

Abstract
The gap junction (GJ) protein connexin32 (Cx32) is expressed by myelinating Schwann cells and oligodendrocytes and is mutated in X-linked Charcot-Marie-Tooth disease. In addition to a demyelinating peripheral neuropathy, some Cx32 mutants are associated with transient or chronic CNS phenotypes. To investigate the molecular basis of these phenotypes, we generated transgenic mice expressing the T55I or the R75W mutation and an IRES-EGFP, driven by the mouse Cnp promoter. The transgene was expressed in oligodendrocytes throughout the CNS and in Schwann cells. Both the T55I and the R75W mutants were localized in the perinuclear cytoplasm, did not form GJ plaques, and did not alter the expression or localization of two other oligodendrocytic GJ proteins, Cx47 and Cx29, or the expression of Cx29 in Schwann cells. On wild type background, the expression of endogenous mCx32 was unaffected by the T55I mutant, but was partly impaired by R75W. Transgenic mice with the R75W mutation and all mutant animals with Gjb1-null background developed a progressive demyelinating peripheral neuropathy along with CNS myelination defects. These findings suggest that Cx32 mutations result in loss of function in myelinated cells without trans-dominant effects on other GJ proteins. Loss of Cx32 function alone in the CNS causes myelination defects.
Introduction
Gap junctions (GJs) are channels that allow the diffusion of ions and small molecules across apposed cell membranes (Bruzzone et al., 1996). In rodents, there are 20 connexin genes, each of which is expressed in subsets of cell types (Willecke et al., 2002). Rodent oligodendrocytes express connexin32 (Cx32) (Scherer et al., 1995), Cx47 (Menichella et al., 2003; Odermatt et al., 2003), and Cx29 (Altevogt et al., 2002). Cx32 is expressed mostly by white matter oligodendrocytes and is localized in the myelin sheath of large diameter fibers, whereas Cx47 is expressed by both white and gray matter oligodendrocytes and forms GJs on cell bodies and proximal processes. Cx29 (Altevogt and Paul, 2004; Kleopa et al., 2004; Li et al., 2004; Kamasawa et al., 2005) and its human ortholog Cx31.3 (Sargiannidou et al., 2008) appear to be restricted to oligodendrocytes that myelinate small caliber fibers, likely forming hemichannels. Cx32 and Cx29 are also expressed by Schwann cells (Scherer et al., 1995; Altevogt et al., 2002).
Cx32 and Cx47 have partially overlapping functions in oligodendrocytes, because mice deficient for either Cx32 or Cx47 develop minimal CNS pathology, whereas double knock-out mice develop severe CNS demyelination (Scherer et al., 1998; Menichella et al., 2003; Odermatt et al., 2003). Both connexins likely mediate GJ coupling of oligodendrocytes to astrocytes through heterotypic coupling: Cx32:Cx30 and Cx47:Cx43 (Nagy et al., 2003; Kamasawa et al., 2005; Orthmann-Murphy et al., 2007b). Cx32 also forms most autologous GJs within the myelin sheath (Rash et al., 2001; Nagy et al., 2003; Altevogt and Paul, 2004; Kamasawa et al., 2005). This network of GJs may serve the spatial buffering of K+ elaborated during the propagation of action potentials (Kamasawa et al., 2005; Menichella et al., 2006). The importance of this network in humans is supported by the finding that recessive mutations in GJC2/GJA12 encoding Cx47 cause Pelizeaus-Merzbacher-like disease, a severe dysmyelinating disorder of the CNS (Uhlenberg et al., 2004; Bugiani et al., 2006; Orthmann-Murphy et al., 2007a).
Hundreds of mutations in GJB1 (encoding Cx32) cause the X-linked form of Charcot-Marie-Tooth disease (CMT1X) (http://www.molgen.ua.ac.be/CMTMutations/default.cfm), a demyelinating peripheral neuropathy (Bergoffen et al., 1993). Evoked potentials demonstrate mild conduction slowing in most patients, indicating subclinical involvement of CNS myelinated axons (Nicholson and Corbett, 1996; Nicholson et al., 1998; Bähr et al., 1999). A subset of Cx32 mutations also cause clinical CNS manifestations including spasticity, hyperactive reflexes, extensor plantar responses, ataxia, or acute reversible encephalopathy (Kleopa et al., 2002; Paulson et al., 2002; Taylor et al., 2003; Kleopa and Scherer, 2006). When expressed in vitro, many of the Cx32 mutants, including all the ones associated with CNS phenotypes, are localized intracellularly in the Golgi or endoplasmic reticulum (ER), with reduced or absent formation of GJ plaques at the cell membrane (Omori et al., 1996; Deschênes et al., 1997; Oh et al., 1997; VanSlyke et al., 2000; Kleopa et al., 2002; Yum et al., 2002; Kleopa et al., 2006). Their intracellular localization raises the possibility of trans-dominant effects on coexpressed GJ proteins, especially Cx47 in oligodendrocytes. However, previously generated Cx32 transgenic mice expressed mutations only in Schwann cells (Huang et al., 2005; Jeng et al., 2006), leaving the cellular mechanisms underlying these CNS phenotypes in CMT1X unclear. Therefore, we generated transgenic mice expressing the T55I and R75W Cx32 mutations in both CNS and PNS. These mutations were chosen because they have been associated with prominent CNS phenotypes preceding the diagnosis of CMT1X (Panas et al., 2001; Taylor et al., 2003), and their in vitro cellular expression is representative of most Cx32 mutants, including ER (T55I) and Golgi (R75W) retention (Kleopa et al., 2002; Yum et al., 2002). Progressive demyelinating neuropathy and mild CNS myelination defects resulted mostly from loss of Cx32 function, and these Cx32 mutants had no discernable effects on either Cx47 or Cx29.
Materials and Methods
Generating transgenic mice.
The human T55I and R75W mutations were generated by site-directed mutagenesis using the QuickChange kit (Stratagene) with mutagenic oligonucleotide primers and PfuTurbo DNA polymerase as previously described (Kleopa et al., 2002; Yum et al., 2002). The human GJB1 open reading frame (ORF) sequence (including the T55I or R75W mutants) along with the downstream IRES-EGFP sequence was amplified by PCR from pIRES2-EGFP construct using the primers PSLN-CLA-F (5′-TA GGATGCATATGG-CGGCCGCCTGCAGCTGGCGCC-3′) and PSLN-SAL-R (5′-AGCTTGGCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAAC-CCTGGCGT-3′). This fragment was ligated downstream of the mouse 2′,3′-cyclic nucleotide phosphodiesterase (CNP) promoter in the pBluescript SK+ vector at the ClaI and SalI sites. The 3.9 kb mouse Cnp promoter (gift from Dr. Vittorio Gallo, Children's National Medical Center, Washington, DC) has been shown to drive expression of lacZ (Gravel et al., 1998) and EGFP (Yuan et al., 2002) both in myelinating Schwann cells and oligodendrocytes. The orientation and the in-frame positioning were confirmed by sequencing analysis. DNA was isolated using Qiagen MaxiPrep kit and the transgene cassette (Fig. 1A) was released from vector sequences by digestion with SalI and AlwNI.
Figure 1.
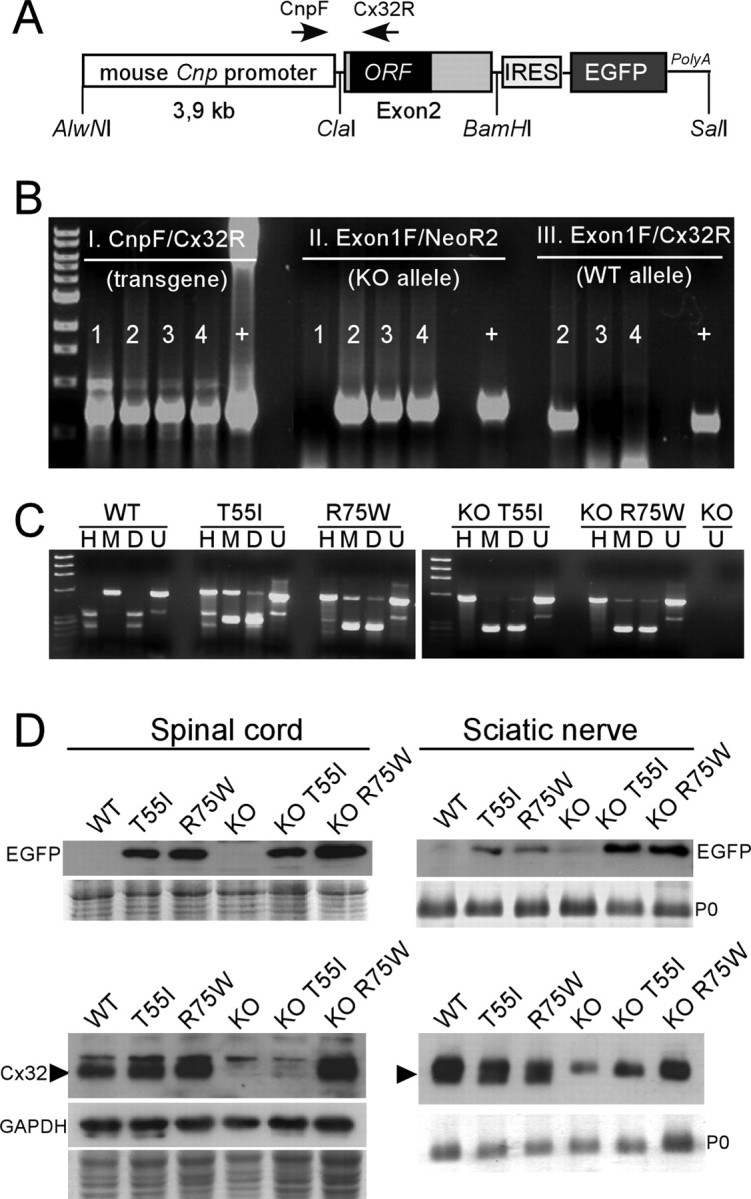
A, The structure of the transgene used to express Cx32 in myelinating cells. The 3.9 kb mouse Cnp promoter is joined upstream of the exon 2 (which contains the ORF) of the human GJB1 gene. The IRES-EGFP sequence was cloned downstream of the GJB1 exon 2 to allow coexpression of the EGFP. The positions of the primers used for PCR screening and restriction sites used for cloning are indicated. B, Triple PCR screening of transgenic lines on WT and Gjb1-null (KO) background. Primer pairs amplifying the transgene (CnpF/Cx32R; shown in A), the neomycin gene that replaced the ORF of Gjb1 in Cx32 KO mice (Exon1F/NeoR2), and the WT Gjb1 allele (Exon1F/Cx32R) were used in combination. Mouse 1 is a male transgenic on WT background (TG+X+Y); mouse 2 is a female transgenic heterozygous KO (TG+X+X−); mouse 3 is TG+X−Y, and mouse 4 is TG+X−X−. “+” is positive control DNA. C, RT-PCR analysis of transgene expression in mouse brain. Mouse and/or human Cx32 cDNA was amplified by RT-PCR (using primers that amplify both) from single brains. The PCR product was digested with MscI (M) (cuts the human Cx32 cDNA) or HhaI (H) (cuts the mouse Cx32 cDNA). D, “Double-cut” with MscI and HhaI; U, uncut. The M-digested human Cx32 cDNA is detectable only in transgenic lines but not in the WT or Cx32 KO mouse, whereas the H-digested mouse Cx32 cDNA is present in WT mouse and in transgenic lines on WT background, but not in the lines on KO background. No human or mouse Cx32 cDNA is present in the Cx32 KO. In the transgenic lines on a WT background, the M-digested product (resulting in two bands of almost same size and therefore merged) gives ∼2-fold higher band intensity than the H-digested product (2 separate bands), indicating that the level of the transgene mRNA is approximately double that of the endogenous/mouse mRNA. D, Immunoblots of spinal cord or sciatic nerve lysates from transgenic lines as well as WT or KO mice, as indicated. Coomassie-stained gels are shown under the blots; P0 is the dense band in the sciatic nerve blots; one blot was reprobed for GAPDH to demonstrate the loading. Note that EGFP (27 kDa band) is detected in the spinal cord and sciatic nerves of all mutants on both WT and KO backgrounds, but not in WT or in KO mice. The specific band for the Cx32 monomer (arrowhead, ∼27 kDa) is detected in the spinal cord and sciatic nerve of WT, T55I, and R75W mice, it is absent from KO mice, reappears in KO R75W mutant mice but is faint (sciatic nerve) or absent (spinal cord) in KO T55I tissues. A faint nonspecific band is present above the Cx32 band in all samples including the KO tissues.
