

Abstract
AIMS
To investigate the influence of food intake on the bioavailability and pharmacodynamic effects of salmon calcitonin (sCT).
METHODS
A single-blind, randomized, partly placebo-controlled study was conducted in 36 healthy postmenopausal female volunteers aged 62–74 years. The influence of food intake on oral dosing with 0.8 mg of sCT at 22.00 h was evaluated for a (i) predose meal at 18.00 h, (ii) predose meal at 20.00 h, (iii) predose meal at 21.00 h, (iv) postdose meal at 22.10 h, (v) no meal, and (vi) meal at 20.00 h and placebo at 22.00 h. Study biomarkers were plasma sCT levels and changes in the bone resorption marker CTX-I (C-terminal telopeptide of collagen type I).
RESULTS
The predose meal at 18.00 and 21.00 h significantly decreased relative oral bioavailability of sCT to 26% [95% confidence interval (CI) 0.09, 0.73 and 0.09, 0.75, P= 0.009 and P= 0.01]. The meal consumed 10 min after dosing decreased the oral bioavailability of sCT to 59% (95% CI 0.21, 1.68), although nonsignificant (P= 0.48). This decreased bioavailability led to lower relative suppression of serum CTX-I, with an AUC of the 4-h efficacy response of −91%–×–hours for those receiving a meal at 18.00 h, compared with −238%–×–hours for fasting subjects. The Dunnett-adjusted difference between these two treatment sequences was 147%–×–hours (95% CI 68, 225) (P= 0.0003). The AUC was comparable among fasting subjects and those consuming a meal 10 min after dosing.
CONCLUSIONS
Postprandial dosing may limit the bioavailability of orally administered sCT. Maximal benefit can be achieved by dosing at least 10 min prior to meal time.
Keywords: bone resorption, calcitonin, CTX-I, oral, pharmacology
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
No orally bioavailable peptides are currently approved by the US Food and Drug Administration or the European Medicines Agency.
Attempts have been made to develop an oral formulation of salmon calcitonin (sCT).
However, the effect of food intake on the bioavailability and efficacy of oral peptide formulations has not been systematically investigated.
WHAT THIS STUDY ADDS
-
We provide evidence that oral doses of sCT may be optimally received in the fasting state or prior to meal intake.
Fasting or preprandial dosing led to improved bioavailability and effect on pharmacodynamic biomarkers of efficacy.
The results may aid other researchers involved in the formulation of oral peptides.
Introduction
Calcitonin, discovered more than 40 years ago [1, 2], exerts potent anti-resorptive effects on bone that are mediated by direct binding of calcitonin to its receptor on osteoclasts [3–6]. Calcitonin is a small, 32-amino-acid peptide hormone produced by parafollicular cells (C cells) in the thyroid gland [7] and secreted in response to excess calcium in serum [8]. Various exogenous sources of calcitonin exist, of which salmon is among the most potent [7]. Calcitonin is approved for the treatment of osteoporosis and other diseases involving accelerated bone turnover [9–11]. Calcitonin administration has been limited to either the subcutaneous or intranasal route [9].
Major obstacles to oral delivery of proteins include the high acid content of the digestive tract and the extensive array of proteases present there. As a result, degradation of most peptides occurs before absorption into the bloodstream. Another barrier to uptake of many macromolecules is poor absorption through biological membranes [12, 13]. An optimal formulation of a medicinal peptide would facilitate transport through the acidic compartment as well as influx across the intestinal membrane.
Attempts have been made to formulate calcitonin for oral administration [12–23]. 8-(N-2-hydroxy-5-chloro-benzoyl)-amino-caprylic acid (5-CNAC), a molecule based on the Eligen technology, has proven useful preclinically and clinically in combination with calcitonin to facilitate calcitonin absorption from the intestinal lumen into the bloodstream [9, 10, 24]. Eligen technology employs low-molecular-weight compounds (termed drug delivery agents or carriers) that interact weakly and noncovalently with proteins, increasing their lipophilicity and, consequently, their ability to cross the gastrointestinal epithelium [25]. The carrier 5-CNAC makes drug available systemically by means of transcellular absorption, a common drug absorption pathway, without compromising the integrity of the intestinal epithelium [26–28]. The 5-CNAC molecule interacts with calcitonin without changing it and forms an insoluble entity at low pH values, thereby reducing degradation by digestive tract peptidases [25]. In the upper part of the intestine at higher pH, the complex dissolves and facilitates intestinal uptake over the nonpolar biological membrane [25]. This oral formulation of 5-CNAC in combination with salmon calcitonin (sCT) has been demonstrated to be safe and efficacious in a 3-month Phase II study in postmenopausal women [9].
Previous studies have indicated that the bioavailability of some drugs is affected by meal timing [29–32] and differs in the fed and fasting states. Small proteins/peptides have been investigated to a much lesser extent than small molecules for the effect of meal timing on oral delivery, and presently there are no orally available peptides approved by the US Food and Drug Administration or the European Medicines Agency.
