

Abstract
Background and purpose:
The critical role of blood supply in the growth of solid tumours makes blood vessels an ideal target for anti-tumour drug discovery. The anti-angiogenic and vascular disrupting activities of C9, a newly synthesized microtubule-depolymerizing agent, were investigated with several in vitro and in vivo models. Possible mechanisms involved in its activity were also assessed.
Experimental approach:
Microtubule-depolymerizing actions were assessed by surface plasmon resonance binding, competitive inhibition and cytoskeleton immunofluorescence. Anti-angiogenic and vascular disrupting activities were tested on proliferation, migration, tube formation with human umbilical vein endothelial cells, and in rat aortic ring, chick chorioallantoic membrane and Matrigel plug assays. Western blots and Rho activation assays were employed to examine the role of Raf-MEK-ERK (mitogen-activated ERK kinase, extracellular signal-regulated kinase) and Rho/Rho kinase signalling.
Key results:
C9 inhibited proliferation, migration and tube formation of endothelial cells and inhibited angiogenesis in aortic ring and chick chorioallantoic membrane assays. C9 induced disassembly of microtubules in endothelial cells and down-regulated Raf-MEK-ERK signalling activated by pro-angiogenic factors. In addition, C9 disrupted capillary-like networks and newly formed vessels in vitro and rapidly decreased perfusion of neovasculature in vivo. Endothelial cell contraction and membrane blebbing induced by C9 in neovasculature was dependent on the Rho/Rho kinase pathway.
Conclusions and implications:
Anti-angiogenic and vascular disruption by C9 was associated with changes in morphology and function of endothelial cells, involving the Raf-MEK-ERK and Rho/Rho kinase signalling pathways. These findings strongly suggest that C9 is a new microtubule-binding agent that could effectively target tumour vasculature.
Keywords: C9, anti-angiogenesis, vascular disrupting effect, microtubule, Raf-MEK-ERK signalling pathway
Introduction
Functional blood vessels provide oxygen and nutrients to solid tumours and serve as the route for metastatic spread, making tumour vasculature an attractive target for anti-tumour therapy (Ferrara and Kerbel, 2005). Two main strategies have been used to target tumour vasculature: anti-angiogenic and vascular disrupting therapies(Denekamp, 1993; Folkman, 1995).
Anti-angiogenic therapy has been thought to be an important component for cancer treatment since Judah Folkman first addressed the concept in 1971 (Folkman, 1971). Bevacizumab (Avastin), a monoclonal antibody against vascular endothelial growth factor (VEGF) was the first angiogenesis inhibitor approved by Food and Drug Administration. Nine other angiogenesis inhibitors have subsequently been approved or used in different countries (Folkman, 2007). Although these anti-angiogenic drugs are active in cancer treatment, as single agents or in combination with other drugs, they have minimal effects on existing tumour blood vessels within well-established tumours and have had limited effects on large tumours (Bergers et al., 1999; Bergers et al., 2003).
Vascular disrupting therapy using vascular disrupting agents (VDAs) has become attractive as it causes a rapid and selective shutdown of the established tumour vasculature by disrupting the tumour endothelium (Thorpe, 2004). Differences in the physiology of immature tumour vasculature versus mature normal vasculature provide an opportunity for the selective disruption of tumour blood flow (Neri and Bicknell, 2005). One of the potential advantages of VDAs is that they can exert greater actions on large experimental tumours than small ones (Landuyt et al., 2000; Siemann and Rojiani, 2005). In addition, VDAs produce central necrosis in experimental tumours, but have no effect on the thin rim of the tumour periphery, where angiogenesis occurs most vigorously.
As such, the development of new compounds with both anti-angiogenic and vascular targeting properties has attracted greater attention (Belleri et al., 2005; Kruczynski et al., 2006). A newly synthesized compound, C9 (Figure 1), was identified during our search for a potential candidate agent with both activities. Preliminary studies with C9 suggested it inhibited microtubule polymerization in vitro (Li et al., 2005). The current study aims to investigate the potential anti-angiogenic activity of C9 and to further characterize its anti-vascular properties and mechanisms of action in vitro and in vivo.
Figure 1.
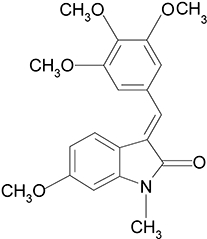
Chemical structure of C9.
Methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) were isolated as previously described (Jaffe et al., 1973), and presence of von Willebrand factor was confirmed by immunofluorescence staining. HUVECs were cultured (37°C, 5% CO2) in M199 medium supplemented with 20% heat-inactivated fetal bovine serum (FBS) containing 30 µg·mL−1 endothelial cell growth supplement, 10 ng·mL−1 epidermal growth factor, 100 units·mL−1 penicillin and 100 µg·mL−1 streptomycin. Cells at passages 3–7 were used for experiments.
Cell proliferation assay
Human umbilical vein endothelial cells (1.0 × 105 cells·mL−1) were seeded onto 96-well plates, attached overnight and subsequently exposed to different concentrations of C9 (0.03, 0.06, 0.13, 0.25, 0.5, 1.0 and 2.0 µmol·L−1) for 12, 24, 36 or 48 h. Cell proliferation was determined by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazalium bromide] assay (Alley et al., 1988). The inhibition rate of cell proliferation was calculated as: growth inhibition (%) = (ODcontrol− ODtreated)/ODcontrol× 100%.
Cell cycle assay
Cells (5.0 × 105) were fixed in 70% ice-cold ethanol, centrifuged, washed with phosphate-buffered solution (PBS), centrifuged, resuspended in 1 mL DNA-staining solution (20 µg·mL−1 RNase, PBS and 20 µg·mL−1 propidium iodide) and incubated (30 min, room temperature, in the dark). Cell samples (at least 1.0 × 105) were then examined by a FACS Calibur analyser. Cell cycle phase analysis was performed by utilizing Modifit software (BD, San Jose, CA, USA).
Cytoskeleton immunofluorescence
Human umbilical vein endothelial cells were seeded on fibronectin-coated chamber slides (10 µg·mL−1) before exposure to C9 (0.5 µmol·L−1, 1 h). Tubulin was visualized with FITC-anti-tubulin monoclonal antibody (Sigma) and F-actin with Alexa Fluor 633 Phalloidin (Molecular Probes, OR, USA). Slides were mounted by using VECTASHIELD (Vector laboratories, CA, USA). Fluorescence images were obtained by using a Lecia TCS confocal microscope (Leica, Deerfield, IL, USA).