The fragment was isolated, purified, and microinjected into the male pronucleus of fertilized oocytes obtained from C57BL/6 mice according to standard protocols. Transgenic progeny was identified by PCR of genomic tail DNA with transgene-specific primers: Cnp1F (5′-TGTGGCTTTGCCCATACATA-3′) and Cx32R (5′-CGCTGTT-GCAGCCAGGCTGG-3′) resulting in a 732bp PCR product (94°C × 5 min, 40 cycles of 94°C × 30 s, 57°C × 30 s, 72°C × 30 s and then 72°C × 7 min) (Fig. 1B-I). Potential founders gave rise to transgenic lines, and each line was screened for the expression of EGFP using immunostaining, FACS analysis of trypsinized brain cells, and immunoblot analysis of tissue lysates (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (data not shown).
The transgenic lines with best expression for each Cx32 mutation were further expanded for analysis. In addition, to generate transgenic mice on Gjb1-null background, male transgenic mice expressing either the T55I or the R75W mutation were bred with female heterozygous Gjb1-null mice (C57BL/6×129) obtained from the European Mouse Mutant Archive, Monterotondo, Italy (originally generated by Prof. Klaus Willecke, University of Bonn, Bonn, Germany). In these mice, the neor gene was inserted in frame into the exon 2 of Gjb1 gene which contains the ORF (Nelles et al., 1996). Genotypes of the offspring were determined using a triple-PCR screening with transgene specific primers (above, Fig. 1B-I), as well as primers for the neor gene (Gjb1-null) (Fig. 1B-II, Exon1F: 5′-GACCACTCCCCCTACACAGA-3′; NeoR2: 5′-CTCGTCCTG-CAGTTCATTCA-3′) resulting in a 721 bp PCR product (94°C × 5 min, 35 cycles of 94°C × 30 s, 56°C × 30 s, 72°C × 30 s and then 72°C × 7 min); and primers specific for the wild-type (WT) Gjb1 mouse gene (Fig. 1B-III, Exon1F and Cx32R, above).
Reverse transcription PCR.
Total RNA was isolated from snap-frozen brains using the TRIZOL reagent (Invitrogen), according the manufacturer's protocol. DNase I (New England Biolabs) treatment was performed and the RNA was quantified by spectrophotometry. RNA (0.5 μg) was used for the synthesis of cDNA by the Taqman Reverse transcription reagents (Applied Biosystems). The cDNA was PCR amplified using primers F (5′-TGAGGCAGGATGAACTGGACAGGT-3′) and R (5′-CACGAAGCAGTCCACTGT-3′) that amplify both endogenous/mouse and transgenic/human Cx32, resulting in a 553 bp PCR product (94°C × 5 min, 40 cycles of 94°C × 30 s, 60°C × 30 s, 72°C × 30 s and then 72°C × 7 min). Digestion of the reverse transcription (RT)-PCR product with MscI (cuts specifically in the human GJB1 ORF) or HhaI (cuts specifically in the mouse Gjb1 ORF) allowed us to determine the relative mRNA levels of endogenous/mouse and transgenic/human Cx32 (Fig. 1C).
Immunoblot analysis.
Fresh tissues were lysed in ice-cold RIPA buffer (10 mm sodium phosphate pH 7.0, 150 mm NaCl, 2 mm EDTA, 50 mm sodium fluoride, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS) containing a mixture of protease inhibitors (Roche). Proteins (50 μg) from tissue lysates were fractionated by 12% SDS-PAGE and then transferred to a Hybond-C extra membrane (GE Healthcare Bio-Sciences), using a semidry transfer unit (GE Healthcare Bio-Sciences). Nonspecific sites on the membrane were blocked with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 1 h at room temperature. Immunoblots were then incubated with either mouse monoclonal anti-Cx32 antibodies (Santa Cruz Biotechnology, diluted 1:500; Zymed, 1:1000; 7C6.C7, 1:10) or with rabbit antisera against Cx32 [#918, diluted 1:10,000 (Ahn et al., 2008)], EGFP (Abcam; diluted 1:10,000), Cx47 [1:20,000 (Orthmann-Murphy et al., 2007a)], or Cx29 (Zymed, 1:500) in 5% milk–TBS-T, at 4°C overnight. To reduce nonspecific binding, the rabbit antiserum against Cx32 (#918) was preincubated with Gjb1-null tissue lysate. Briefly, brain and liver tissue from Gjb1-null mice was homogenized in RIPA buffer. The homogenate was incubated in acetone and after a spin the pellet was air dried. The Cx32 antiserum was preincubated with this powder for 1 h at room temperature before immunoblotting. After washing, immunoblots were incubated with an anti-mouse or anti-rabbit HRP-conjugated secondary antiserum (Jackson ImmunoResearch Laboratories, diluted 1:5000 and 1:10,000 respectively) in 5% milk-TBS-T, for 1 h. The bound antibody was visualized by enhanced chemiluminescence system (ECL Plus, GE Healthcare Bio-Sciences).
Immunohistochemistry.
Four-month-old mice from all genotypes were anesthetized with Avertin according to institutionally approved protocols and then transcardially perfused with PBS followed by fresh 4% paraformaldehyde (PFA) in 0.1 m PBS. Tissues were harvested and further fixed for 30 min and then cryoprotected in 20% sucrose in PBS overnight. Ten μm thick sections were thaw-mounted onto glass slides, permeabilized in cold acetone (−20°C for 10 min), and incubated at room temperature with blocking solution of 5% bovine serum albumin (BSA) containing 0.5% Triton-X for 1 h. The primary antibodies diluted in blocking solution were incubated overnight at 4°C: mouse monoclonal antibodies against Cx32 (Zymed, 1:50), Cx47 (Zymed, 1:500), GFAP (Sigma, 1:200), RIP (Millipore Bioscience Research Reagents, 1:1200), NeuN (Millipore Bioscience Research Reagents, 1:400), and MBP (Abcam, 1:500), as well as rabbit antisera against Cx47 (Invitrogen, 1:500), Cx29 (Zymed, 1:300), and EGFP (Invitrogen, 1:2000). Sections were then washed in PBS and incubated with fluorescein- and rhodamine-conjugated donkey cross-affinity purified secondary antibodies (Jackson ImmunoResearch Laboratories, 1:100) for 1 h at room temperature. Cell nuclei were visualized with 4′,6′-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich). Slides were mounted with Dako Fluorescent Mounting Medium and images were photographed under a Zeiss fluorescence microscope with a digital camera using the Zeiss Axiovision software (Carl Zeiss MicroImaging). Where appropriate, we obtained images with comparable exposure times to allow better comparison between different genotypes.
Electron microscopy.
After anesthesia, 2-, 4- and 8-month-old mice from all genotypes as well as WT littermates were transcardially perfused with 2.5% glutaraldehyde in 0.1 m PB and the lumbar spinal cords as well as the femoral, sciatic and optic nerves were dissected and further fixed overnight at 4°C, then osmicated, dehydrated, and embedded in Araldite resin. Transverse semithin sections (1 μm) were obtained and stained with alkaline toluidine blue. Ultrathin sections (80–100 nm) were counterstained with lead citrate and uranyl acetate before being examined in a JEOL JEM-1010 transmission electron microscope.
Morphometric analysis.