Bone resorption can be assed by biochemical markers. Bone resorption by osteoclasts is mainly mediated by the cysteine protease cathepsin K degrading collagen type I, which is the major protein in bone [33]. The protease activity of cathepsin K results in a specific degradation fragment of collagen type I, CTX-I (C-terminal telo-peptide of collagen type I) [33, 34]. CTX-I fragments have been extensively used as a surrogate measure of bone resorption for in vitro, preclinical and clinical studies [33, 35].
Because of the short half-life of calcitonin in serum, one potential path to optimizing the clinical benefits of calcitonin could be to administer treatment when bone resorption reaches maximal levels, i.e. during the evening [9, 29, 36]. Diurnal variation is a principal parameter of bone turnover, in which postprandial decreases in bone resorption are observed [36]. Bone resorption during the night may account for >75% of total resorbed calcium. However, evening dosing, rather than morning dosing in the fasting state, may introduce the potential for food–drug interactions. Presently, it is unknown how food intake may affect the pharmacokinetics and pharmacodynamics of sCT.
The aim of the current study was to investigate the influence of food intake on the absorption and pharmacodynamic effects of sCT, as measured by an effect on pharmacodynamic biomarkers of efficacy, CTX-I, in addition to levels of sCT in plasma.
Materials and methods
Drug substance
SMC021 A/C is an oral formulation of sCT. The investigational drug consisted of 0.8 mg of recombinant sCT and 200 mg of 5-CNAC, a unimolecular enhancer of gastrointestinal peptide absorption developed by Emisphere Technology, Inc. (Cedar Knolls, NJ, USA). The sCT-5-CNAC formulation was provided by Novartis (Basel, Switzerland).
Study population
Healthy, ambulatory female volunteers aged 55–75 years who had undergone natural or surgical menopause at least 5 years before entering the study were eligible for participation. Subjects were also required not to have diseases or to be taking medications known to affect bone metabolism.
Study design
A single-blind, randomized, partly placebo-controlled study was conducted to evaluate the effect of dosing with 0.8 mg of recombinant sCT administered at 22.00 h and the influence of food intake. Each subject was randomly assigned to two dosing sessions, separated by a wash-out interval of ≥48 h. The times of dosing and meal intake are shown in Table 1. The study followed a balanced, cross-over design, with two subjects allocated to each of the following treatment sequences: i–iv; iv–i; i–v; v–i; i–vi; vi–i; ii–iv; iv–ii; ii–v; v–ii; ii–vi; vi–ii; iii–iv; iv–iii; iv–v; v–iv; iv–vi; and vi–iv. The subjects fasted from noon on the day of dosing. During the study period, the meal was served at the scheduled time point only, with no other food consumed; however, intake of water was allowed at all times. The meal was similar in caloric content and nutrient distribution for all subjects.
Table 1.
Meal and dosing schedule
| Group | Treatment | Time of dosing | Time of meal | Time of meal relative to dosing |
|---|---|---|---|---|
| I | sCT | 22.00 h | 18.00 h | −4 h |
| II | sCT | 22.00 h | 20.00 h | −2 h |
| III | sCT | 22.00 h | 21.00 h | −1 h |
| IV | sCT | 22.00 h | 22.10 h | +10 min |
| V | sCT | 22.00 h | 02.00 h | No meal |
| VI | Placebo | 22.00 h | 20.00 h | −2 h |
Time of meal in relation to the time of dosing of oral salmon calcitonin (sCT).
For the evaluation of the pharmacokinetics, blood samples were collected immediately prior to dosing (time 0 min) and at intervals of 5, 10, 15, 30, 45, 60, 90 and 120 min after dosing. At each of these time points, 5 ml of blood was collected in lithium heparin tubes and placed on ice until centrifugation within 60 min of venepuncture. The plasma samples were stored at −80°C until analysis. For the evaluation of pharmacodynamics, blood samples were collected immediately prior to dosing (time 0 min), and at intervals of 30, 60, 120, 180 and 240 min after dosing. At each of these time points, 5 ml of blood was collected in uncoated tubes and left at room temperature for 15–30 min before centrifugation within 180 min of venepuncture. The serum samples were stored at −20°C until analysis.
The concentration of plasma sCT was measured by a chemiluminescence-based assay that has been described previously [10]. Levels below the lower limit of quantification of 2.5 pg ml−1 were assigned the value of 2.5 pg ml−1. The assay was a two-site immunometric type employing two antibodies, one biotinylated and the other acridium labelled. Specificity has been tested against synthetic fragments of sCT and against human as well as eel calcitonin, with no interaction found over the range of the standard curve. The quality control samples, ranging from 2.5 to 700 pg ml−1, were prepared daily and measured in three to five replicates. The overall accuracy and precision (CV) of the control samples measured on 11 days were, respectively, 101.3 and 10.1% for the 2.5 pg ml−1 concentration and 94.3 and 6.0% for the 700 pg ml−1 concentration. The sCT assay does not react with degraded sCT.