Surface plasmon resonance (SPR)-binding assay
Tubulin-binding assays were performed by using an SPR technology-based Biacore 3000 instrument (Biacore AB, Uppsala, Sweden) as described by Li et al., (2007). Briefly, tubulin was covalently immobilized on a CM5 sensor chip (BIAcore), and indicated concentrations of the compound were dissolved in the running buffer. The equilibrium dissociation constant (KD) was derived by fitting the data by using the 1:1 Langmuir binding model based on BIAevaluation 3.1 software.
Competitive inhibition assay
Colchicine binding to tubulin
The tubulin-colchicine complex was formed by incubating 3 µmol·L−1 tubulin with colchicine (30 min, 37°C, excitation 365 nm, emission 435 nm). C9 (0–50 µmol·L−1) or vinblastine (0–50 µmol·L−1) was then added to the preformed tubulin-colchicine complex (60 min, 37°C), and fluorescence was measured by using a Hitachi F-2500 spectrofluorimeter (n= 3).
Vinblastine binding to tubulin
Binding of the fluorescent conjugate of vinblastine (BODIPY FL-vinblastine; Molecular probes Inc., Eugene, OR, USA) to tubulin will increase fluorescence at 490 nm excitation wavelength (Gupta et al., 2004). Tubulin (3 µmol·L−1) was incubated with various concentrations of C9 (45 min, 37°C) or vinblastine, then incubated with BODIPY FL-vinblastine (2 µmol·L−1, 20 min, 37°C). Data were analysed by a Hitachi F-4500 fluorimeter.
Cell migration assay
Human umbilical vein endothelial cell migration was determined in a transwell Boyden chamber (Costar, MA, USA) (Osusky et al., 2004). Briefly, cell suspensions (5 × 105 cells·mL−1) with different concentrations of C9 (0.25, 0.5, 1.0 and 2.0 µmol·L−1) or control was added to the upper compartment of the chamber. The lower compartment contained 20% FBS M199 medium and the same concentrations of C9 or control. After incubation (8 h, 37°C), the inhibition of migration was calculated thus: inhibition of migration =[1 − (Acompound− Ablank)/(Acontrol− Ablank)]× 100%.
Tube formation assay
Matrigel (55 µL·well−1) was used to coat 96-well plates, allowed to solidify (37°C, 1 h) (Soeda et al., 2000), prior to seeding with HUVECs (1 × 105 cells·mL−1) that were then cultured (37°C, 6 h) in M199 medium with 20% FBS containing C9 (0.25, 0.5, 1.0 and 2.0 µmol·L−1) or control.
For investigation of neovessel disruption, HUVECs were seeded on Matrigel (9.3 mg·mL−1) and left to align for 24 h (Kruczynski et al., 2006). The formed capillary-like structures or cords were exposed to C9 (0.5 µmol·L−1) then examined with an inverted phase contrast microscope (DP70, Olympus, Japan). The number of the tubes was quantified from five random fields (Ashton et al., 1999). The inhibition of tube formation was calculated thus: inhibition of tube formation =[1 − (tubesC9/tubescontrol)]× 100%.
Aortic ring assay
All animal procedures and experiments complied with the ethical Guidelines for the care and use of animals and were approved by the Institute animal reviews boards of Shanghai Institute of Materia Medica, Chinese Academy of Sciences. Aortic rings obtained from 6-week-old Sprague–Dawley rats (Nicosia and Ottinetti, 1990; Belleri et al., 2005) were embedded with 30 µL Matrigel, incubated with M199 medium, with or without 10% FBS, and various concentrations of C9 (0.5, 1.0 and 2.0 µmol·L−1), then photographed on day 7 with an inverted phase contrast microscope (DP70, Olympus, Japan). The degree of endothelial cell sprouting from aorta samples was analysed with computer image analysis (Image pro-plus 4.5; Media Cybernetics, LP), with inhibition rate calculated as: inhibition rate of sprouting areas of the aortic ring =[1 − (sprouting areasC9/sprouting areascontrol)]× 100%.
Rings cultured for 7 days were used in disruption assays of neo-formed vessels (Belleri et al., 2005). Neovessels were photographed at different time points after treatment with C9 (0.5 µmol·L−1, 24 h).
Chick chorioallantoic membrane (CAM) assay
Fertilized chicken eggs were incubated (7 days) in a humidified egg incubator Roll-X base (Lyon Electric Company, CA, USA) (Ribatti et al., 2000). The chorioallantoic membrane (CAM) was treated with various concentrations of C9 or 0.01% dimethyl sulphoxide (v/v) continuously for 48 h, then evaluated and recorded by stereomicroscopic photography (MS5, Leica, Switzerland). Angiogenesis was quantified by counting the number of blood vessel branch points in each photograph under the coverslips; at least 10 viable embryos were tested for each treatment. The CAM inhibition rate was measured as: inhibition of CAM =[1 − (branchesC9/branchescontrol)]× 100%.
In vivo Matrigel plug assay
A Matrigel plug assay was employed as previously described (Micheletti et al., 2003), with some modifications. Briefly, basic fibroblast growth factor (bFGF; 100 ng per pellet) was embedded in a Matrigel pellet (0.5 mL) and s.c. injected into C57BL/6N mice. On day 7, when functional vessels were formed and evident in the plug, C9 (200 mg·kg−1, i.p.) was injected into mice for 1 or 3 h before necropsy. Mice were injected i.v. with FITC-conjugated dextran (100 µL, Sigma, St. Louis, MO, USA) 1 h before necropsy. The Matrigel was removed and FITC-positive vessels were analysed by fluorescence microscopy (BX51, Olympus, Japan) at 488 nm.
Western blot analysis
Human umbilical vein endothelial cells (5 × 106 cells·mL−1) were treated, and cell lysates were prepared as previously described (Tan et al., 2004). Equal amounts of protein were separated on SDS-PAGE gels and transferred to nitrocellulose membranes, incubated (overnight, 4°C) with primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies (1:2000). Detection was performed by using ECL Plus Western Blotting Detection System.
Rho activation assay
Rho activation was analysed by using Rho assay kits (Upstate Biotechnology). Briefly, HUVECs grown in 100 mm diameter dishes were serum-starved (2 h) followed by pretreatment with vehicle, Y27632 (10 µmol·L−1) or taxol (0.2 µmol·L−1) for 30 min, then exposed to C9 (0.5 µmol·L−1, 1 h). Cells were then rinsed with ice-cold PBS buffer and lysed (lysis buffer, 500 µL per plate), following the manufacturer's protocol. Cell lysates were clarified by centrifugation (14 000×g, 4°C, 5 min), supernatants incubated with glutathione S-transferase (GST)-Rhotekin-Rho-binding domain (RBD) (GST-RBD) beads (4°C, 45 min), and washed three times with the wash buffer. Bound Rho proteins were detected by Western blotting using a monoclonal antibody against Rho. RBD-bound Rho was normalized to the total amount of Rho in cell lysates for comparison of Rho activity in different samples (Ren et al., 1999).