Semithin sections of femoral and sciatic nerves as well as anterior lumbar roots were visualized with 20× and 63× objective lenses and captured with AxioHR camera. Images of whole nerve transverse sections were obtained at a 200× final magnification; a series of partially overlapping fields covering the cross-sectional area of the nerve were captured at a 630× final magnification. Electron micrographs of optic nerve and spinal cord were obtained and scanned, and overlapping images were transferred to a computer and used for analysis. Morphometric data were collected using ImageJ 1.4 (NIH). The cross-sectional area of ∼90 myelinated fibers and their axons (randomly picked, regardless of their size) was measured for each femoral and sciatic nerve. This was achieved by identifying the borders of each myelinated fiber and its respective axon after defining a threshold gray-scale level for axons and myelin profiles. The diameters of each fiber and the respective axon were obtained using the following formula: diameter = 2 × √(area/π). We obtained the g-ratios by dividing the axonal diameter by the fiber diameter. Axons were classified into three categories according to their diameters: 1–2.99 μm, small; 3–6.99 μm, medium; ≥7 μm, large.
We also examined the degree of abnormal myelination in transverse semithin sections of the femoral nerve in all transgenic lines. All demyelinated, remyelinated, and normally myelinated axons were counted using the following criteria: axons larger than 1 μm without a myelin sheath were considered demyelinated; axons with myelin sheaths that were <10% of the axonal diameter and/or axons that were surrounded by “onion bulbs” (circumferentially arranged Schwann cell processes and extracellular matrix) were considered remyelinated; the other myelinated axons were considered normally myelinated.
Furthermore, we measured the CNS myelin fraction in semithin sections of the spinal cord dorsal and ventral funiculus and in ultrathin sections of the optic nerve in each transgenic line using a modified method to estimate the density of myelinated fibers and myelin sheaths (Tang and Nyengaard, 1997; Sutor et al., 2000). Images of semithin sections captured at 630× final magnification following the same processing and microscopy settings (above) were imported into Photoshop (Adobe Systems) and a transparent counting grid was placed on the image. All intersections of the grid lines hitting white matter, myelinated fibers and myelin sheaths were counted separately. The volume density of the myelinated fibers in the white matter, Vv(nf/wm), was estimated with the formula: Vv(nf/wm) = ∑P(nf)/∑P(wm), where ∑P(nf) is the total number of points hitting myelinated fibers in the white matter, and ∑P(wm) is the total number of points hitting white matter. The volume density of the myelin sheaths in the white matter, Vv(m/wm), was estimated with the formula: Vv(m/wm) = ∑P(m)/∑P(wm), where ∑P(m) is the total number of points hitting the myelin sheaths in the white matter. On average, 392 or more points hitting the white matter (n = 2 or 3 different sections) per animal were counted.
Statistical analysis.
We compared the proportion of abnormally myelinated fibers in femoral motor nerves from different genotypes as well as the volume density of myelin in the lumbar spinal cord dorsal and ventral funiculi with the Mann–Whitney test (significance level: p = 0.05) using the Minitab 15 statistical software.
Results
Generation of Cx32 mutant mice on wild type and Gjb1-null background
We used a mouse Cnp promoter to drive the expression of T55I and R75W mutants in myelinating glia (Gravel et al., 1998; Yuan et al., 2002). Three T55I (24, 30, and 32) and two R75W (3 and 17) lines with stable transmission of the transgene were identified by PCR of genomic DNA (Fig. 1A,B). Because Cx32 antibodies do not distinguish transgenic/human Cx32 protein from endogenous/mouse Cx32, we immunoblotted and immunostained tissues for EGFP, which is expressed from an IRES in the transgene (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In all transgenic lines, both Schwann cells and oligodendrocytes expressed EGFP, but expression was higher in the T55I-30 and R75W-3 lines, which were used for all experiments in this report (hereafter designated as T55I and R75W). By immunoblotting, the total level of Cx32 in the T55I and R75W lines on a WT background was only mildly increased compared with WT animals (Fig. 1D), indicating that mutant Cx32 protein was not grossly overexpressed. This was repeated at least 3 times, using both spinal cord and brain samples, with similar results.
We investigated these issues further by crossing transgene-positive (TG+) male mice with Gjb1-knock-out (KO) females, producing equal proportions of TG+ and TG− male progeny on a KO background (hereafter designated KO T55I and KO R75W). We compared the expression of Cx32 mRNA from adult brains with RT-PCR, combined with restriction enzymes specific for the cDNA derived from the transgenic/human construct or the endogenous/mouse Gjb1 gene (Scherer et al., 2005; Jeng et al., 2006). This analysis showed that transgenic mice had two- to threefold higher levels of Cx32 mRNA than did WT mice (Fig. 1C). This was performed twice with similar results. No obvious behavioral abnormalities were observed in Cx32 mutant mice either on WT or KO background for up to 18 months old.
Oligodendrocytes and Schwann cells express Cx32 mutants
To determine whether the transgene was specifically expressed in oligodendrocytes, we immunostained different CNS areas (including spinal cord, brainstem, cerebellum, cerebrum, and optic nerve) with antibodies to EGFP, combined with cell markers for oligodendrocytes (RIP), astrocytes (GFAP), and neurons (NeuN). EGFP colocalized with RIP in oligodendrocytes, but was distinct from GFAP- or NeuN-positive cells (Fig. 2A–C) (data not shown). To determine the extent of transgene expression, we double labeled sections from the same CNS areas with antibodies to Cx47, which is expressed in the cell bodies of most if not all oligodendrocytes (Menichella et al., 2003; Odermatt et al., 2003; Kleopa et al., 2004). In multiple CNS regions, for both T55I and R75W lines, almost all Cx47-positive cells also expressed EGFP, on both a WT and a KO background (Fig. 2D–F; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This was repeated at least 3 times per line, with similar results. EGFP expression was also localized to the perinuclear cytoplasm of myelinating Schwann cells in both T55I and R75W lines (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Immunoblot analysis of CNS and sciatic nerve lysates confirmed that EGFP was expressed in transgenic but not in WT or KO mice, and at similar levels between the selected T55I and R75W lines (Fig. 1D), in keeping with the RT-PCR results.
Figure 2.
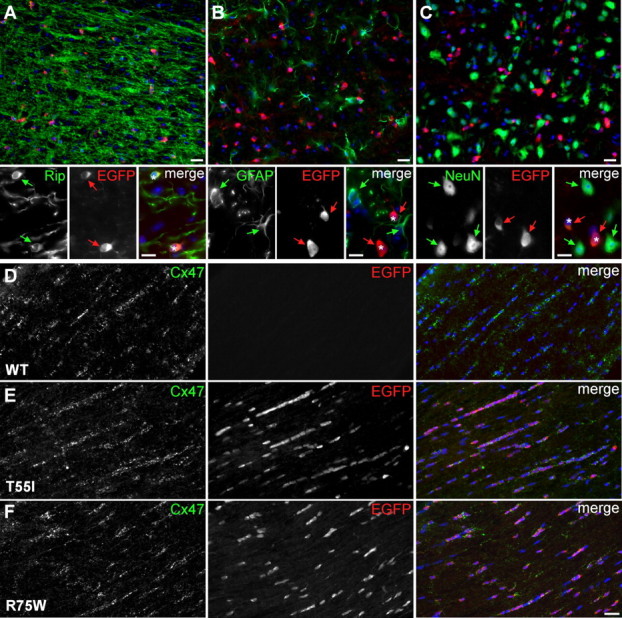
Expression of the transgene in oligodendrocytes of CMT1X mice. A–F, These are images of sections of spinal cords (A–C) or optic nerves (D–F) from 4-month-old WT mice or mutant mice in a WT background, double labeled with antibodies against EGFP (red) and cell markers or Cx47 (green) as indicated. Nuclei are visualized with DAPI (blue). In spinal cords, EGFP-positive cells (red arrows and asterisks in merged images) are Rip-positive oligodendrocytes (green arrows in A), and not GFAP-positive astrocytes (green arrows in B) or NeuN-positive neurons (green arrows in C). In optic nerves, EGFP is expressed in Cx47-positive oligodendrocytes in the T55I and R75W lines, but not in WT mice. Scale bars, 20 μm; in insets, 10 μm.
T55I and R75W mutants are localized in the perinuclear cytoplasm
The above analysis demonstrated that the transgene was widely expressed. To determine whether the mutant proteins themselves were expressed, we examined their localization in KO T55I and KO R75W mice. In both lines, Cx32-immunoreactivity was detected in the perinuclear cytoplasm of oligodendrocytes throughout the CNS, including the white and gray matter of the spinal cord, brainstem, cerebellum, cerebrum, corpus callosum, and optic nerve (Fig. 3; supplemental Figs. 4, 5, available at www.jneurosci.org as supplemental material) (data not shown). Thus, like EGFP, almost all oligodendrocytes expressed the mutant Cx32, including a subpopulation that normally lack Cx32-immunoreactivity, such as oligodendrocytes in the corpus callosum and optic nerve (Kleopa et al., 2004). Furthermore, in all of these locations, GJ plaques were not seen, indicating that neither T55I nor R75W traffic properly to the cell membrane, as in transfected cells (Kleopa et al., 2002; Yum et al., 2002). Compared with the T55I mutant, the R75W mutant appeared to have stronger cytoplasmic staining in most CNS areas (Figs. 3, 4) (data not shown); this was also seen by immunoblot analysis: KO R75W mice had more Cx32 than did KO T55I mice (Fig. 1D) (data not shown). Given the comparable levels of Cx32 mRNA expression in the two lines (Fig. 1C), these findings suggest that oligodendrocytes more rapidly degrade the T55I mutant.
Figure 3.

Mutant Cx32 does not alter the localization of other gap junction proteins in oligodendrocytes. A–I, These are images of longitudinal sections through the white matter of spinal cords from Cx32 KO mice (A–C) as well as T55I (D–F) and R75W (G–I) mutant mice in a KO background, as indicated. Sections are double labeled with mouse monoclonal antibodies (green) against Cx47 (A, D, G) or Cx32 (B, C; E, F; H, I) and rabbit antisera (red) against GFP (A, D, G), Cx47 (B, E, H), or Cx29 (C, F, I). Nuclei are labeled with DAPI (blue). In mutant mice (D, G), EGFP-positive oligodendrocytes (asterisks) express Cx47, which forms numerous GJ plaques at the perikaryon and proximal processes (green arrowheads), as in Cx32 KO mice (A). Cx32 is absent from Cx32 KO mice (B, C), and mutant Cx32 is localized in the cytoplasm of oligodendrocytes in both KO T55I and KO R75W lines (green arrows in E, F and H, I); in the same cells Cx47 again appears to be normally localized, forming GJ plaques (red arrowheads in B, E, H). Cx29 is also normally localized along thin myelinated fibers (red arrows) in both mutant lines despite the presence of the Cx32 mutants (F, I), as it does in Cx32 KO mice (C). Scale bars (including insets), 10 μm.
