

Abstract
Polyarteritis nodosa is a necrotizing vasculitis of medium-sized arteries of unknown origin. Hypertension is present in 30% of patients with polyarteritis nodosa. In those cases, high renin levels are thought to be secondary to renal involvement. The present study was performed to identify causal factors of polyarteritis nodosa. In cyp1a1ren-2 transgenic rats, vasculitis of medium-sized arteries resembling classical polyarteritis nodosa can be induced. In this model, oral administration of indole-3-carbinol (I3C) activates the liver-specific cyp1a1 promoter, leading to prorenin expression in a dose-dependent manner. After the first 6 weeks of chronic induction with 0.125% I3C, the mean arterial pressure reached a plateau of about 170 mmHg. Ten out of 11 I3C-treated rats, which were chronically instrumented with a telemetric device to measure blood pressure, developed polyarteritis nodosa within 10 weeks of I3C treatment. I3C alone or instrumentation alone did not cause polyarteritis nodosa. The angiotensin-converting enzyme inhibitor captopril completely prevented the development of polyarteritis nodosa, indicating that local angiotensin II generation is a pathogenetic factor in this model. The renin–angiotensin system can play a primary role in the development of polyarteritis nodosa in rats.
Keywords: polyarteritis nodosa, angiotensin, inflammation, prorenin
Introduction
Polyarteritis nodosa is a multi-system, necrotizing vasculitis of medium-sized arteries. In humans, the involvement of the visceral and renal arteries is characteristic. In the pathogenesis of polyarteritis nodosa, a role of viral antigens is documented for the hepatitis B virus (HBV)-positive type, which is seen as a distinct form of polyarteritis nodosa and is treated with anti-viral therapy [1]. The pathogenesis of the HBV-negative forms of polyarteritis nodosa is still unknown.
Patients usually present with weakness, malaise, weight loss, headache, abdominal pain, testicular pain and myalgias. Despite improvements in the therapy of polyarteritis nodosa, about 10% of patients still die from fulminant polyarteritis, with a median survival time of 3 months after diagnosis [2]. In more than 30% of the patients, hypertension is observed. Cases have been reported with renin-dependent hypertension. In these cases, hypertension was believed to be secondary to polyarteritis-associated inflammation of renal arteries [3–5].
Recently, the cyp1a1ren-2 transgenic rat has been generated as a transgenic model of renin-dependent hypertension [6]. In these rats, occasionally, cases have been observed with nodular infiltration of mesenteric vessels, as is typical for polyarteritis nodosa (Peters, B. unpublished observations). In cyp1a1ren-2 transgenic rats, the expression of the ren-2 renin transgene is under the control of the promoter of the cyp1a1 gene, which can be activated by oral administration of various xenobiotics, including the non-toxic aryl hydrocarbon compound indole-3-carbinol (I3C). Thus, the model permits the induction of renin expression predominantly in the liver. The administration of I3C results in dose-dependent elevations of plasma prorenin levels [7]. Various degrees of hypertension can be induced, ranging from mild to severe hypertension. Long-term administration of 0.125% I3C results in stable chronic hypertension in cyp1a1ren-2 transgenic rats, with the mean arterial pressures around 170 mmHg. At high concentrations, I3C induces malignant hypertension within 2 weeks and animals present with severe hypertensive cardiac and renal damage [6, 8, 9].
The renin–angiotensin system is an important mediator of volume and blood pressure homeostasis and also a modulator of inflammatory processes. Angiotensin II (Ang II) causes contraction of vascular smooth muscle cells and activates nicotinamide adenine dinucleotide (phosphate) (NAD(P)H) oxidase, thus enhancing the production of superoxide anions. Furthermore, Ang II activates p38 mitogen-activated protein kinase, JNK and nuclear factor (NF)-κB [10]. It is therefore conceivable that the activation of the renin–angiotensin system is not only a consequence of polyarteritis nodosa but might, in fact, be a causal factor in polyarteritis nodosa.
The present study addresses the following questions: Whether polyarteritis nodosa can reproducibly be induced in cyp1a1ren-2 transgenic rats? Which conditions promote its development? Whether the renin–angiotensin system is a primary cause of polyarteritis nodosa?