Statistical analysis
All results are expressed as mean ± SD. Statistical significance was assessed by two-tailed Student's t-test.
Materials
C9, (2H-Indol-2-one, 1, 3-dihydro-6-methoxy-1-methyl-3-[(3, 4, 5-trimethoxyphenyl) methylene]-, (3E)-(9CI) (Figure 1), was prepared with commercially available 6-methoxy-2-indolinone as previously described (Li et al., 2005; Pandit et al., 2006). The stock solution (10−2 mol·L−1) was made in dimethyl sulphoxide. Type I collagenases, endothelial cell growth supplement, epidermal growth factor, MTT, DAPI, propidium iodide, tubulin, taxol, Y27632, colchicine and antibodies to von Willebrand factor were purchased from Sigma Aldrich (St. Louis, MO, USA); M199 medium and FBS from Gibco (Grand Island, NY, USA); VEGF and bFGF from R&D systems (Minneapolis, MN, USA); Matrigel from BD Biosciences (San Jose, CA, USA); antibodies to phosphorylated form of the VEGF receptor (KDR), FGF receptor (FGFR)1, c-Raf, extracellular signal-regulated kinase (ERK), mitogen-activated ERK kinase (MEK), Akt and myosin light chain2 (MLC2) from Cell Signaling Technology (Danvers, MA, USA); antibody to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) from KangChen Bio-tech (Shanghai); secondary antibodies from Calbiochem (San Diego, CA, USA), and ECL Plus Western Blotting Detection System was purchased from GE Healthcare (UK).
Results
C9 as a tubulin-depolymerizing agent
C9 disrupts the cytoskeleton and changes the morphology of endothelial cells
C9 disrupted tubulin polymerization with an IC50 of approximately 13 µmol·L−1 (Pandit et al., 2006). The effects of C9 on microtubule and F-actin cytoskeleton organization were examined by immunofluorescence. As shown in Figure 2A, C9 led to a dramatic disruption of the HUVEC cytoskeleton, producing a diffuse microtubule network and an increase in actin stress fibres and membrane blebbing. At the same time, cell morphology was significantly changed, with rounded and retracted shape following exposure to C9 (see later).
Figure 2.
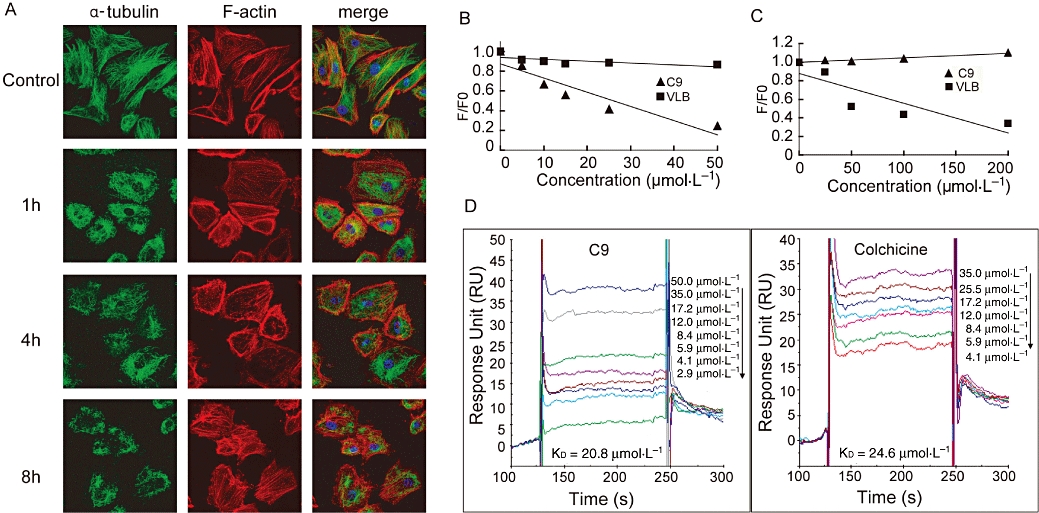
C9 is a microtubule-binding agent. (A) Effects of C9 on the endothelial cell cytoskeleton. Human umbilical vein endothelial cells were seeded onto coverslips coated with fibronectin and incubated overnight prior to treatment (1, 4 and 8 h, with or without 0.5 µmol·L−1 C9). Cells were then fixed and stained for microtubules (green) and F-actin (red); fluorescence images were viewed by using a Leica TCS confocal microscope (43×). (B) Effects of C9 on the colchicine-binding site. Tubulin (3 µmol·L−1) was incubated with colchicine to form tubulin-colchicine binding complexes prior to treatment (37°C, 60 min) with C9 (5, 10, 15, 25 and 50 µmol·L−1) or vinblastine (VLB; 5, 10, 15, 25 and 50 µmol·L−1). (C) Effects of C9 on the Vinca alkaloid site. Tubulin (3 µmol·L−1) was incubated (37°C, 45 min) with various concentrations of C9 (25, 50, 100 and 200 µmol·L−1) or vinblastine (VLB; 25, 50, 100 and 200 µmol·L−1) prior to treatment with BODIPY FL-vinblastine (20 min, 37°C). (D) C9 or colchicine tubulin-binding affinity by surface plasmon resonance assay. C9 (2.9, 4.1, 5.9, 8.4, 12.0, 17.2, 35.0 and 50.0 µmol·L−1) or colchicine (4.1, 5.9, 8.4, 12.0, 17.2, 24.5 and 35.0 µmol·L−1) were prepared as an analyte and then injected into a Biacore 3000 system following tubulin immobilization on the chip.
C9 induces G2/M phase arrest
Given the correlation between G2/M phase arrest and cytoskeletal dynamics, we next investigated the effect of C9 on the cell cycle. Our data showed that treatment with C9 (24 h) induced a dose-dependent reduction in the proportion of cells in G0/G1, with a concomitant accumulation of cells arrested in G2/M (Table 1).
Table 1.
C9 blocks the cell cycle in human umbilical vein endothelial cells at the G2/M phase
| C9 (µmol·L−1) | |||||
|---|---|---|---|---|---|
| 0 | 0.25 | 0.5 | 1.0 | 2.0 | |
| G1 phase (%) | 67.4 ± 6.1 | 66.3 ± 4.4 | 54.6 ± 5.2 | 51.1 ± 1.5 | 48.3 ± 11.4 |
| S phase (%) | 17.1 ± 1.5 | 11.3 ± 9.4 | 10.6 ± 9.2 | 11.7 ± 10.3 | *4.8 ± 6.9 |
| G2 phase (%) | 15.5 ± 6.2 | 22.4 ± 5.1 | 34.8 ± 12.3 | *37.2 ± 9.5 | *46.9 ± 9.1 |
Data shown in the table are percentage of cells in the phases of the cell cycle indicated, as measured by FACS (see Methods). Incubation with increasing concentrations of C9 decreased the proportion of cells in the S phase and increased that in the G2 phase.