Figure 4.
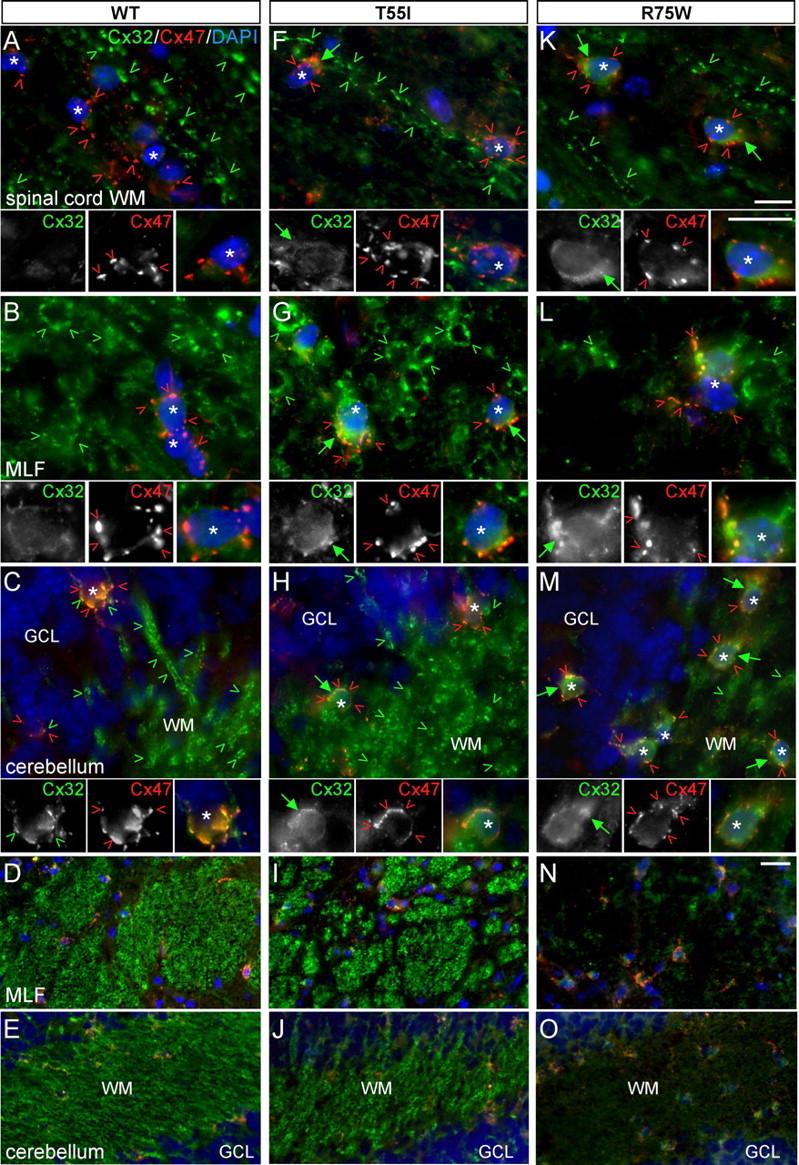
The R75W mutant but not the T55I mutant alters the expression of endogenous mCx32 on WT background. A–O, These are images of the indicated CNS regions, longitudinal sections of spinal cord white matter (WM) (A, F, K), transverse sections of the medial longitudinal fasciculus (MLF) (B, G, L, and low magnification in D, I, N), and the cerebellar white mater (C, H, M, and low magnification in E, J, O), from WT mice (A–E) as well as T55I (F–J) and R75W (K–O) mutant mice in a WT background. Sections were double labeled with a mouse monoclonal antibody against Cx32 (green) and a rabbit antiserum against Cx47 (red). Cell nuclei are visualized with DAPI (blue). In WT mice (A–E), endogenous/mouse Cx32-immunoreactivity is seen along large myelinated fibers and occasionally in perikarya of cerebellar oligodendrocytes (C, green arrowheads); T55I mice (F–J) have the same pattern of Cx32-immunoreactivity except that oligodendrocyte somata have more pronounced Cx32-immunoreactivity (likely the T55I mutant protein). In contrast, in R75W mice (K–O), Cx32-immunoreactivity is strongly reduced in white matter tracts; this is particularly evident in low-magnification images of MLF (N) and cerebellar white matter (O). In addition, oligodendrocytes (asterisks) have stronger Cx32-immunoreactivity in their perinuclear cytoplasm (likely the R75W mutant protein) than WT or T55I mutant mice. Despite the presence of mutant Cx32, Cx47 is normally expressed in the perikarya and proximal processes of oligodendrocytes (red arrowheads) in all of these CNS areas, as in the WT CNS. Scale bars: in A–C, F–H, K–M (and insets), 10 μm; in D, E, I, J, N, O, 20 μm.
T55I and R75W mutants do not affect the expression of Cx29 and Cx47
To determine whether T55I and R75W affect the localization of other connexins, we double labeled for Cx32 and Cx47 or Cx29. In all CNS regions we examined, Cx47 formed GJ plaques at the perikarya and proximal processes of all oligodendrocytes comparable to KO or WT mice (Fig. 3; supplemental Fig. 4, available at www.jneurosci.org as supplemental material) (data not shown). Similarly, Cx29 was normally localized along thinly myelinated fibers in the spinal cord white matter, brainstem, cerebellum, corpus callosum, and optic nerves comparable to KO or WT mice (Fig. 3; supplemental Fig. 5, available at www.jneurosci.org as supplemental material) (data not shown). Furthermore, immunoblot analysis of CNS tissues showed comparable levels of Cx47 and Cx29 in T55I and R75W mutants, Gjb1-null mice, and WT mice (see Fig. 6) (data not shown). These immunostaining and immunoblot experiments were repeated at least 3 times per line, with similar results. Thus, neither mutant appeared to have a trans-dominant effect on other connexins expressed by oligodendrocytes.
Figure 6.
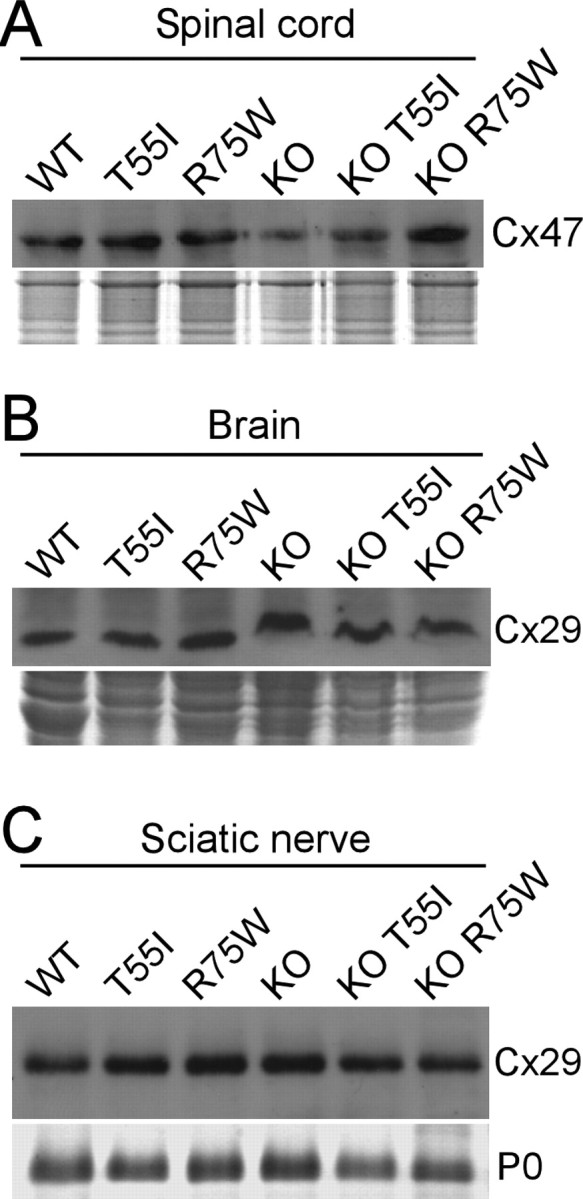
Cx32 mutants do not affect the expression of other gap junction proteins in myelinating cells. A–C, Immunoblot analysis of Cx47 in the spinal cords (A) and Cx29 in the brains (B) and sciatic nerves (C) of Cx32 WT, Cx32 KO, and T55I or R75W mutant mice either on WT or on KO background, as indicated. In all cases, the levels are similar. Coomassie-stained gels are shown under the blots; P0 is the dense band in the sciatic nerve blot.
We also examined these mutations on a WT background to look for possible dominant effects of T55I and R75W on endogenous/mouse Cx32. As previously described (Kleopa et al., 2004), Cx32 immunoreactivity in WT mice was most prominent along large myelinated fibers of the white matter and in gray matter oligodendrocytes, forming GJ plaques in cell bodies and proximal processes (Fig. 4A–E; supplemental Fig. 6, available at www.jneurosci.org as supplemental material) (data not shown). This expression pattern was also found in the T55I mutant mice (Fig. 4F–J; supplemental Fig. 6, available at www.jneurosci.org as supplemental material) (data not shown). In contrast, the R75W mutant mice had diminished Cx32-positive GJ plaques in most CNS white matter areas in which Cx32 expression is normally prominent, including the myelinated tracts in the spinal cord, the medial longitudinal fasciculus (MLF), the cerebellar white matter, and the lateral olfactory tract (Fig. 4K–O; supplemental Fig. 6I–L, available at www.jneurosci.org as supplemental material) (data not shown). Because total Cx32 was not reduced in R75W spinal cord and brain compared with the T55I mutant or WT mice (Fig. 1D) (data not shown), these findings suggest that the R75W mutant retains endogenous WT Cx32 in the perinuclear cytoplasm, where R75W is found in KO mice. In contrast, the localization of Cx47 and Cx29 did not appear to be perturbed in the R75W mutant line (Fig. 4; supplemental Fig. 6, available at www.jneurosci.org as supplemental material) (data not shown), indicating that the R75W mutant may specifically affect the localization of Cx32.