Materials and methods
Animals
Cyp1a1ren-2 transgenic rats were bred at the University of Greifswald. The animals were housed in a temperature- and humidity-controlled room with lights on from 06:00 a.m. to 06:00 p.m. Unless otherwise indicated, the rats received tap water and standard rat chow (Ssniff, Soest, Germany) ad libitum. Body weight was recorded twice weekly, and fluid uptake was averaged over a 2-day period. All procedures were approved by a governmental committee on animal welfare, the guidelines of which are in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Experimental protocol
Thirty adult male cyp1a1ren-2 transgenic rats were randomly assigned to the following groups: group ‘Con’ (n= 3) remained untreated, group ‘TM’ (n= 6) received transmitters only, group ‘I3C’ (n= 6) was treated with 0.125% I3C, group ‘ShamTM+I3C’ (n= 3) was sham-operated and received 0.125% I3C, group ‘TM+I3C’ (n= 6) received transmitters and was treated with 0.125% I3C and group ‘TM+I3C+Cap’ (n= 6) received transmitters and was treated with 0.125% I3C as well as with the angiotensin-converting enzyme inhibitor captopril. Radiotelemetric devices were implanted when the animals were 15–16 weeks old. Body weight before the operation was 349±17 g. Immediately after the implantation of transmitters or at a corresponding age in animals that did not receive transmitters, the rats were placed on an experimental diet (EF 1/80; Ssniff, Soest, Germany) to which NaCl was added to yield a final NaCl content of 1.0%. After 2 weeks of recovery from surgery, the body weight of the operated rats was still lower than the body weight of the rats without operation (TM: 342±13 g, ShamTM+I3C: 329±10 g, TM+I3C: 347±4 g, TM+I3C+Cap: 335±12 g, versus control: 375±8 g and I3C: 369±21 g). At this time point, the experimental protocol was started. I3C (Sigma, Deisenhofen, Germany) was added to the diet to yield a final concentration (dry weight:dry weight) of 0.125%. The transgene inducer was given for 10 weeks. Rats in the Con and TM groups continued to receive the experimental diet without I3C. Rats in the TM+I3C+Cap group additionally received captopril in the drinking water. Captopril treatment was started simultaneously with the application of I3C and also lasted for 10 weeks. The dose of captopril was adjusted so as to accomplish a blood pressure reduction. The captopril concentration in the drinking fluid was increased from 2 mg/100 ml at the beginning to 25 mg/100 ml towards the end of the treatment so as to keep blood pressure in the normotensive range, despite rising prorenin levels throughout the protocol. Captopril concentration in the drinking fluid was the same for all rats of the TM+I3C+Cap group at any given time point.
Radiotelemetry
Radiotelemetric devices (TA11PA-C40, DSI-Transoma Medical, St. Paul, MN, USA) were implanted under ether anaesthesia via an abdominal mid-line incision under aseptic conditions. The transmitters were fixed within the upper abdominal cavity [11]. During the recording period, data on arterial pressure, heart rate and locomotor activity were acquired with LabPro software version 3.11 (DSI-Transoma Medical) at 10-min. intervals. Telemetric data are given as 24-hrs mean values. In three rats (Sham TM + I3C), the transmitters were implanted into the abdominal cavity without inserting the catheter into the aorta.
Histology
At the end of the experiment, the rats were anaesthetized with ether and killed, organs were taken and all nodular changes in the vasculature of the mesentericum were identified. For histological investigations, one half of the left and right cardiac ventricles, mesenteric vessels, abdominal aorta, pancreas, kidney and brain were fixed in 4% formalin, embedded in paraffin, sectioned at a thickness of 4 μm and stained with haematoxylin and eosin. Furthermore, sections of the mesenteric vessels were stained with Elastica van Gieson to identify the arteries. Sections of the kidneys were also stained with Jones methenamine silver method or acid fuchsin orange G stain (AFOG) or the periodic acid–Schiff reaction was used. For immunohistochemistry, 4-μm-thick paraffin slices of the mesenteric vessels were incubated with anti-CD3 (Dako, Hamburg, Germany) in a 1:50 dilution. Binding of the primary antibody was visualized using the View DAB Detection kit (Ventana, Vreden, Germany), according to the manufacturer’s protocol. Furthermore, the chloracetate esterase reaction was applied on different sections.