The serum CTX-I test is a sandwich enzyme enzyme-linked immunosorbent assay (ELISA) employing two monoclonal antibodies, both recognizing the C-telopeptide of the α1-chain in type I collagen [37]. The monoclonal antibodies, i.e. MAb F1103 and MAb F12, recognize the eight-amino-acid sequence EKAHD-β-GGR, where D- β-G denotes an isomerized bond between aspartate and glycine, and both antibodies require the presence of a free C-terminal arginine for binding. Cathepsin K, secreted by the osteoclast, is responsible for the proteolytic cleavage exposing the free C-terminal arginine [33]. The sandwich construction assures that only cross-linked di-peptides, i.e. EKAHD-β-GGR × EKAHD-β-GGR, are measured by the serum CTX-I ELISA. The measuring range is 0.020–3.380 ng ml−1; in this range, the intra- and interassay CVs are <3.0 and <10.9%, respectively, and the dilution recovery is 103%. The reference ranges {mean [95% confidence interval (CI)]} for postmenopausal women, premenopausal women, and men are 0.439 ng ml−1 (0.142, 1.351), 0.287 ng ml−1 (0.112, 0.738) and 0.294 ng ml−1 (0.115, 0.748), respectively, according to the manufacturer (Immunodiagnostic Systems Nordic, Herlev, Denmark) [37].
The study was conducted in accordance with Helsinki Declaration II version II, approved by the local Danish Ethical Committees, and conducted in Ballerup, Denmark. The trial registration number is EudraCT: 2004-001916-30. As the screening period of the study was in December 2004 and the study conducted in January 2005, this is prior to the policy on clinical trials initiated after 1 July 2007, and the study is not registered at http://www.clinicaltrials.gov.
Written informed consent was obtained from all participants.
Statistical analysis
The trapezoidal method was applied for calculation of the area under the concentration–time curve (AUC) of plasma sCT and changes in serum CTX after dosing. The relative value of serum CTX was calculated as a percentage of the individual predose value. The relative change in serum CTX was determined as 100% minus the relative value of serum CTX. The AUC of plasma sCT, the time course data for plasma sCT, serum CTX-I concentrations, and relative values of serum CTX-I were logarithmically transformed to obtain normality and symmetry of variances. Comparison between treatment sequences for AUC of plasma sCT and changes in serum CTX was performed in a linear mixed effect model, with response as the variable, treatment sequences (five levels: i, ii, iii, iv and v) and dosing sequence (two levels: 1 and 2) as fixed effects, and subject as a random effect. The significance level of the pairwise multiple comparisons of treatment sequences i, ii, iii and iv against group v was Dunnett adjusted. A difference was considered significant if the P-value was <5%. Statistical calculations were performed with the SAS software package (release 9.1; SAS Institute Inc., Cary, NC, USA).
Results
The demographic characteristics of the 36 study participants are given in Table 2. The subjects were aged between 62 and 74 years and had passed menopause between 6 and 31 years previously. One subject assigned to treatment group ii did not attend her final session because of eczema. The remaining 35 subjects were present for all planned study visits.
Table 2.
Demographic characteristics of study subjects (n= 36)
| Age (years) | 68.2 (62.4–74.3) |
| Years since menopause | 18 (6–31) |
| Body mass index (kg m−2) | 25.9 (20.3–39.4) |
| Serum CTX-I (ng ml−1) | 0.57 (0.25–1.48) |
Demographic of the study subjects. Values given are mean (range).
The effect of meal on plasma SCT and S-CTX-I
The effect of a meal on the plasma levels of sCT was investigated as described in Table 1. The maximum concentration of plasma sCT was reached 15 min after dosing, with an average maximal plasma sCT concentration of 75 pg ml−1. After 120 min, the concentration of plasma sCT was below the detection limit of the assay, as presented in Figure 1. Dosing after meal intake resulted in a significant reduction in the maximum concentration of sCT and the AUC in the 2-h period after dosing, as summarized in Table 3. The predose meal at 18.00 and 21.00 h significantly decreased relative oral bioavailability of sCT to 26% (95% CI 0.09, 0.73 and 0.09, 0.75, P= 0.009 and P= 0.01) compared with that of the dose in the fasting state. The predose meal at 20.00 h decreased relative oral bioavailability to 35% (95% CI 0.12, 1.04) (P= 0.06) and the meal consumed 10 min after dosing decreased the oral bioavailability of sCT to 59% (95% CI 0.21, 1.68) (P= 0.48).
Figure 1.