We also examined these issues in myelinating Schwann cells. As in oligodendrocytes, T55I and R75W were localized in the perinuclear cytoplasm in the KO T55I and KO R75W lines and did not form GJ plaques at paranodes and incisures (Fig. 5), where Cx32 is normally localized (Scherer et al., 1995). As in oligodendrocytes, there appeared to be more perinuclear Cx32-immunoreactivity in the KO R75W than in the KO T55I mutant mice; this was supported by immunoblot analysis, suggesting that the T55I mutant was more rapidly degraded (Fig. 1D) (data not shown). In a WT background, R75W (but not T55I) reduced Cx32 immunoreactivity at the paranodes and incisures (Fig. 5F), whereas the total amount of Cx32 in immunoblots of sciatic nerves lysates was unchanged (Fig. 1D), suggesting that, as in oligodendrocytes, the R75W mutant retains endogenous WT Cx32 in the perinuclear cytoplasm. Neither T55I nor R75W appeared to alter the localization of Cx29 in either a KO or a WT background (Fig. 5), and the amount of Cx29 was unchanged (Fig. 6). Thus, as in oligodendrocytes, the R75W but not the T55I mutant has dominant effects on the endogenous Cx32, but neither T55I nor R75W impairs the expression of Cx29.
Figure 5.

Cx32 mutants do not affect the expression of Cx29 in Schwann cells but the R75W mutant alters the expression of endogenous Cx32. A–F, These are images of teased fibers from adult sciatic nerves of Cx32 KO (A), WT (D), or T55I and R75W mutant mice on KO background (B, C) or Cx32 WT background (E, F), double stained with a monoclonal antibody against Cx32 (green) and a rabbit antiserum against Cx29 (red), as indicated. Schwann cell nuclei (asterisks) are stained with DAPI. Cx32 is absent in KO mice (A), and is localized in the perinuclear cytoplasm of Schwann cells of the T55I and R75W mutants on KO background (green arrows in B, C and E, F). Cx32-immunoreactivity appears stronger in the R75W mutant compared with T55I (B, C). In a WT background, endogenous Cx32 is normally localized at paranodes (green arrowheads) and incisures (green carets); this staining is maintained in T55I mutant mice, but appears reduced in the R75W mouse, with a corresponding increase in perinuclear staining (F). In all cases, Cx29 is properly localized at juxtaparanodes (red arrowheads) and incisures (red carets). Scale bars, 10 μm.
T55I and R75W mutant mice develop progressive demyelinating neuropathy and a subtle CNS myelin defect
To determine whether T55I or R75W affects myelination, we examined epoxy sections from peripheral nerves (sciatic and femoral), spinal cord, and optic nerves from T55I and R75W mice in a WT and a KO background at 2, 4 and 8 months of age (Figs. 7, 8). On a WT background, the R75W but not the T55I mice have more abnormally myelinated femoral motor fibers (either demyelinated or remyelinated) compared with WT mice (Table 1 and Fig. 7G). On a KO background, both KO T55I and KO R75W mutants have more abnormally myelinated fibers than do KO mice (Table 1, Fig. 7H). Whereas g-ratios were not significantly different between T55I and R75W and WT controls, or KO T55I and KO R75W and KO mice, axon profiles obtained from sciatic nerves at 8 months of age indicated a reduction in the number of large caliber (>7 μm diameter) fibers in nerves from all KO lines as well as in the R75W mutant on WT background (supplemental Fig. 7, available at www.jneurosci.org as supplemental material) (data not shown). Thus, the T55I and the R75W mutants may have deleterious effects beyond those of a null Gjb1 allele itself, and the R75W mutation appears to have a dominant effect.
Figure 7.
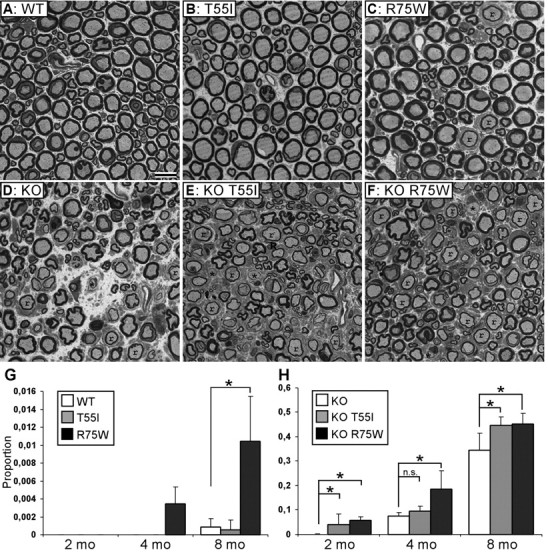
Cx32 mutant mice develop a progressive demyelinating peripheral neuropathy. A–F, Photomicrographs of semithin sections of femoral motor branches from 8-month-old WT (A), KO (D), as well as T55I and R75W Cx32 mutant mice on WT (B, C) or KO (E, F) background, as indicated. Myelinated axons appear normal in WT and T55I mice (A, B), whereas there are some remyelinated axons (r) in R75W mice (C). In all KO lines (D–F), there many remyelinated (r) and some demyelinated (*) axons. Scale bar, 10 μm. G, H, Quantitative analysis of abnormally myelinated axons in femoral nerves from 2-, 4-, and 8-month-old transgenic mice on WT background (G) and on KO background (H). The average proportion of abnormal fibers is shown; those data are shown in Table 1. On a WT background, the R75W but not the T55I mutant causes significant myelination defects compared with WT (G). All KO lines (H) have progressively increasing number of abnormal fibers with age; the R75W (significant at all ages) and the T55I mutants (significant at 2 and 8 months of age) have more abnormal fibers than do simple KO.
Figure 8.
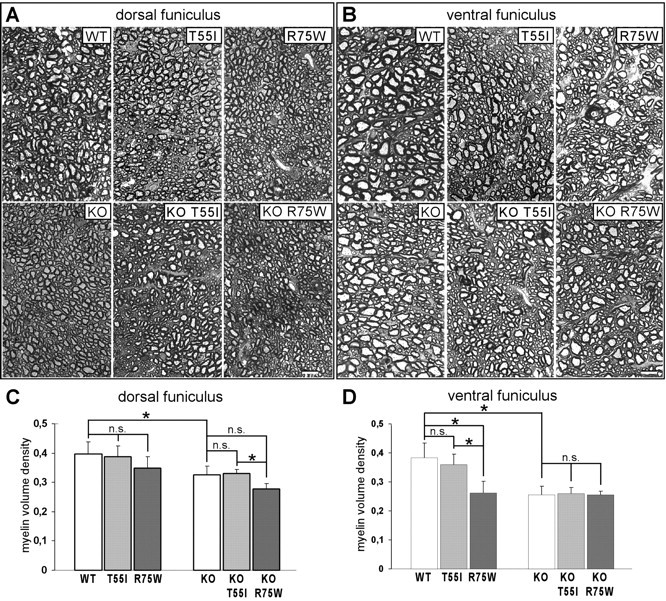
Spinal cord myelination defects in Cx32 mutant mice. A–D, Myelin volume density was measured in semithin sections from the dorsal columns (C) and ventral funiculus (D) of 8 month groups (n = 4 in each group); representative images of dorsal columns (A) and ventral funiculi (B) are shown. Note that myelin density is significantly reduced in all lines on KO background compared with WT in both white matter areas. Furthermore, the R75W mutants on WT background show reduced myelin volume density in both areas compared with WT or T55I mutants, more severe in the ventral funiculus (similar to the KO). There are no significant differences between KO and mutants on KO background except for reduced myelin volume density in the KO R75W mice compared with KO T55I mice in dorsal funiculus (see also Table 2). Scale bars, 10 μm.
Table 1.
Quantitative analysis of abnormally myelinated fibers in the femoral motor nerve from different age groups on wild-type and cx32-null background
| Genotype and age group | Demyelinated | Remyelinated | Total fibers counted | Proportion abnormal (average ± STDV) | Mann–Whitney test |
|---|---|---|---|---|---|
| WT background | |||||
| 2 mo WT (n = 4) | 0 | 0 | 1770 | 0 | |
| 2 mo T55I (n = 4) | 0 | 0 | 1268 | 0 | |
| 2 mo R75W (n = 4) | 0 | 0 | 1575 | 0 | |
| 4 mo WT (n = 3) | 0 | 0 | 1235 | 0 | |
| 4 mo T55I (n = 3) | 0 | 0 | 1380 | 0 | |
| 4 mo R75W (n = 3) | 2 | 3 | 1383 | 0.0034 ± 0.002 | |
| 8 mo WT (n = 4) | 1 | 1 | 2411 | 0.0008 ± 0.001 | |
| 8 mo T55I (n = 4) | 0 | 1 | 2057 | 0.0005 ± 0.001 | p = 0.86 (WT) |
| 8 mo R75W (n = 4) | 4 | 15 | 1951 | 0.0104 ± 0.005 | p = 0.029* (WT) |
| p = 0.03* (T55I) | |||||
| Cx32-null background | |||||
| 2 mo KO (n = 4) | 0 | 1 | 1336 | 0.0006 ± 0.001 | |
| 2 mo KO T55I (n = 4) | 16 | 32 | 1194 | 0.0403 ± 0.044 | p = 0.026* (KO) |
| 2 mo KO R75W (n = 3) | 19 | 41 | 974 | 0.0575 ± 0.015 | p = 0.04* (KO) |
| p = 0.37 (KO T55I) | |||||
| 4 mo KO (n = 4) | 36 | 78 | 1443 | 0.0763 ± 0.014 | |
| 4 mo KO T55I (n = 3) | 25 | 62 | 914 | 0.0968 ± 0.020 | p = 0.21 (KO) |
| 4 mo KO R75W (n = 4) | 70 | 244 | 1715 | 0.1858 ± 0.075 | p = 0.03* (KO) |
| p = 0.11 (KO T55I) | |||||
| 8 mo KO (n = 4) | 77 | 454 | 1545 | 0.3439 ± 0.070 | |
| 8 mo KO T55I (n = 4) | 74 | 600 | 1514 | 0.4453 ± 0.035 | p = 0.03* (KO) |
| 8 mo KO R75W (n = 3) | 54 | 435 | 1085 | 0.4523 ± 0.044 | p = 0.05* (KO) |
| p = 0.86 (KO T55I) |
The average proportion of abnormally myelinated fibers counted in all mice in each genotype and age group (n = number of animals studied per group), as well as p values of statistical comparison between transgenic mice of the same background and age with Mann–Whitney test, are indicated.
