat straital cultures. Eur J Pharmacol 465:15–22. doi:10.1016/S0014-2999(03)01495-X [DOI] [PubMed] [Google Scholar]
- Osakada F, Hashino A, Kume T, Katsuki H, Kaneko S, Akaike A (2004) α-Tocotrienol provides the most potent neuroprotection among vitamin E analogs on cultured striatal neurons. Neuropharmacol 47:904–915. doi:10.1016/j.neuropharm.2004.06.029 [DOI] [PubMed] [Google Scholar]
- Peter ME, Krammer PH (2003) The CD95 (APO-1/Fas) DISC and beyond. Cell Death Differ 10:26–35. doi:10.1038/sj.cdd.4401186 [DOI] [PubMed] [Google Scholar]
- Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S (2005) Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 352:2379–2388. doi:10.1056/NEJMoa050151 [DOI] [PubMed] [Google Scholar]
- Roy S, Lado BH, Khanna S, Sen CK (2002) Vitamin E sensitive genes in the developing rat fetal brain: a high-density oligonucleotide microarray analysis. FEBS Lett 530:17–23. doi:10.1016/S0014-5793(02)03309-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ (1997) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med 336(17):1216–1222. doi:10.1056/NEJM199704243361704 [DOI] [PubMed] [Google Scholar]
- Sastry PS, Rao KK (2000) Apoptosis and the Nervous System. J Neurochem 74:1–20. doi:10.1046/j.1471-4159.2000.0740001.x [DOI] [PubMed] [Google Scholar]
- Sen CK, Khanna S, Roy S, Packer L (2000) Molecular basis of vitamin E: tocotrienol potently inhibits glutamate-induced pp60c-src kinase activation and death of HT4 neuronal cells. J Biol Chem 275:13049–13055. doi:10.1074/jbc.275.17.13049 [DOI] [PubMed] [Google Scholar]
- Shabnam MS, Srinivasam R, Wali A, Majumdar S, Joshi K, Behera D (2004) Expression of p53 protein and the apoptotic regulatory molecules Bcl-2, Bcl-XL, and Bax in locally advanced squamous cell carcinoma of the lung. Lung Cancer 45:181–188. doi:10.1016/j.lungcan.2004.01.021 [DOI] [PubMed] [Google Scholar]
- Srinivas G, Kusumakumary P, Nair MK, Panicker KR, Pillai MR (2000) Mutant p53 protein, Bcl/Bax ratios and apoptosis in paediatric acute lymphoblastic leukaemia. J Cancer Res Clin Oncol 126:62–67. doi:10.1007/s004320050010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava JK, Gupta S (2006) Tocotrienol-rich fraction of palm oil induces cell cycle arrest and apoptosis selectively in human prostate cancer cells. Biochem Biophys Res Commun 346:447–453. doi:10.1016/j.bbrc.2006.05.147 [DOI] [PubMed] [Google Scholar]
- Sylvester PW (2007) Vitamin E and apoptosis. Vitam Horm 76:329–356. doi:10.1016/S0083-6729(07)76012-0 [DOI] [PubMed] [Google Scholar]
- Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267:1456–1462. doi:10.1126/science.7878464 [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB (1997) Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 91:627–637. doi:10.1016/S0092-8674(00)80450-X [DOI] [PubMed] [Google Scholar]
- Vervaart P, Knight KR (1996) Oxidative stress and the cell. Clin Biochem Rev 17:3–16 [Google Scholar]
- Wali VB, Sylvester PW (2007) Synergistic antiproliferative effects of gamma-tocotrienol and statin treatment on mammary tumor cells. Lipids 42:1113–1123. doi:10.1007/s11745-007-3102-0 [DOI] [PubMed] [Google Scholar]
- Wang JY, Shum AY, Ho YJ (2003) Oxidative neurotoxicity in rat cerebral cortex neurons: synergistic effects of H2O2 and NO on apoptosis involving activation of p38 mitogen-activated protein kinase and caspase-3. J Neurosci Res 72:508–519. doi:10.1002/jnr.10597 [DOI] [PubMed] [Google Scholar]
- Wang X, Quinn PJ (2000) The location and function of vitamin E in membranes. Mol Membr Biol 17:143–156. doi:10.1080/09687680010000311 [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Niki E, Noguchi N (2003) Comparative study on the action of tocopherols and tocotrienols as antioxidants: chemical and physical effects. Chem Phys Lipids 123:63–75. doi:10.1016/S0009-3084(02)00164-0 [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Saito Y, Jones LS, Shigeri Y (2007) Chemical preactivities and physical effects in comparison between tocopherols and tocotrienols: physiological significance and prospects as antioxidants. J Biosci Bioeng 104:439–445. doi:10.1263/jbb.104.439 [DOI] [PubMed] [Google Scholar]
- Zhang B, Tanaka J, Yang L, Yang L, Sakanaka M, Hata R, Maeda N, Mitsuda N (2004) Protective effect of vitamin E against focal brain ischemia and neuronal death through induction of target genes of hypoxia-inducible factor-1. Neuroscience 126:433–440. doi:10.1016/j.neuroscience.2004.03.057 [DOI] [PubMed] [Google Scholar]
- Zingg JM (2007) Modulation of signal transduction by vitamin E. Mol Aspects Med 28:481–506. doi:10.1016/j.mam.2006.12.009 [DOI] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, Wang X (1999) An Apaf-1 cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274:11549–11556. doi:10.1074/jbc.274.17.11549 [DOI] [PubMed] [Google Scholar]