Significantly different from corresponding value in control cultures (C9 at 0 µmol·L−1); Student's t-test. At least 105 cells were assessed from three separate cultures.
C9 binds to the colchicine-binding site of tubulin
Most microtubule-targeting agents interact with either the Vinca alkaloid (Downing, 2000) or the colchicine site (Ravelli et al., 2004) of tubulin. We examined whether C9 exerted its activity through binding to either of these two sites. Colchicine is weakly fluorescent in aqueous solution but becomes strongly fluorescent upon binding to tubulin (Hastie, 1991). As expected, no decrease in fluorescence was noted following vinblastine, which is known to bind to tubulin at the Vinca alkaloid site. Conversely, 5–50 µmol·L−1 C9 competed with colchicine to bind to tubulin, releasing colchicine from the complexes with consequent decrease of fluorescence (75.6% at 50 µmol·L−1), suggesting that C9 competed for the same tubulin-binding site as colchicine (Figure 2B).
We next investigated whether C9 inhibited BODIPY FL-vinblastine binding to tubulin using a BODIPY FL-vinblastine competition-binding assay. While we observed no inhibitory effect of C9, the positive control vinblastine (50 µmol·L−1) decreased the fluorescence by 47.8%. These results suggested that C9 binds with tubulin through the colchicine site and not the vinblastine-binding site (Figure 2C).
C9 exhibits high binding affinity for tubulin
We subsequently investigated the binding affinity of C9 for tubulin. SPR sensor kinetic analysis data demonstrated that the C9 equilibrium dissociation constant KD was 20.8 µmol·L−1, compared with 24.6 µmol·L−1 for colchicine, suggesting C9 and colchicine have similar binding affinities for tubulin (Figure 2D).
Anti-angiogenic activity of C9
C9 inhibits endothelial cell proliferation
Angiogenesis is a complex process that includes the proliferation, migration and tube formation of endothelial cells. We first tested the inhibitory effect of C9 on endothelial cell proliferation by MTT assay. As shown in Figure 3A, C9 treatment (48 h) had little effect on cell growth at 0.03, 0.06, 0.1, 0.2 and 0.5 µmol·L−1 and exhibited slight growth inhibition at 1.0 and 2.0 µmol·L−1 (11.8% and 34.9% respectively). Therefore, concentrations below 2 µmol·L−1 C9 were used to assess the anti-angiogenic effects in HUVECs.
Figure 3.
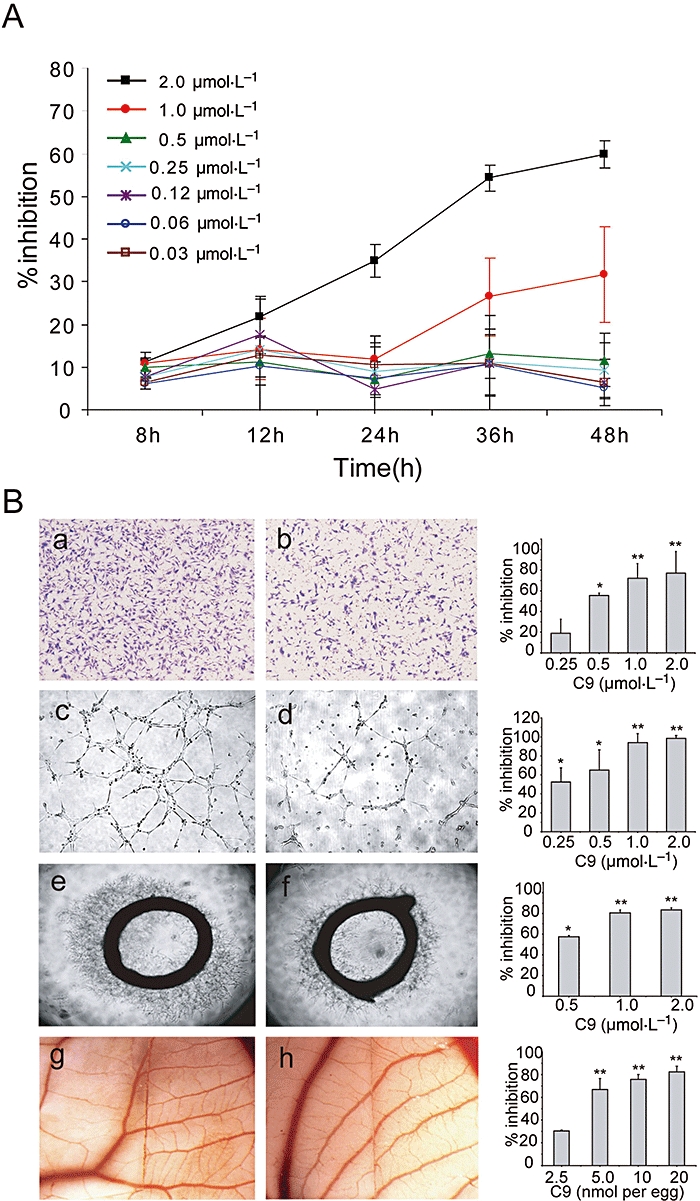
C9 inhibited angiogenesis. (A) C9 inhibited endothelial cell proliferation. Human umbilical vein endothelial cells were incubated overnight then exposed to different concentrations of C9 (0.03, 0.06, 0.12, 0.25, 0.5, 1.0 and 2.0 µmol·L−1) and cultured for 8, 12, 24, 36 and 48 h. Inhibition of proliferation was measured by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazalium bromide] assay. (B) Inhibitory effect of C9 on cell migration, tube formation, aortic ring and chick chorioallantoic membrane assays. Representative images and summary data (bar graphs on right hand side) showing inhibition of migration (a and b), tube formation (c and d), angiogenesis in aortic ring (e and f) and chick chorioallantoic membrane assays (g and h). Cells were treated with vehicle (a, c, e and g) or 0.5 µmol·L−1 C9 (b, d and f) or 5 nmol per egg C9 (h) (see Methods); magnification: 100× (a, b, c and d), 40× (e and f), 1.6× (g and h). Results are expressed as mean ± SD; n= 3; *P < 0.01 versus control, **P < 0.005 versus control.