* indicates significant results.
Light and electron microscopic examination of the optic nerve and lumbar spinal cord at 2, 4, and 8 months, did not reveal any abnormalities of CNS myelin (supplemental Fig. 8A,B, available at www.jneurosci.org as supplemental material) (data not shown). Even the g-ratios of 8-month-old mice showed no significant differences between genotypes in these two CNS areas (supplemental Fig. 8C, available at www.jneurosci.org as supplemental material) (data not shown). Based on the work of Sutor et al. (2000), we also measured the volume density of myelin in the dorsal and ventral funiculus in sections of lumbar spinal cord from all 8-month-old mice in each line. This analysis showed that volume density of myelinated fibers was significantly reduced in KO mice as well as in KO T55I and KO R75W transgenic mice compared with WT mice (Table 2; Fig. 8). Furthermore, the volume density of R75W mutants on a WT background was significantly reduced compared with WT or T55I mutants on WT background; in the ventral funiculus, this reduction approached that measured in the KO lines. At 4-months, however, there were no statistically significant differences in the myelin volume density between any of these genotypes in the spinal cord (data not shown). Furthermore, we found no significant differences in myelin volume density of the optic nerve (which has low expression of Cx32) between 8-month-old mice from all lines either in a WT or a KO background (data not shown). Together, these results indicate that a loss of Cx32 in oligodendrocytes results in progressive myelination defects in white matter areas in which Cx32 expression is physiologically prominent.
Table 2.
Quantitative analysis of spinal cord myelination in 8-month-old mice on wild type and cx32-null background
| Spinal cord dorsal funiculus |
Spinal cord ventral funiculus |
|||||||
|---|---|---|---|---|---|---|---|---|
| Volume density of myelinated fibers | Volume density of myelin | Volume density of myelinated fibers | Volume density of myelin | |||||
| WT (n = 4) | 0.6944 ± 0.067 | 0.3978 ± 0.041 | 0.6619 ± 0.076 | 0.3820 ± 0.052 | ||||
| T55I (n = 4) | 0.6818 ± 0.033 | p = 0.88 (WT) | 0.3882 ± 0.036 | p = 0.88 (WT) | 0.6010 ± 0.045 | p = 0.19 (WT) | 0.3593 ± 0.037 | p = 0.66 (WT) |
| R75W (n = 4) | 0.6199 ± 0.069 | p = 0.19 (WT)p = 0.19 (T55I) | 0.3497 ± 0.038 | p = 0.11 (WT)p = 0.38 (T55I) | 0.4748 ± 0.053 | p = 0.03* (WT)p = 0.03* (T55I) | 0.2623 ± 0.039 | p = 0.03* (WT) p = 0.03* (T55I) |
| KO (n = 4) | 0.5726 ± 0.014 | p = 0.03* (WT) | 0.3263 ± 0.028 | p = 0.03* (WT) | 0.4997 ± 0.055 | p = 0.03* (WT) | 0.2549 ± 0.030 | p = 0.03* (WT) |
| KO T55I (n = 4) | 0.5584 ± 0.021 | p = 0.31 (KO) | 0.3297 ± 0.014 | p = 1.00 (KO) | 0.5074 ± 0.032 | p = 0.88 (KO) | 0.2603 ± 0.019 | p = 1.0 (KO) |
| KO R75W (n = 4) | 0.5315 ± 0.046 | p = 0.19 (KO)p = 0.56 (KO T55I) | 0.2727 ± 0.017 | p = 0.06 (KO)p = 0.03* (KO T55I) | 0.5012 ± 0.044 | p = 0.88 (KO)p = 0.88 (KO T55I) | 0.2544 ± 0.015 | p = 0.88 (KO) p = 0.77 (KO T55I) |
The average ± SD results for each genotype group are shown (numbers of mice per group are indicated). p values obtained with the Mann–Whitney test are shown for all comparisons (the group with which comparison was made is indicated in parentheses).
* indicates significant results.
Discussion
We have generated the first transgenic mice that express Cx32 mutants in oligodendrocytes (in addition to Schwann cells), and find pathological changes in both cell types. In myelinating Schwann cells, R75W had dominant effects on endogenous Cx32 resulting in a mild demyelination in a WT background and a more severe demyelinating neuropathy in a KO background. In oligodendrocytes, the R75W but not the T55I mutant had subtle effects on CNS myelin in WT background, and both mutants had no additional effects in a KO background. Like a null allele of Gjb1, neither the T55I nor the R75W mutant appears to affect Cx29 or Cx47. Thus, the loss of Cx32 function appears to be the main effect of the T55I and R75W mutants, in both the PNS and the CNS.
T55I and R75W are mislocalized but do not have trans-dominant interactions with Cx29 or Cx47
We found that T55I and R75W are mislocalized in myelinating Schwann cells and oligodendrocytes. Thus, like the R142W, G280S, and S281x mutants (Huang et al., 2005; Jeng et al., 2006), the localization of T55I and R75W in heterologous cells (Kleopa et al., 2002; Yum et al., 2002) predicts their localization in myelinating Schwann cells, and as we show here (for T55I and R75W), in oligodendrocytes, too. In heterologous cells, many Cx32 mutants appear to be retained in the ER or Golgi (Kleopa et al., 2002; Yum et al., 2002). ER-retained mutants are probably misfolded, and are subsequently degraded by proteosomes (VanSlyke et al., 2000; Kleopa et al., 2002; Thomas et al., 2004). Why some mutants appear to be retained in the Golgi, however, is unknown, as the Golgi is not known to modify Cx32. Cx43 oligomerizes in the trans-Golgi network (Musil and Goodenough, 1993), whereas Cx32 can form hexamers in the ER (Maza et al., 2005). However, Cx32 is also prenylated (Huang et al., 2005), and this posttranslational modification is accomplished on the cytoplasmic surface of the ER and Golgi (Silvius, 2002; Wright and Philips, 2006).
Based on the finding that mice lacking both Cx32 and Cx47 have a much more severe phenotype than mice lacking either one alone (Menichella et al., 2003; Odermatt et al., 2003) we hypothesized that the Cx32 mutants that are associated with transient CNS phenotypes interact directly with Cx47. Further, because these “CNS mutants” appear to be retained in the ER or Golgi (Kleopa et al., 2002; Yum et al., 2002) we thought that they would interact directly with Cx47 and lead to its mislocalization in the ER/Golgi and diminish its localization at GJs. Neither the R75W mutant nor the T55I mutant, however, appeared to impair the expression of Cx29 or Cx47 in oligodendrocytes, or the expression of Cx29 in Schwann cells, extending the previous finding that R142W had no effect on the expression of Cx29 in Schwann cells (Jeng et al., 2006). Furthermore, our recent in vitro studies failed to show dominant effects of the same Cx32 mutants on Cx31.3, the human ortholog of Cx29 (Sargiannidou et al., 2008). Whereas we could show that mostly the R75W had dominant effects on WT Cx32, this effect is not clinically relevant, since only one GJB1 allele is expressed in each cell (Scherer et al., 1998). These results suggest that T55I and R75W cause loss of Cx32 function and do not have trans-dominant effects on Cx47 or Cx31.3.
The lack of trans-dominant effects of Cx32 mutants is in keeping with the evidence that Cx29 and Cx32 are distinctly localized in myelinating glia (Kleopa et al., 2004), and do not appear to interact in vitro (Ahn et al., 2008). Similarly, Cx32 and Cx47 show only partial overlap in oligodendrocytes (Kleopa et al., 2004) and to not appear to interact in vitro (unpublished observations). The lack of interaction between Cx32 and Cx29 is in keeping with the findings that Cx29 is normally localized in Cx32 KO mice (Meier et al., 2004) and Cx32 is normally localized in Cx29 KO (Altevogt and Paul, 2004). Here, we do not find an altered expression of Cx29 or Cx47 in the CNS of Cx32 KO mice, in keeping with previous observations (Nagy et al., 2003).
The best examples of trans-dominant effects between a mutant connexin and a different WT connexin are for those connexins that normally oligomerize to form heteromeric hemichannels, such as Cx31 and Cx30.3 in the skin (Plantard et al., 2003) and Cx26 and Cx30 in the ear (Forge et al., 2003; Marziano et al., 2003; Yum et al., 2007). Trans-dominant selectivity of different Cx26 mutants (Thomas et al., 2004) and the cell-specific pattern of connexin expression (Das Sarma et al., 2001) may account for some of the complicated phenotypes observed with dominant Cx26 mutants associated with hearing loss and various skin disorders. Although there are examples of trans-dominant interactions between compatible connexins forming heteromeric hemichannels, it remains to be shown that a disease results from dominant interactions of a mutant with a different WT connexin that does not normally interact.
Pathological effects of T55I and R75W in the PNS and CNS
A demyelinating neuropathy was the major pathological finding in our transgenic mice. In Schwann cells, its effects were similar to those described in transgenic mice expressing the R142W mutant exclusively in Schwann cells (Jeng et al., 2006): a demyelinating neuropathy that predominantly affects motor axons, starting after 2 months of age and progressing with time, but always milder than that seen in KO mice (Anzini et al., 1997; Scherer et al., 1998; Scherer et al., 2005). In a KO background, the R75W mutant appeared to be retained in the perinuclear cytoplasm, and did not accumulate in the incisures and paranodes; in a WT background, the R75W resulted in diminished Cx32 (presumably WT Cx32) in the incisures and paranodes. Both of these effects were found for R142W (Jeng et al., 2006).