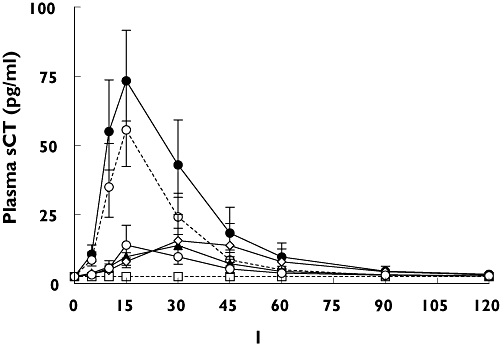
Time course of plasma salmon calcitonin (sCT) in each meal and treatment sequence in the 2 h following dosing with one single dose of 0.8 mg of oral sCT at 22.00 h. Values given are geometric mean ± 1 SEM. I: Meal at 18:00 (—○—); II: Meal at 20:00 (—◊—); III: Meal at 21:00 ( ); IV: Meal at 22:10 (--○--); V: No meal (—•—); VI: Placebo (--□--)
); IV: Meal at 22:10 (--○--); V: No meal (—•—); VI: Placebo (--□--)
Table 3.
Oral bioavailability of salmon calcitonin (sCT) based on AUC0–2 h of plasma sCT
| IMeal at 18.00 h | IIMeal at 20.00 h | IIIMeal at 21.00 h | IVMeal at 22.10 h | VNo meal | |
|---|---|---|---|---|---|
| Mean (pg ml−1 min−1) | 795 | 1098 | 812 | 1824 | 3110 |
| [95% CLM] | [454, 1394] | [527, 2291] | [495, 1332] | [1241, 2682] | [1850, 5227] |
| P-value | 0.009 | 0.06 | 0.01 | 0.48 | – |
| Relative oral bioavailability | 0.26 | 0.35 | 0.26 | 0.59 | = 1.00 |
| [95% CI] | [0.09, 0.73] | [0.12, 1.04] | [0.09, 0.75] | [0.21, 1.68] |
Oral bioavailability of sCT based on AUC0–2 h of plasma sCT. The dose was given at 22.00 h. Values of AUC0–2 h of plasma sCT are given as geometric mean and 95% confidence interval of mean (CLM). The P-values represent the Dunnett-adjusted significance of the multiple comparisons of the treatment sequence i, ii, iii and iv against meal treatment sequences v (no meal). The relative oral bioavailability is the bioavailability relative to treatment sequences v (no meal), and the Dunnett-adjusted 95% confidence interval (CI) in the relative bioavailability from treatment sequence v (no meal).
In accordance with the absorption of sCT, the effect of a meal on bone resorption, as measured by the pharmacodynamic biomarkers of efficacy CTX-I, revealed a similar time and meal dependency (Figures 2 and 3). Overall, evening dosing resulted in significant suppression of serum CTX-I. Dosing 1 h before or 10 min after meal intake resulted in smaller and nonsignificant decreases in CTX-I compared with no meal, as summarized in Table 4. However, dosing 4 or 2 h after meal intake resulted in significantly lower reductions in CTX-I compared with that of no meal, as summarized in Table 4.
Figure 2.
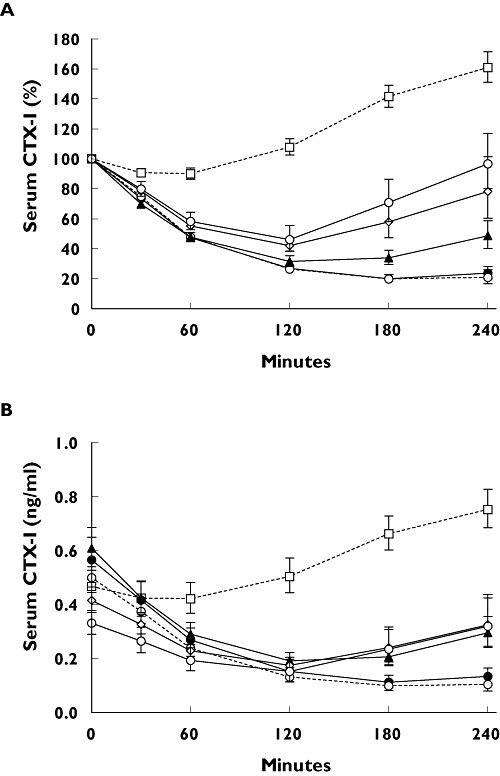
Time course of serum C-terminal telopeptide of collagen type I (CTX-I) in the 4 h following one single dose of 0.8 mg of oral salmon calcitonin (sCT) in each treatment sequence. (A) The value relative to predose value; (B) serum CTX-I in absolute levels. Values given are geometric mean ± 1 SEM. I: Meal at 18:00 (—○—); II: Meal at 20:00 (—◊—); III: Meal at 21:00 ( ); IV: Meal at 22:10 (--○--); V: No meal (—•—); VI: Placebo (--□--)
); IV: Meal at 22:10 (--○--); V: No meal (—•—); VI: Placebo (--□--)
Figure 3.