C9 blocks migration of endothelial cells
Endothelial cell migration, which occurs through chemotaxis, is necessary for angiogenesis. As shown in Figure 3B, treatment with 0.25–2.0 µmol·L−1 C9 (8 h, a and b) significantly reduced the number of migrating cells in a concentration-dependent manner, compared with controls. When endothelial cells were exposed to 0.5 µmol·L−1 C9 for 8 h, only a marginal proliferation inhibitory effect was observed (9.9% ± 2.4), but the migration of endothelial cells was strongly inhibited, suggesting that inhibition of migration by C9 did not result from its cytotoxity.
C9 impairs capillary tube formation
We further evaluated the effects of C9 on the formation of functional tubes utilizing HUVECs plated on a Matrigel substratum. In the control group, stimulation with 20% FBS resulted in the rapid alignment of HUVECs and formation of tube-like structures, within 6 h (Figure 3B, c). C9 suppressed FBS-stimulated tube formation in a concentration-dependent manner, suggesting C9 interfered with the ability of HUVECs to form capillary-like tubes in vitro, one of the important traits of endothelial cells.
C9 decreased vessel sprouting from aortic rings and reduced newly formed blood vessels embedded in Matrigel
The effects of C9 on microvessel sprouting from vascular tissues were tested by using an ex vivo rat aortic ring assay, which mimics several processes in angiogenesis, including endothelial cell proliferation, migration and tube formation. Microvessels were observed in the control group on days 2–3, with the number and length of microvessels increasing with prolonged culture time (Figure 3B, e and f). Treatment with C9 resulted in a decrease of capillary sprouting in a dose-dependent manner, with a microvessel growth inhibition of almost 60% noted after exposure to 0.5 µmol·L−1 C9 for 7 days.
C9 reduces neovascularization in CAM assay
CAM assays were used to examine the effects of C9 in vivo as they provide a unique model to investigate the process of new blood vessel formation and effects of anti-angiogenic agents. Compared with the control group, neovascularization of CAM treated with C9 (5 nmol per egg) was dramatically decreased, an inhibitory effect that was also concentration-dependent (Figure 3B, g and h). These results suggested that C9 inhibited angiogenesis in vitro, ex vivo and in vivo, in a concentration-dependent manner.
C9 anti-angiogenic activity is mediated through down-regulation of Raf-MEK-ERK signalling molecules
We further examined the mechanisms of anti-angiogenic activity of C9 treatment. VEGF and bFGF stimulate the proliferation, migration and differentiation of endothelial cells via binding to their receptors and receptor activation. We evaluated the effects of C9 on endothelial cell-specific receptors for VEGF (KDR) or bFGF (FGFR1) and their downstream signalling pathways. HUVECs were exposed to various concentrations of C9 for 2 h after 24 h serum starvation and subsequently stimulated (10 min) with VEGF (50 ng·mL−1) or bFGF (25 ng·mL−1). The phosphorylation of KDR and FGFR1 and overall KDR and FGFR1 expression levels in cells (Figure 4A,B) were not affected by pretreatment with C9 (1 µmol·L−1 and 2 µmol·L−1). However, phosphorylation of downstream molecules Raf, MEK and ERK1/2 was down-regulated in a concentration-dependent manner (0.25, 0.5, 1.0 and 2.0 µmol·L−1). Interestingly, effects on Akt phosphorylation were only noted at high concentrations (2 µmol·L−1). In all cases, total steady state protein levels remained unchanged. These results suggested that reduction by C9 of phosphorylation in the Raf-MEK-ERK signalling pathway, contributed to the anti-angiogenic effects of this compound.
Figure 4.
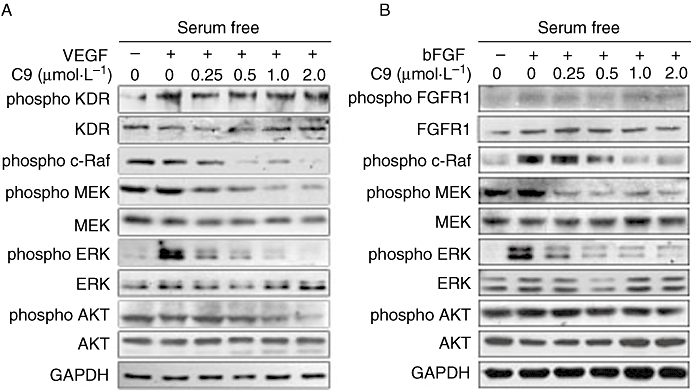
C9 down-regulated the Raf-MEK-ERK signalling pathway induced by VEGF and bFGF. Serum-starved cells were treated with different concentrations of C9 for 2 h, followed by the addition of 50 ng·mL−1 VEGF or 25 ng·mL−1 bFGF (10 min), then assayed by Western blotting (n= 3). bFGF, basic fibroblast growth factor; ERK, extracellular signal-regulated kinase; FGFR1, FGF receptor 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; KDR, receptor for vascular endothelial growth factor; MEK, mitogen-activated ERK kinase; VEGF, vascular endothelial growth factor.
Vascular disrupting effect of C9
We then tested the vascular targeting effects of C9 utilizing in vitro, ex vivo and in vivo models. HUVECs seeded on Matrigel will form a network, which is typically completed within 24 h (Figure 5A). The addition of C9 (3 h) to these newly formed capillary-like structures disrupted the network, after 3 h and this effect increased over the following 9 h (Figure 5A).
Figure 5.