The CNS myelin defects, in contrast, were subtle. In both the T55I and R75W mutants in a KO background, and to a lesser degree in R75W mutants in WT background, the chief finding was a diminished myelinated fiber and myelin volume density, particularly in white matter areas with prominent Cx32 expression, such as the ventral and dorsal funiculus of the spinal cord (Kleopa et al., 2004). Similar abnormalities were previously described in the neocortex of Cx32 KO mice that were >6 months old (Sutor et al., 2000). How the loss of Cx32 function leads to these CNS myelination defects remains unknown. They could result from a cell autonomous role of Cx32 in the myelin sheath itself (e.g., transport of metabolites used to synthesize the myelin sheath) or they could depend on astrocyte-oligodendrocyte interactions that are mediated by heterotypic Cx30:Cx32 channels (Nagy et al., 2003; Altevogt and Paul, 2004). We did not detect any frank pathological changes in CNS myelin or g-ratio alterations in our mutant mice, in keeping with previous observations in the neocortex (Sutor et al., 2000) and in the optic nerve (Scherer et al., 1995) of Cx32 KO mice. Reduced myelin density may result from a combination of changes including a shift to smaller diameter axons, mild reduction of myelin thickness, and mild reduction in the number of myelinated axons (Sutor et al., 2000). During development, Cx32 expression in the CNS starts around postnatal day 5–7 and peaks around postnatal day 20 (Dermietzel et al., 1989; Scherer et al., 1995; Nadarajah et al., 1997). This course matches that of oligodendrocyte differentiation and onset of myelination around postnatal day 10 (Parnavelas et al., 1983), suggesting that Cx32 may be involved in the maturation of CNS myelin sheaths. However, we found significant abnormalities in Cx32 KO mice at 8 but not at 4 months of age, which indicates that degenerative mechanisms likely contribute to these CNS myelination defects.
Although less severe, the PNS and CNS alterations caused by the R75W mutant in WT background are similar to those found in KO mice, and indicate that this mutant has dominant-negative effects on WT Cx32. The immunostaining of Cx32 protein in the PNS and CNS directly support this interpretation. The immunoblot analysis indicates that the T55I mutant is degraded to a much higher degree than the R75W mutant, thus the retained R75W mutant is more likely to interact with WT Cx32 and reduce its trafficking to the cell membrane. The exacerbated demyelinating neuropathy found in T55I KO and especially in R75W KO mice, however, cannot be the result of a dominant-negative effect on Cx32. The R142W mutant has similar effects (Jeng et al., 2006), but how R75W and R142W exacerbate demyelination in KO mice remains unknown. Impairment of Golgi dynamics caused by retention is unlikely since in the R142W mutants (Jeng et al., 2006) as well as in our mice no obvious effects were detected in the synthesis or trafficking of other proteins, including Cx29. Another consideration is that overexpression of the transgene may have nonphysiological toxic effects, and the effects of similarly expressed WT Cx32 in KO oligodendrocytes remain to be determined. Overexpression of WT Cx32 in Schwann cells at levels 12-fold higher than the endogenous expression caused myelin splitting without demyelination, but these changes were not seen in mice with six- to sevenfold expression levels (Jeng et al., 2006). Thus, it is unlikely that overexpression of the transgene in our mice at levels two- to threefold of the endogenous could have contributed significantly to the pathology we describe in addition to mutation-specific effects.
Clinical correlations
Our results do not support the idea that dominant-negative effects play a role in the pathogenesis of CMT1X, either in the peripheral nerves or in the CNS. This conclusion is in keeping with results of a large clinical study of CMT1X patients with various mutations, including the deletion of the GJB1 gene, who had a similar phenotype, suggesting that most GJB1 mutations cause neuropathy through loss of normal Cx32 function (Shy et al., 2007). Furthermore, the relative severity of demyelinating or axonal features in peripheral nerve biopsies are not associated with particular GJB1 mutations (Hahn et al., 2000; Nakagawa et al., 2001; Hattori et al., 2003). If GJB1 mutations do not have clinically relevant dominant effects, then treatment for CMT1X may be feasible with gene replacement strategies. The mouse models presented here provide a useful tool to test such therapies.
Footnotes
This work was supported by the National Multiple Sclerosis Society (United States) (Grant RG3457A2/1 to K.A.K.), the Cyprus Research Promotion Foundation (Grant Health/0506/04 to K.A.K.), the Cyprus Telethon (grant to K.A.K.), and the National Institutes of Health (Grant RO1 NS55284 to S.S.S.). We thank Dr. Vittorio Gallo for the Cnp promoter construct, Prof. Klaus Willecke for Gjb1-knock-out mice, and Thalia Michael for technical assistance.
References
- Ahn M, Lee J, Gustafsson A, Enriquez A, Lancaster E, Sul JY, Haydon PG, Paul DL, Huang Y, Abrams CK, Scherer SS. Cx29 and Cx32, two connexins expressed by myelinating glia, do not interact and are functionally distinct. J Neurosci Res. 2008;86:992–1006. doi: 10.1002/jnr.21561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt BM, Paul DL. Four classes of intercellular channels between glial cells in the CNS. J Neurosci. 2004;24:4313–4323. doi: 10.1523/JNEUROSCI.3303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL. Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci. 2002;22:6458–6470. doi: 10.1523/JNEUROSCI.22-15-06458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzini P, Neuberg DH, Schachner M, Nelles E, Willecke K, Zielasek J, Toyka KV, Suter U, Martini R. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin32. J Neurosci. 1997;17:4545–4551. doi: 10.1523/JNEUROSCI.17-12-04545.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähr M, Andres F, Timmerman V, Nelis ME, Van Broeckhoven C, Dichgans J. Central visual, acoustic, and motor pathway involvement in a Charcot-Marie-Tooth family with an Asn205Ser mutation in the connexin32 gene. J Neurol Neurosurg Psychiatry. 1999;66:202–206. doi: 10.1136/jnnp.66.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- Bugiani M, Al Shahwan S, Lamantea E, Bizzi A, Bakhsh E, Moroni I, Balestrini MR, Uziel G, Zeviani M. GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy. Neurology. 2006;67:273–279. doi: 10.1212/01.wnl.0000223832.66286.e4. [DOI] [PubMed] [Google Scholar]
- Das Sarma J, Meyer RA, Wang F, Abraham V, Lo CW, Koval M. Multimeric connexin interactions prior to the trans-Golgi network. J Cell Sci. 2001;114:4013–4024. doi: 10.1242/jcs.114.22.4013. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Traub O, Hwang TK, Beyer E, Bennett MV, Spray DC, Willecke K. Differential expression of three gap junction proteins in developing and mature brain tissues. Proc Natl Acad Sci U S A. 1989;86:10148–10152. doi: 10.1073/pnas.86.24.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes SM, Walcott JL, Wexler TL, Scherer SS, Fischbeck KH. Altered trafficking of mutant connexin32. J Neurosci. 1997;17:9077–9084. doi: 10.1523/JNEUROSCI.17-23-09077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forge A, Marziano NK, Casalotti SO, Becker DL, Jagger D. The inner ear contains heteromeric channels composed of cx26 and cx30 and deafness-related mutations in cx26 have a dominant negative effect on cx30. Cell Commun Adhes. 2003;10:341–346. doi: 10.1080/cac.10.4-6.341.346. [DOI] [PubMed] [Google Scholar]
- Gravel M, Di Polo A, Valera PB, Braun PE. Four-kilobase sequence of the mouse CNP gene directs spatial and temporal expression of lacZ in transgenic mice. J Neurosci Res. 1998;53:393–404. doi: 10.1002/(SICI)1097-4547(19980815)53:4<393::AID-JNR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Hahn AF, Ainsworth PJ, Naus CCG, Mao J, Bolton CF. Clinical and pathological observations in men lacking the gap junction protein connexin 32. Muscle Nerve. 2000;23:S39–S48. [PubMed] [Google Scholar]
- Study Group for Hereditary Neuropathy in Japan. Hattori N, Yamamoto M, Yoshihara T, Koike H, Nakagawa M, Yoshikawa H, Ohnishi A, Hayasaka K, Onodera O, Baba M, Yasuda H, Saito T, Nakashima K, Kira J, Kaji R, Oka N, Sobue G. Demyelinating and axonal features of Charcot-Marie-Tooth disease with mutations of myelin-related proteins (PMP22, MPZ and Cx32): a clinicopathological study of 205 Japanese patients. Brain. 2003;126:134–151. doi: 10.1093/brain/awg012. [DOI] [PubMed] [Google Scholar]
- Huang Y, Sirkowski EE, Stickney JT, Scherer SS. Prenylation-defective human connexin32 mutants are normally localized and function equivalently to wild-type connexin32 in myelinating Schwann cells. J Neurosci. 2005;25:7111–7120. doi: 10.1523/JNEUROSCI.1319-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeng LJ, Balice-Gordon RJ, Messing A, Fischbeck KH, Scherer SS. The effects of a dominant connexin32 mutant in myelinating Schwann cells. Mol Cell Neurosci. 2006;32:283–298. doi: 10.1016/j.mcn.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Kamasawa N, Sik A, Morita M, Yasumura T, Davidson KG, Nagy JI, Rash JE. Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: implications for ionic homeostasis and potassium siphoning. Neuroscience. 2005;136:65–86. doi: 10.1016/j.neuroscience.2005.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleopa KA, Scherer SS. Molecular genetics of X-linked Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;8:107–122. doi: 10.1385/nmm:8:1-2:107. [DOI] [PubMed] [Google Scholar]