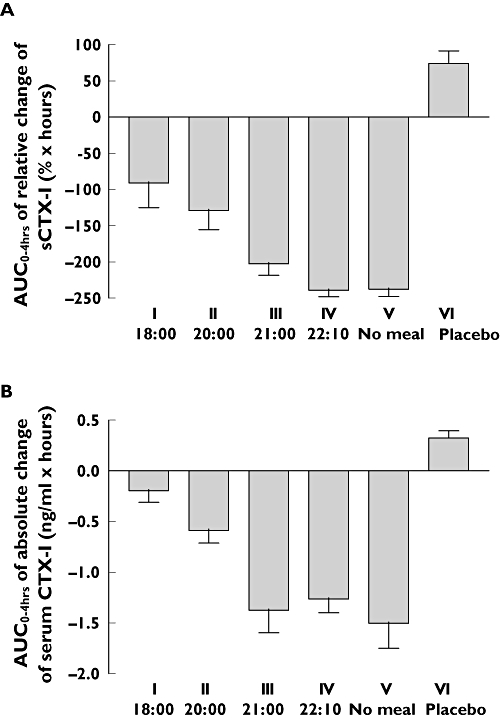
AUC of relative change (A) and absolute change (B) in serum C-terminal telopeptide of collagen type I (CTX-I) in the 4 h following one single dose of 0.8 mg of oral salmon calcitonin (sCT) in each treatment sequence. Values given are mean ± 1 SEM. Analysis of variance (i–ii–iii–iv–v) of panel A: P < 0.001
Table 4.
Pharmacodynamic effect on serum CTX-I
| IMeal at 18.00 h | IIMeal at 20.00 h | IIIMeal at 21.00 h | IVMeal at 22.10 h | VNo meal | |
|---|---|---|---|---|---|
| Mean (% h−1) | −91 | −130 | −202 | −235 | −238 |
| [95% CLM] | [−135, −47] | [−177, −84] | [−247, −158] | [−279, −190] | [−282, −193] |
| Difference (% h−1) | 147 | 107 | 35 | 3 | – |
| [95% CI] | [68, 225] | [27, 187] | [−43, 114] | [−77, 83] | |
| P-value | 0.0003 | 0.008 | 0.58 | 1.00 | – |
| Mean (ng ml−1 h−1) | −0.40 | −0.52 | −1.21 | −1.25 | −1.47 |
| [95% CLM] | [−0.74, −0.07] | [−0.87, −0.17] | [−1.55, −0.88] | [−1.57, −0.92] | [−1.79, −1.15] |
| Difference (ng ml−1 h−1) | 1.06 | 0.94 | 0.25 | 0.22 | – |
| [95% CI] | [0.55, 1.58] | [0.42, 1.47] | [−0.26, 0.77] | [−0.32, 0.76] | |
| P-value | <0.0001 | 0.0005 | 0.51 | 0.66 | – |
Pharmacodynamic effect of salmon calcitonin (sCT) based on AUC0–4 h of serum CTX-I. The dose was given at 22.00 h. Values of AUC0–4 h of serum CTX-I are given as mean and 95% confidence interval of mean (CLM). The differences are the differences of AUC0–4 h from treatment sequences v (no meal) and the Dunnett-adjusted 95% confidence interval (CI). The P-values represent the Dunnett-adjusted significance of the multiple comparisons of the treatment sequence i, ii, iii and iv against meal treatment sequences v (no meal).
As presented in Figure 2A, the initial reduction in CTX-I levels was comparable for all treatment sequences. The highest suppression was found in the treatment sequence that had no evening meal (v) or received the sCT dose before the meal (iv). The AUC in the 4-h period of change in the relative values of serum CTX-I reached −238 (% × h) and −235 (% × h). Doses given after a meal resulted in smaller serum CTX-I reductions: i.e. the nadir reached in all treatment sequences was nearly the same as that reached in the treatment sequence that received the sCT dose before a meal, but importantly, the return to baseline started earlier in the treatment sequences dosed after a meal, resulting in a smaller AUC (Figures 2 and 3). Furthermore, the suppression of serum CTX-I was more variable in subjects dosed after than in those dosed before a meal.
Discussion
Calcitonin is approved for the treatment of osteoporosis [9]. Recently, a new oral formulation of calcitonin in combination with the carrier molecule 5-CNAC has been tested clinically [9, 10]. Because of the small intestinal uptake of peptides, the bioavailability of the oral formulation is limited, even in the presence of carrier molecules. Therefore, an optimal approach to inhibiting bone resorption transiently may be to ensure maximal effects at times of peak bone resorption, e.g. during the night [29, 36]. However, late-day dosing may be complicated by food intake, potentially resulting in food–drug interactions and, as a consequence, attenuation of plasma calcitonin concentrations and impaired drug efficacy.