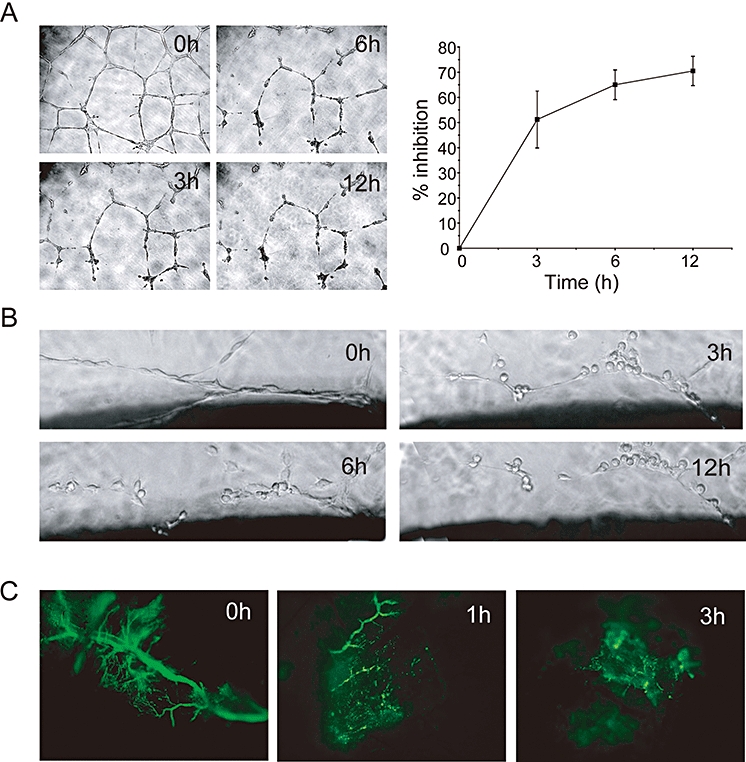
Vascular targeting activity of C9. (A) Neovasculature network disruption assay: cells were plated on Matrigel and allowed to form capillary-like networks for 24 h. These networks were then exposed to C9 (0.5 µmol·L−1, 12 h). C9 treatment resulted in a time-dependent inhibition. Magnification, 100×. (B) Aortic ring assay; aortic rings were cultured for 7 days and then treated with C9 (0.5 µmol·L−1) for 12 h. Magnification, 200×. (C) In vivo Matrigel plug assay. Fluorescence microscopy image of FITC-dextran perfusing the neovasculature before (0 h) and 1 and 3 h following C9 treatment (200 mg·kg−1, i.p.), as described in Methods. The clearly visible blood vessels at 0 h were almost totally disrupted 3 h after i.p. injection of C9. Magnification, 200×.
In the aortic ring assay, vessels grow continuously and become bushy by day 7. Time course experiments showed that formed vessels were cleaved following treatment with C9 (0.5 µmol·L−1) in a time-dependent manner (Figure 5B).
We further evaluated the vascular disruptive effects of C9 in vivo using Matrigel plug assays, in which a pellet of Martigel containing bFGF was implanted s.c. After 7 days of implantation, fluorescent dextran was injected i.v. to assess the function of the newly formed vasculature. As shown in Figure 5C, in the control group, functional blood vessels were perfused by FITC-dextran. However, C9 treatment (1 h) resulted in a rapid reduction of vessels and the perfusion of FITC-dextran almost disappeared after 3 h of treatment. Taken together, these data indicated that C9 showed not only anti-angiogenic, but also vascular-disrupting activities in vitro and in vivo.
Morphological changes in HUVECs induced by C9
Cell morphology is an important indicator of endothelial cell behaviour (Schmidt et al., 2006). We found that cells became rounded and retracted, accompanied by membrane blebbing following treatment of C9 (Figure 6A). Taxol, a microtubule-polymerizing agent, can reverse the effect of microtubule-depolymerizing agents (Taraboletti et al., 2005). Pretreatment with taxol resulted in the rounded or retracted cells becoming extended. In addition, taxol pretreatment attenuated the disruptive effects of C9 on existing HUVEC networks in Matrigel, with inhibition decreasing markedly (Figure 6B).
Figure 6.
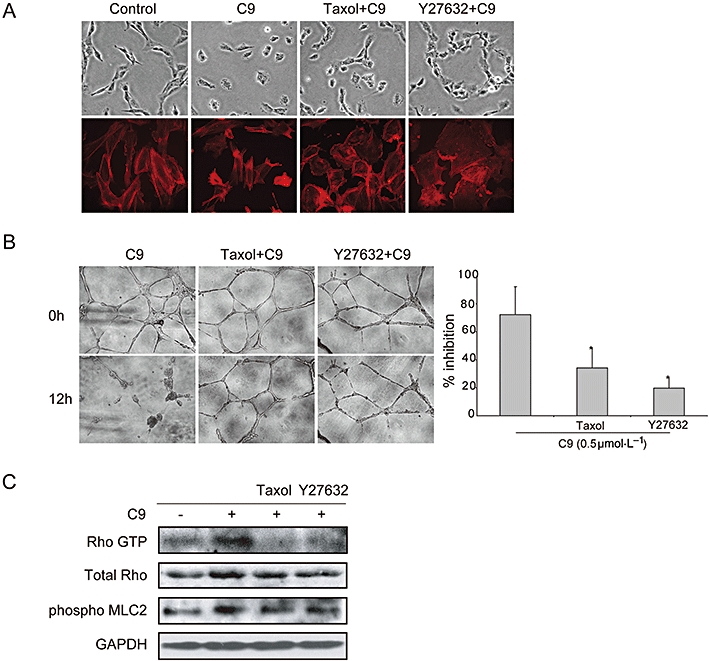
Microtubule disassembly contributed to the vascular disrupting activity of C9. (A) The change in endothelial cell morphology induced by C9 was attenuated by taxol and Y27632. Endothelial cells were pretreated with vehicle control (30 min), taxol (0.2 µmol·L−1, 30 min) or Y27632 (10 µmol·L−1, 30 min), then incubated with C9 (0.5 µmol·L−1, 1 h). Cells were examined with an inverted microscope (40×) or stained for F-actin (red); fluorescence images were obtained by a fluorescence microscope (43×). (B) Effects of C9 on newly formed vessels were antagonized by taxol and Y27632. HUVECs were seeded onto Matrigel to form a functional network, then pretreated with or without taxol (0.2 µmol·L−1, 30 min) or Y27632 (10 µmol·L−1, 30 min) and then exposed to C9 (0.5 µmol·L−1, 12 h); magnification, 100×. (C) C9 activated Rho GTPase and increased the phosphorylation of MLC2, which was antagonized by taxol and Y27632. Serum-starved HUVECs were treated with control or taxol (0.2 µmol·L−1, 30 min) or Y27632 (10 µmol·L−1, 30 min), prior to analysis of Rho activity. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HUVECs, human umbilical vein endothelial cells; MLC2, myosin light chain2.
The Rho/Rho kinase signalling pathway is a critical regulator of the cytoskeleton and of cell behaviour. We therefore examined the effect of the Rho kinase inhibitor Y27632 on C9-driven events. We found that Y27632 attenuated cell morphology changes induced by C9. Similarly, pretreatment with Y27632 protected HUVEC networks from disruption by C9, with a striking reduction in inhibition (Figure 6B). C9 was further noted to activate Rho GTPase and induce phosphorylation of MLC2; both of these effects were reversed by pretreatment with Y27632 (Figure 6C).