- Kleopa KA, Yum SW, Scherer SS. Cellular mechanisms of connexin32 mutations associated with CNS manifestations. J Neurosci Res. 2002;68:522–534. doi: 10.1002/jnr.10255. [DOI] [PubMed] [Google Scholar]
- Kleopa KA, Orthmann JL, Enriquez A, Paul DL, Scherer SS. Unique distribution of gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia. 2004;47:346–357. doi: 10.1002/glia.20043. [DOI] [PubMed] [Google Scholar]
- Kleopa KA, Zamba-Papanicolaou E, Alevra X, Nicolaou P, Georgiou DM, Hadjisavvas A, Kyriakides T, Christodoulou K. Phenotypic and cellular expression of two novel connexin32 mutations causing CMT1X. Neurology. 2006;66:396–402. doi: 10.1212/01.wnl.0000196479.93722.59. [DOI] [PubMed] [Google Scholar]
- Li X, Ionescu AV, Lynn BD, Lu S, Kamasawa N, Morita M, Davidson KG, Yasumura T, Rash JE, Nagy JI. Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience. 2004;126:611–630. doi: 10.1016/j.neuroscience.2004.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marziano NK, Casalotti SO, Portelli AE, Becker DL, Forge A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum Mol Genet. 2003;12:805–812. doi: 10.1093/hmg/ddg076. [DOI] [PubMed] [Google Scholar]
- Maza J, Das Sarma J, Koval M. Defining a minimal motif required to prevent connexin oligomerization in the endoplasmic reticulum. J Biol Chem. 2005;280:21115–21121. doi: 10.1074/jbc.M412612200. [DOI] [PubMed] [Google Scholar]
- Meier C, Dermietzel R, Davidson KG, Yasumura T, Rash JE. Connexin32-containing gap junctions in Schwann cells at the internodal zone of partial myelin compaction and in Schmidt–Lanterman incisures. J Neurosci. 2004;24:3186–3198. doi: 10.1523/JNEUROSCI.5146-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci. 2006;26:10984–10991. doi: 10.1523/JNEUROSCI.0304-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1065–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Jones AM, Evans WH, Parnavelas JG. Differential expression of connexins during neocortical development and neuronal circuit formation. J Neurosci. 1997;17:3096–3111. doi: 10.1523/JNEUROSCI.17-09-03096.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Ionescu AV, Lynn BD, Rash JE. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: implications from normal and connexin32 knockout mice. Glia. 2003;44:205–218. doi: 10.1002/glia.10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M, Takashima H, Umehara F, Arimura K, Miyashita F, Takenouchi N, Matsuyama W, Osame M. Clinical phenotype in X-linked Charcot-Marie-Tooth disease with an entire deletion of the connexin 32 coding sequence. J Neurol Sci. 2001;185:31–37. doi: 10.1016/s0022-510x(01)00454-3. [DOI] [PubMed] [Google Scholar]
- Nelles E, Bützler C, Jung D, Temme A, Gabriel HD, Dahl U, Traub O, Stümpel F, Jungermann K, Zielasek J, Toyka KV, Dermietzel R, Willecke K. Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9565–9570. doi: 10.1073/pnas.93.18.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson G, Corbett A. Slowing of central conduction in X-linked Charcot-Marie-Tooth neuropathy shown by brain auditory evoked responses. J Neurol Neurosurg Psychiatry. 1996;61:43–46. doi: 10.1136/jnnp.61.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson GA, Yeung L, Corbett A. Efficient neurophysiological selection of X-linked Charcot-Marie-Tooth families. Neurology. 1998;51:1412–1416. doi: 10.1212/wnl.51.5.1412. [DOI] [PubMed] [Google Scholar]
- Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Büssow H, Schilling K, Steinhäuser C, Willecke K. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23:4549–4559. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Ri Y, Bennett MV, Trexler EB, Verselis VK, Bargiello TA. Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron. 1997;19:927–938. doi: 10.1016/s0896-6273(00)80973-3. [DOI] [PubMed] [Google Scholar]
- Omori Y, Mesnil M, Yamasaki H. Connexin 32 mutations from X-linked Charcot-Marie-Tooth disease patients: functional defects and dominant negative effects. Mol Biol Cell. 1996;7:907–916. doi: 10.1091/mbc.7.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthmann-Murphy JL, Enriquez AD, Abrams CK, Scherer SS. Loss-of-function connexin47 mutations cause Pelizaeus-Merzbacher-like disease. Mol Cell Neurosci. 2007a;34:629–641. doi: 10.1016/j.mcn.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthmann-Murphy JL, Freidin M, Fischer E, Scherer SS, Abrams CK. Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J Neurosci. 2007b;27:13949–13957. doi: 10.1523/JNEUROSCI.3395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panas M, Kalfakis N, Karadimas C, Vassilopoulos D. Episodes of generalized weakness in two sibs with the C164T mutation of the connexin 32 gene. Neurology. 2001;57:1906–1908. doi: 10.1212/wnl.57.10.1906. [DOI] [PubMed] [Google Scholar]
- Parnavelas JG, Luder R, Pollard SG, Sullivan K, Lieberman AR. A qualitative and quantitative ultrastructural study of glial cells in the developing visual cortex of the rat. Philos Trans R Soc Lond B Biol Sci. 1983;301:55–84. doi: 10.1098/rstb.1983.0022. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Garbern JY, Hoban TF, Krajewski KM, Lewis RA, Fischbeck KH, Grossman RI, Lenkinski R, Kamholz JA, Shy ME. Transient central nervous system white matter abnormality in X-linked Charcot-Marie-Tooth disease. Ann Neurol. 2002;52:429–434. doi: 10.1002/ana.10305. [DOI] [PubMed] [Google Scholar]
- Plantard L, Huber M, Macari F, Meda P, Hohl D. Molecular interaction of connexin 30.3 and connexin 31 suggests a dominant-negative mechanism associated with erythrokeratodermia variabilis. Hum Mol Genet. 2003;12:3287–3294. doi: 10.1093/hmg/ddg364. [DOI] [PubMed] [Google Scholar]
- Rash JE, Yasumura T, Dudek FE, Nagy JI. Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J Neurosci. 2001;21:1983–2000. doi: 10.1523/JNEUROSCI.21-06-01983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargiannidou I, Ahn M, Enriquez AD, Peinado A, Reynolds R, Abrams C, Scherer SS, Kleopa KA. Human oligodendrocytes express Cx31.3: function and interactions with Cx32 mutants. Neurobiol Dis. 2008;30:221–233. doi: 10.1016/j.nbd.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Deschênes SM, Xu YT, Grinspan JB, Fischbeck KH, Paul DL. Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci. 1995;15:8281–8294. doi: 10.1523/JNEUROSCI.15-12-08281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ. Connexin32-null mice develop a demyelinating peripheral neuropathy. Glia. 1998;24:8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8::aid-glia2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Messing A, Willecke K, Fischbeck KH, Jeng LJ. Transgenic expression of human connexin32 in myelinating Schwann cells prevents demyelination in connexin32-null mice. J Neurosci. 2005;25:1550–1559. doi: 10.1523/JNEUROSCI.3082-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shy ME, Siskind C, Swan ER, Krajewski KM, Doherty T, Fuerst DR, Ainsworth PJ, Lewis RA, Scherer SS, Hahn AF. CMT1X phenotypes represent loss of GJB1 gene function. Neurology. 2007;68:849–855. doi: 10.1212/01.wnl.0000256709.08271.4d. [DOI] [PubMed] [Google Scholar]
- Silvius JR. Mechanisms of Ras protein targeting in mammalian cells. J Membr Biol. 2002;190:83–92. doi: 10.1007/s00232-002-1026-4. [DOI] [PubMed] [Google Scholar]
- Sutor B, Schmolke C, Teubner B, Schirmer C, Willecke K. Myelination defects and neuronal hyperexcitability in the neocortex of connexin 32-deficient mice. Cereb Cortex. 2000;10:684–697. doi: 10.1093/cercor/10.7.684. [DOI] [PubMed] [Google Scholar]
- Tang Y, Nyengaard JR. A stereological method for estimating the total length and size of myelin fibers in human brain white matter. J Neurosci Methods. 1997;73:193–200. doi: 10.1016/s0165-0270(97)02228-0. [DOI] [PubMed] [Google Scholar]
- Taylor RA, Simon EM, Marks HG, Scherer SS. The CNS phenotype of X-linked Charcot-Marie-Tooth disease: more than a peripheral problem. Neurology. 2003;61:1475–1478. doi: 10.1212/01.wnl.0000095960.48964.25. [DOI] [PubMed] [Google Scholar]
- Thomas T, Telford D, Laird DW. Functional domain mapping and selective trans-dominant effects exhibited by Cx26 disease-causing mutations. J Biol Chem. 2004;279:19157–19168. doi: 10.1074/jbc.M314117200. [DOI] [PubMed] [Google Scholar]
- Uhlenberg B, Schuelke M, Rüschendorf F, Ruf N, Kaindl AM, Henneke M, Thiele H, Stoltenburg-Didinger G, Aksu F, Topaloðlu H, Nürnberg P, Hübner C, Weschke B, Gärtner J. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004;75:251–260. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanSlyke JK, Deschenes SM, Musil LS. Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol Biol Cell. 2000;11:1933–1946. doi: 10.1091/mbc.11.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Güldenagel M, Deutsch U, Söhl G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2002;383:725–737. doi: 10.1515/BC.2002.076. [DOI] [PubMed] [Google Scholar]
- Wright LP, Philips MR. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res. 2006;47:883–891. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- Yuan X, Chittajallu R, Belachew S, Anderson S, McBain CJ, Gallo V. Expression of the green fluorescent protein in the oligodendrocyte lineage: a transgenic mouse for developmental and physiological studies. J Neurosci Res. 2002;70:529–545. doi: 10.1002/jnr.10368. [DOI] [PubMed] [Google Scholar]
- Yum SW, Kleopa KA, Shumas S, Scherer SS. Diverse trafficking abnormalities of Connexin32 mutants causing CMTX. Neurobiol Dis. 2002;11:43–52. doi: 10.1006/nbdi.2002.0545. [DOI] [PubMed] [Google Scholar]
- Yum SW, Zhang J, Valiunas V, Kanaporis G, Brink PR, White TW, Scherer SS. Human connexin26 and connexin30 form functional heteromeric and heterotypic channels. Am J Physiol Cell Physiol. 2007;293:C1032–C1048. doi: 10.1152/ajpcell.00011.2007. [DOI] [PubMed] [Google Scholar]