In the present study, we have clearly demonstrated that postprandial oral dosing resulted in severely impaired calcitonin uptake – as much as a 74% reduction – and correspondingly hampered drug efficacy as measured by a biochemical marker of bone resorption, CTX-I. This was observed with dosing 1, 2 and 4 h after meal intake. In contrast, preprandial dosing by 10 min before meal time limited and almost reversed this reduction. Previous studies have indicated that the bioavailability of some drugs is heavily influenced by the timing of meals [29–32] and may be different in the fasting state. The current study is in alignment with previous research in documenting the detrimental effects on the uptake of certain drugs when administrated either concomitantly with or after a meal [29]. The data reported here are the first from a systematic investigation of the effect of meal timing on the uptake of small peptides, such as calcitonin. This investigation may be important for the general understanding of oral delivery of small peptides and consequently may aid in the development of other peptide drugs. However, we cannot rule out that the present findings are specific to the carrier molecule and formulation of sCT studied.
A possible discrepancy was found between the pharmacokinetic concentrations of sCT in plasma and the pharmacodynamic effects evidenced by bone resorption, such that the effect on bone resorption was sustained as plasma concentration declined. This protracted pharmacodynamic effect of sCT relative to plasma concentration was best observed after 4 h, which typically corresponds to an indirect pharmacodynamic model [38]. Pharmacodynamic relationships are usually characterized by a sigmoid shape and saturate beyond a certain level of dose exposure, where the pharmacodynamic profile may be perfectly consistent with the pharmacokinetic profile. This relationship has previously been reported for this oral formulation of calcitonin in a 3-month safety and efficacy study [9, 10]. In the present short-term study, this relationship was not observed. This may be attributable to the fact that sCT acts on CTX production by osteoclasts, and blood CTX concentrations reflect a convolution of production and elimination over time, which have their own kinetic determinants. For example, if CTX production is eliminated sharply, a time interval is needed for gradual change in blood CTX levels. This delayed response has previously been described for other drugs, such as warfarin, and is best described by indirect pharmacodynamic models [38]. In addition, the protracted effect on bone resorption may in part be related to the direct effect on osteoclasts, resulting in osteoclastic morphological transformations prolonging the inhibition of bone resorption; this effect has been seen in in vitro studies [3, 5, 39].
The presented data are of major importance in identifying the optimal dosing regimen for future clinical trials with oral calcitonin. The efficacy of the novel oral formation of calcitonin used in this study may be improved by preprandial dosing, possibly in the evening, when bone resorption peaks.
Conclusion
The current study has clearly demonstrated that postprandial dosing limits the bioavailability of oral sCT (SMC021) and, consequently, its efficacy as measured by a biochemical marker of bone resorption. The rapid absorption of sCT into plasma suggests that maximal benefits with respect to inhibiting the nocturnal rise in bone resorption can be achieved by dosing at least 10 min prior to meal intake.
Competing interests
LC is a full-time employee of Novartis.
Editorial assistance was provided by BioScience Communications.
REFERENCES
- 1.Copp DH, Cameron EC, Cheney BA, Davidson AG, Henze KG. Evidence for calcitonin – a new hormone from the parathyroid that lowers blood calcium. Endocrinology. 1962;70:638–49. doi: 10.1210/endo-70-5-638. [DOI] [PubMed] [Google Scholar]
- 2.Kumar MA, Foster GV, MacIntyre I. Further evidence for calcitonin. A rapid-acting hormone which lowers plasma-calcium. Lancet. 1963;35:480–2. doi: 10.1016/s0140-6736(63)90224-1. [DOI] [PubMed] [Google Scholar]
- 3.Chambers TJ, Moore A. The sensitivity of isolated osteoclasts to morphological transformation by calcitonin. J Clin Endocrinol Metab. 1983;57:819–24. doi: 10.1210/jcem-57-4-819. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki H, Nakamura I, Takahashi N, Ikuhara T, Matsuzaki K, Isogai Y, Hori M, Suda T. Calcitonin-induced changes in the cytoskeleton are mediated by a signal pathway associated with protein kinase A in osteoclasts. Endocrinology. 1996;137:4685–90. doi: 10.1210/endo.137.11.8895334. [DOI] [PubMed] [Google Scholar]