Discussion
Anticancer therapies focused on tumour vasculature are classified into anti-angiogenic and vascular disrupting therapies, with many successful compounds having been employed clinically. Microtubules, one of the major dynamic and structural cellular components, are essential for cell function and are, consequently, an attractive target not only for cancer chemotherapy, but also as agents targeting tumour vasculature (Jordan and Wilson, 1998). Microtubule-destabilizing agents, such as combretastatin and vinblastine, are also described as VDAs, with observed anti-angiogenic activity, even at low concentrations (Vincent et al., 2005). Consistent with this notion, the vascular targeting and anti-angiogenic activity of C9 were clearly observed in vitro, but this compound showed limited inhibition of endothelial cell proliferation, at non-toxic concentrations. All these suggested that the effect of C9 on the already formed vasculature and angiogenesis was related to its effects on endothelial cell function, rather than on cell proliferation.
Much ongoing research has attempted to elucidate the molecular bases of the anti-angiogenic and vascular targeting activity of microtubule-binding agents (Hotchkiss et al., 2002; Mabjeesh et al., 2003; Bijman et al., 2006), while it is widely accepted that the main effects of such agents result from their alteration of microtubule dynamics. In the present study, the inhibitory effects of C9 on microtubule dynamics and its affinity for tubulin have been clearly identified, as C9 disrupted the microtubule cytoskeleton through binding to tubulin via the colchicine site (but not the vinblastine-binding site). These results together indicated that impairment of microtubule function might be responsible for the anti-angiogenic or vascular targeting effects of C9. The attenuation of the effects of C9 by pretreatment with taxol, a microtubule-polymerizing agent, provides further support for this mechanism of action.
Evidence has suggested that Raf-MEK-ERK signalling molecules are required for angiogenesis (Gollob et al., 2006) and the microtubule inhibitors are known to elicit distinct and specific effects on mitogen-activated protein kinase-mediated signalling pathways (Tan et al., 2004). In our study, C9 decreased the activation of Raf-MEK-ERK signalling molecules stimulated by either VEGF or bFGF or FBS, but with no effects noted on phosphorylation of KDR or FGFR1. These results strongly suggested that the anti-angiogenic activities of C9 were partly mediated through the Raf-MEK-ERK1/2 signalling pathway.
Although the vascular targeting mechanisms have not, up to date, been clearly elucidated (Hotchkiss et al., 2002; Mabjeesh et al., 2003; Bijman et al., 2006), it has been suggested that changes in endothelial cell morphology are associated with vascular targeting activities, as cell contraction or retraction may cause an increase in vascular resistance and obstruction of tumour blood flow (Kanthou and Tozer, 2002). Such changes are critically regulated by the Rho/Rho kinase signalling pathway. In the current study, Y27632, a Rho kinase inhibitor, attenuated cell morphology changes induced by C9. Similarly, pretreatment with Y27632 protected cord networks from disruption by C9. Consistent with the results of these morphological and immunofluorescence assays, C9 also activated Rho GTPase and the phosphorylation of MLC2, which was reversed by pretreatment with Y27632. All these findings suggest that C9 disrupted newly formed vessels via a Rho/Rho kinase pathway.
Crosstalk between the microtubule and the actin cytoskeleton is essential for the regulation of many cellular functions. Our findings that taxol reversed the effects of C9 on Rho GTPase and MLC2 phosphorylation indicated that the morphological and vascular disrupting activity of C9 was associated with the disruption of microtubules and actin reorganization. Taken together, these results suggested the C9 induced microtubule disassembly in endothelial cells and actin reorganization through the Rho/Rho kinase pathway; and it also suggested that its vascular disruption effect resulted from changes in endothelial cell morphology.
In conclusion, we demonstrated for the first time that C9, a newly synthesized microtubule-binding agent, exhibited both anti-angiogenic and vascular disrupting effects in vivo and in vitro. The Raf-MEK-ERK pathway and microtubule disruption were found to be associated with the anti-angiogenic activity of C9. Endothelial cell contraction and membrane blebbing involving the Rho/Rho kinase-dependent pathway were likely to be responsible for its vascular disrupting activity.
Acknowledgments
We thank Li-Juan Lu and Yong Xi for Matrigel plug assays and Dr Li Du for SPR-binding assays. This work was supported by grants from the national Natural Science Foundation of China (NSFC) (No. 30572201, No. 30721005 and No. 30730103).
Glossary
Abbreviations:
- bFGF
basic fibroblast growth factor
- CAM
chick chorioallantoic membrane
- ERK
extracellular signal-regulated kinase
- FGFR1
FGF receptor 1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- KDR
receptor for vascular endothelial growth factor
- MEK
mitogen-activated ERK kinase
- MLC2
myosin light chain2
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazalium bromide
- VEGF
vascular endothelial growth factor
Statement of conflict of interest
None.
References
- Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, et al. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- Ashton AW, Yokota R, John G, Zhao S, Suadicani SO, Spray DC, et al. Inhibition of endothelial cell migration, intercellular communication, and vascular tube formation by thromboxane A(2) J Biol Chem. 1999;274:35562–35570. doi: 10.1074/jbc.274.50.35562. [DOI] [PubMed] [Google Scholar]
- Belleri M, Ribatti D, Nicoli S, Cotelli F, Forti L, Vannini V, et al. Antiangiogenic and vascular-targeting activity of the microtubule-destabilizing trans-resveratrol derivative 3,5,4′-trimethoxystilbene. Mol Pharmacol. 2005;67:1451–1459. doi: 10.1124/mol.104.009043. [DOI] [PubMed] [Google Scholar]
- Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science. 1999;284:808–812. doi: 10.1126/science.284.5415.808. [DOI] [PubMed] [Google Scholar]
- Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijman MN, van Nieuw Amerongen GP, Laurens N, van Hinsbergh VW, Boven E. Microtubule-targeting agents inhibit angiogenesis at subtoxic concentrations, a process associated with inhibition of Rac1 and Cdc42 activity and changes in the endothelial cytoskeleton. Mol Cancer Ther. 2006;5:2348–2357. doi: 10.1158/1535-7163.MCT-06-0242. [DOI] [PubMed] [Google Scholar]