- 5.Shyu JF, Shih C, Tseng CY, Lin CH, Sun DT, Liu HT, Tsung HC, Chen TH, Lu RB. Calcitonin induces podosome disassembly and detachment of osteoclasts by modulating Pyk2 and Src activities. Bone. 2007;40:1329–42. doi: 10.1016/j.bone.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 6.Sorensen MG, Henriksen K, Schaller S, Henriksen DB, Nielsen FC, Dziegiel MH, Karsdal MA. Characterization of osteoclasts derived from CD14+ monocytes isolated from peripheral blood. J Bone Miner Metab. 2007;25:36–45. doi: 10.1007/s00774-006-0725-9. [DOI] [PubMed] [Google Scholar]
- 7.Sexton PM, Findlay DM, Martin TJ. Calcitonin. Curr Med Chem. 1999;6:1067–93. [PubMed] [Google Scholar]
- 8.Karsdal MA, Sondergaard BC, Arnold M, Christiansen C. Calcitonin affects both bone and cartilage: a dual action treatment for osteoarthritis? Ann NY Acad Sci. 2007;1117:181–95. doi: 10.1196/annals.1402.041. [DOI] [PubMed] [Google Scholar]
- 9.Tanko LB, Bagger YZ, Alexandersen P, Devogelaer JP, Reginster JY, Chick R, Olson M, Benmammar H, Mindeholm L, Azria M, Christiansen C. Safety and efficacy of a novel salmon calcitonin (sCT) technology-based oral formulation in healthy postmenopausal women: acute and 3-month effects on biomarkers of bone turnover. J Bone Miner Res. 2004;19:1531–8. doi: 10.1359/JBMR.040715. [DOI] [PubMed] [Google Scholar]
- 10.Bagger YZ, Tanko LB, Alexandersen P, Karsdal MA, Olson M, Mindeholm L, Azria M, Christiansen C. Oral salmon calcitonin induced suppression of urinary collagen type II degradation in postmenopausal women: a new potential treatment of osteoarthritis. Bone. 2005;37:425–30. doi: 10.1016/j.bone.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 11.Karsdal MA, Tanko LB, Riis BJ, Sondergard BC, Henriksen K, Altman RD, Qvist P, Christiansen C. Calcitonin is involved in cartilage homeostasis: is calcitonin a treatment for OA? Osteoarthritis Cartilage. 2006;14:617–24. doi: 10.1016/j.joca.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Lee YH, Sinko PJ. Oral delivery of salmon calcitonin. Adv Drug Deliv Rev. 2000;42:225–38. doi: 10.1016/s0169-409x(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 13.Van den Mooter G. Colon drug delivery. Expert Opin Drug Deliv. 2006;3:111–25. doi: 10.1517/17425247.3.1.111. [DOI] [PubMed] [Google Scholar]
- 14.Streubel A, Siepmann J, Bodmeier R. Gastroretentive drug delivery systems. Expert Opin Drug Deliv. 2006;3:217–33. doi: 10.1517/17425247.3.2.217. [DOI] [PubMed] [Google Scholar]
- 15.Shareef MA, Khar RK, Ahuja A, Ahmad FJ, Raghava S. Colonic drug delivery: an updated review. AAPS PharmSci. 2003;5:E17. doi: 10.1208/ps050217. [DOI] [PubMed] [Google Scholar]
- 16.Smoum R, Rubinstein A, Srebnik M. Chitosan– pentaglycine–phenylboronic acid conjugate: a potential colon-specific platform for calcitonin. Bioconjug Chem. 2006;17:1000–7. doi: 10.1021/bc050357y. [DOI] [PubMed] [Google Scholar]
- 17.Bernkop-Schnurch A, Hoffer MH, Kafedjiiski K. Thiomers for oral delivery of hydrophilic macromolecular drugs. Expert Opin Drug Deliv. 2004;1:87–98. doi: 10.1517/17425247.1.1.87. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Fuentes M, Torres D, Alonso MJ. New surface-modified lipid nanoparticles as delivery vehicles for salmon calcitonin. Int J Pharm. 2005;296:122–32. doi: 10.1016/j.ijpharm.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 19.Lamprecht A, Yamamoto H, Takeuchi H, Kawashima Y. pH-sensitive microsphere delivery increases oral bioavailability of calcitonin. J Control Release. 2004;98:1–9. doi: 10.1016/j.jconrel.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Guggi D, Kast CE, Bernkop-Schnurch A. In vivo evaluation of an oral salmon calcitonin-delivery system based on a thiolated chitosan carrier matrix. Pharm Res. 2003;20:1989–94. doi: 10.1023/b:pham.0000008047.82334.7d. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Chow D, Heiati H, Shen WC. Reversible lipidization for the oral delivery of salmon calcitonin. J Control Release. 2003;88:369–80. doi: 10.1016/s0168-3659(03)00008-7. [DOI] [PubMed] [Google Scholar]
- 22.Sakuma S, Suzuki N, Sudo R, Hiwatari K, Kishida A, Akashi M. Optimized chemical structure of nanoparticles as carriers for oral delivery of salmon calcitonin. Int J Pharm. 2002;239:185–95. doi: 10.1016/s0378-5173(02)00113-8. [DOI] [PubMed] [Google Scholar]
- 23.Torres-Lugo M, Peppas NA. Transmucosal delivery systems for calcitonin: a review. Biomaterials. 2000;21:1191–6. doi: 10.1016/s0142-9612(00)00011-9. [DOI] [PubMed] [Google Scholar]
- 24.Sondergaard BC, Ostergaard S, Christiansen C, Karsdal MA. The effect of oral calcitonin on cartilage turnover and surface erosion in the ovariectomized rat model. Arthritis Rheum. 2007;2007:2674–8. doi: 10.1002/art.22797. [DOI] [PubMed] [Google Scholar]
- 25.Malkov D, Angelo R, Wang HZ, Flanders E, Tang H, Gomez-Orellana I. Oral delivery of insulin with the Eligen technology: mechanistic studies. Curr Drug Deliv. 2005;2:191–7. doi: 10.2174/1567201053586001. [DOI] [PubMed] [Google Scholar]
- 26.Mustata G, Dinh SM. Approaches to oral drug delivery for challenging molecules. Crit Rev Ther Drug Carrier Syst. 2006;23:111–35. doi: 10.1615/critrevtherdrugcarriersyst.v23.i2.20. [DOI] [PubMed] [Google Scholar]
- 27.Mlynek GM, Calvo LJ, Robinson JR. Carrier-enhanced human growth hormone absorption across isolated rabbit intestinal tissue. Int J Pharm. 2000;197:13–21. doi: 10.1016/s0378-5173(99)00322-1. [DOI] [PubMed] [Google Scholar]
- 28.Wu SJ, Robinson JR. Transcellular and lipophilic complex-enhanced intestinal absorption of human growth hormone. Pharm Res. 1999;16:1266–72. doi: 10.1023/a:1014809916407. [DOI] [PubMed] [Google Scholar]
- 29.Gertz BJ, Clemens JD, Holland SD, Yuan W, Greenspan S. Application of a new serum assay for type I collagen cross-linked N-telopeptides: assessment of diurnal changes in bone turnover with and without alendronate treatment. Calcif Tissue Int. 1998;63:102–6. doi: 10.1007/s002239900497. [DOI] [PubMed] [Google Scholar]
- 30.Lentz KA, Quitko M, Morgan DG, Grace JE, Jr, Gleason C, Marathe PH. Development and validation of a preclinical food effect model. J Pharm Sci. 2007;96:459–72. doi: 10.1002/jps.20767. [DOI] [PubMed] [Google Scholar]
- 31.Karara AH, Dunning BE, McLeod JF. The effect of food on the oral bioavailability and the pharmacodynamic actions of the insulinotropic agent nateglinide in healthy subjects. J Clin Pharmacol. 1999;39:172–9. doi: 10.1177/00912709922007606. [DOI] [PubMed] [Google Scholar]
- 32.Sostek MB, Chen Y, Andersson T. Effect of timing of dosing in relation to food intake on the pharmacokinetics of esomeprazole. Br J Clin Pharmacol. 2007;64:386–90. doi: 10.1111/j.1365-2125.2007.02889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaller S, Henriksen K, Hoegh-Andersen P, Sondergaard BC, Sumer EU, Tanko LB, Qvist P, Karsdal MA. In vitro ex vivo, and in vivo methodological approaches for studying therapeutic targets of osteoporosis and degenerative joint diseases: how biomarkers can assist? Assay Drug Dev Technol. 2005;3:553–80. doi: 10.1089/adt.2005.3.553. [DOI] [PubMed] [Google Scholar]
- 34.Schaller S, Henriksen K, Sveigaard C, Heegaard AM, Hélix N, Stahlhut M, Ovejero MC, Johansen JV, Solberg H, Andersen TL, Hougaard D, Berryman M, Shiødt CB, Sørensen BH, Lichtenberg J, Christophersen P, Foged NT, Delaissé JM, Engsig MT, Karsdal MA. The chloride channel inhibitor n53736 prevents bone resorption in ovariectomized rats without changing bone formation. J Bone Miner Res. 2004;19:1144–53. doi: 10.1359/JBMR.040302. [DOI] [PubMed] [Google Scholar]
- 35.Ravn P, Hosking D, Thompson D, Cizza G, Wasnich RD, McClung M, Yates AJ, Bjarnason NH, Christiansen C. Monitoring of alendronate treatment and prediction of effect on bone mass by biochemical markers in the early postmenopausal intervention cohort study. J Clin Endocrinol Metab. 1999;84:2363–8. doi: 10.1210/jcem.84.7.5847. [DOI] [PubMed] [Google Scholar]
- 36.Schlemmer A, Hassager C, Jensen SB, Christiansen C. Marked diurnal variation in urinary excretion of pyridinium cross-links in premenopausal women. J Clin Endocrinol Metab. 1992;74:476–80. doi: 10.1210/jcem.74.3.1740479. [DOI] [PubMed] [Google Scholar]
- 37.Rosenquist C, Fledelius C, Christgau S, Pedersen BJ, Bonde M, Qvist P, Christiansen C. Serum CrossLaps One Step ELISA. First application of monoclonal antibodies for measurement in serum of bone-related degradation products from C-terminal telopeptides of type I collagen. Clin Chem. 1998;44:2281–9. [PubMed] [Google Scholar]
- 38.Jusko WJ, Ko HC. Physiologic indirect response models characterize diverse types of pharmacodynamic effects. Clin Pharmacol Ther. 1994;56:406–19. doi: 10.1038/clpt.1994.155. [DOI] [PubMed] [Google Scholar]
- 39.Murrills RJ, Shane E, Lindsay R, Dempster DW. Bone resorption by isolated human osteoclasts in vitro: effects of calcitonin. J Bone Miner Res. 1989;4:259–68. doi: 10.1002/jbmr.5650040219. [DOI] [PubMed] [Google Scholar]