- Denekamp J. Review article: angiogenesis neovascular proliferation and vascular pathophysiology as targets for cancer therapy. Br J Radiol. 1993;66:181–196. doi: 10.1259/0007-1285-66-783-181. [DOI] [PubMed] [Google Scholar]
- Downing KH. Structural basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu Rev Cell Dev Biol. 2000;16:89–111. doi: 10.1146/annurev.cellbio.16.1.89. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Folkman J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med. 1995;333:1757–1763. doi: 10.1056/NEJM199512283332608. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- Gollob JA, Wilhelm S, Carter C, Kelley SL. Role of Raf kinase in cancer: therapeutic potential of targeting the Raf/MEK/ERK signal transduction pathway. Semin Oncol. 2006;33:392–406. doi: 10.1053/j.seminoncol.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Gupta K, Bishop J, Peck A, Brown J, Wilson L, et al. Antimitotic antifungal compound benomyl inhibits brain microtubule polymerization and dynamics and cancer cell proliferation at mitosis, by binding to a novel site in tubulin. Biochemistry. 2004;43:6645–6655. doi: 10.1021/bi036112v. [DOI] [PubMed] [Google Scholar]
- Hastie SB. Interactions of colchicine with tubulin. Pharmacol Ther. 1991;51:377–401. doi: 10.1016/0163-7258(91)90067-v. [DOI] [PubMed] [Google Scholar]
- Hotchkiss KA, Ashton AW, Mahmood R, Russell RG, Sparano JA, Schwartz EL. Inhibition of endothelial cell function in vitro and angiogenesis in vivo by docetaxel (Taxotere): association with impaired repositioning of the microtubule organizing center. Mol Cancer Ther. 2002;1:1191–1200. [PubMed] [Google Scholar]
- Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Wilson L. Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr Opin Cell Biol. 1998;10:123–130. doi: 10.1016/s0955-0674(98)80095-1. [DOI] [PubMed] [Google Scholar]
- Kanthou C, Tozer GM. The tumor vascular targeting agent combretastatin A-4-phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood. 2002;99:2060–2069. doi: 10.1182/blood.v99.6.2060. [DOI] [PubMed] [Google Scholar]
- Kruczynski A, Poli M, Dossi R, Chazottes E, Berrichon G, Ricome C, et al. Anti-angiogenic, vascular-disrupting and anti-metastatic activities of vinflunine, the latest vinca alkaloid in clinical development. Eur J Cancer. 2006;42:2821–2832. doi: 10.1016/j.ejca.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Landuyt W, Verdoes O, Darius DO, Drijkoningen M, Nuyts S, Theys J, et al. Vascular targeting of solid tumours: a major ‘inverse’ volume-response relationship following combretastatin A-4 phosphate treatment of rat rhabdomyosarcomas. Eur J Cancer. 2000;36:1833–1843. doi: 10.1016/s0959-8049(00)00173-8. [DOI] [PubMed] [Google Scholar]
- Li PK, Xiao Z, Hu Z, Pandit B, Sun Y, Sackett DL, et al. Conformationally restricted analogs of Combretastatin A-4 derived from SU5416. Bioorg Med Chem Lett. 2005;15:5382–5385. doi: 10.1016/j.bmcl.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Li W, Shao Y, Hu L, Zhang X, Chen Y, Tong L, et al. BM6, a new semi-synthetic vinca alkaloid, exhibits its potent in vivo anti-tumor activities via its high binding affinity for tubulin and improved pharmacokinetic profiles. Cancer Biol Ther. 2007;6(5):787–794. doi: 10.4161/cbt.6.5.4006. [DOI] [PubMed] [Google Scholar]
- Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–375. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- Micheletti G, Poli M, Borsotti P, Martinelli M, Imberti B, Taraboletti G, et al. Vascular-targeting activity of ZD6126, a novel tubulin-binding agent. Cancer Res. 2003;63:1534–1537. [PubMed] [Google Scholar]
- Neri D, Bicknell R. Tumour vascular targeting. Nat Rev Cancer. 2005;5:436–446. doi: 10.1038/nrc1627. [DOI] [PubMed] [Google Scholar]
- Nicosia RF, Ottinetti A. Growth of microvessels in serum-free matrix culture of rat aorta. A quantitative assay of angiogenesis in vitro. Lab Invest. 1990;63:115–122. [PubMed] [Google Scholar]
- Osusky KL, Hallahan DE, Fu A, Ye F, Shyr Y, Geng L. The receptor tyrosine kinase inhibitor SU11248 impedes endothelial cell migration, tubule formation, and blood vessel formation in vivo, but has little effect on existing tumor vessels. Angiogenesis. 2004;7:225–233. doi: 10.1007/s10456-004-3149-y. [DOI] [PubMed] [Google Scholar]
- Pandit B, Sun Y, Chen P, Sackett DL, Hu Z, Rich W, et al. Structure-activity-relationship studies of conformationally restricted analogs of combretastatin A-4 derived from SU5416. Bioorg Med Chem. 2006;14:6492–6501. doi: 10.1016/j.bmc.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, et al. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribatti D, Vacca A, Roncali L, Dammacco F. The chick embryo chorioallantoic membrane as a model for in vivo research on anti-angiogenesis. Curr Pharm Biotechnol. 2000;1:73–82. doi: 10.2174/1389201003379040. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Wenzel D, Thorey I, Sasaki T, Hescheler J, Timpl R, et al. Endostatin influences endothelial morphology via the activated ERK1/2-kinase endothelial morphology and signal transduction. Microvasc Res. 2006;71:152–162. doi: 10.1016/j.mvr.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Siemann DW, Rojiani AM. The vascular disrupting agent ZD6126 shows increased antitumor efficacy and enhanced radiation response in large, advanced tumors. Int J Radiat Oncol Biol Phys. 2005;62:846–853. doi: 10.1016/j.ijrobp.2005.02.048. [DOI] [PubMed] [Google Scholar]
- Soeda S, Kozako T, Iwata K, Shimeno H. Oversulfated fucoidan inhibits the basic fibroblast growth factor-induced tube formation by human umbilical vein endothelial cells: its possible mechanism of action. Biochim Biophys Acta. 2000;1497:127–134. doi: 10.1016/s0167-4889(00)00052-5. [DOI] [PubMed] [Google Scholar]
- Tan WF, Zhang XW, Li MH, Yue JM, Chen Y, Lin LP, et al. Pseudolarix acid B inhibits angiogenesis by antagonizing the vascular endothelial growth factor-mediated anti-apoptotic effect. Eur J Pharmacol. 2004;499:219–228. doi: 10.1016/j.ejphar.2004.07.063. [DOI] [PubMed] [Google Scholar]
- Taraboletti G, Micheletti G, Dossi R, Borsotti P, Martinelli M, Fiordaliso F, et al. Potential antagonism of tubulin-binding anticancer agents in combination therapies. Clin Cancer Res. 2005;11:2720–2726. doi: 10.1158/1078-0432.CCR-04-1616. [DOI] [PubMed] [Google Scholar]
- Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004;10:415–427. doi: 10.1158/1078-0432.ccr-0642-03. [DOI] [PubMed] [Google Scholar]
- Vincent L, Kermani P, Young LM, Cheng J, Zhang F, Shido K, et al. Combretastatin A4 phosphate induces rapid regression of tumor neovessels and growth through interference with vascular endothelial-cadherin signaling. J Clin Invest. 2005;115:2992–3006. doi: 10.1172/JCI24586. [DOI] [PMC free article] [PubMed] [Google Scholar]