Histological investigations were performed in a blinded fashion by an investigator who was unaware of the study groups.
Blood sampling and determinations of renin, prorenin and autoantibodies
On the day before induction and after 3 and 6 weeks, 1.5 ml of ethylenediaminetetraacetic acid (EDTA)-blood were collected from the retro-orbital plexus under light ether anaesthesia. Plasma active renin (PRC) levels were determined enzymatically by the capacity of the enzyme to generate angiotensin I (Ang I), as described previously [12]. Plasma prorenin levels were determined after trypsin activation [12]. Serum was diluted 1/100 in PBS and tested for autoantibodies (anti-neutrophil cytoplasmatic antibodies [ANCA] and anti-nuclear antibodies [ANA]) by indirect immunofluorescence. NOVA Lite IFA-ANCA slides (5 × 6 wells) and -ANA slides (INOVA Diagnostic, San Diego, CA, USA) were used as antigens and polyclonal goat-anti-rat IgG-FITC (DIANOVA, Hamburg, Germany) in a dilution of 1/100 was used as the detection antibody.
Statistical analyses
Data are given as means ± standard deviation, except for prorenin levels, which are given as medians. The differences in blood pressure levels and renin levels between the groups were evaluated by a one-way anova, followed by Bonferroni’s all pairwise post-hoc comparisons. For the differences in weight gain and prorenin levels, anova on ranks (Kruskal–Wallis), followed by Dunn’s all pairwise post-hoc comparisons, was performed. P-values of less than 0.05 were accepted to indicate statistical significance. All statistical analyses were performed using the SigmaStat software (SPSS, Inc., Chicago, IL, USA).
Results
Increase of blood pressure by I3C and its prevention by captopril
In cyp1a1ren-2 transgenic rats, prorenin-dependent hypertension was induced. Without I3C treatment, the blood pressure in cyp1a1ren-2 transgenic rats was in the normal range and was stable (TM: 105±11 mmHg, the average of 10 weeks; Fig. 1). In I3C-treated rats (TM+I3C), the mean arterial pressure increased continuously until it reached a plateau of about 170 mm Hg at week 6 (Fig. 1). The angiotensin-converting enzyme inhibitor captopril prevented partially the I3C-induced increase in the mean arterial pressure to a mean of 125 mmHg in TM+I3C+Cap rats, but still it was higher than in the TM group (P < 0.05).
Fig 1.

Effect of captopril treatment on the mean arterial pressure in cyp1a1ren-2 transgenic rats receiving indole-3-carbinol (I3C). TM: non-treated rats with transmitter, TM+I3C: I3C-treated rats with transmitter and TM+I3C+Cap: I3C-treated rats with transmitter and captopril; n= 6 per group. Time 0 marks the start of treatment with I3C and captopril, respectively. *P < 0.01 versus TM; §P < 0.001 versus TM+I3C.
Body weight
One clinical criterion for polyarteritis nodosa is weight loss. During the 10-week protocol, there were no statistically significant differences in weight gain between the following groups: Con: 18 ± 3%, TM: 18 ± 7%, I3C: 11 ± 3% and Sham TM + I3C: 12 ± 4% (n.s.; Fig. 2). In contrast, TM +I3C rats kept loosing weight as a clinical sign of polyarteritis nodosa (TM + I3C: –15 ± 6%; P < 0.001 versus all other groups). The difference in the body weight changes between TM+I3C rats and all other groups was statistically significant already 6 weeks after the start of I3C treatment. Captopril prevented the weight loss. The weight gain in captopril-treated rats was not different from control groups (TM + I3C + Cap: 18 ± 6%, n.s.; Fig. 2).
Fig 2.

Weight loss in rats with polyarteritis nodosa and its prevention by captopril. Con: untreated control rats (n= 3), TM: non-treated rats with transmitter (n= 6), TM+I3C: I3C-treated rats with transmitter (n= 6), Sham TM+I3C: sham-operated I3C-treated rats (n= 3) and TM+I3C+Cap: I3C-treated rats with transmitter and captopril (n= 6). Time 0 is the start of treatment with I3C and captopril and the initial weight of 100%, respectively. *P < 0.05.
Histology
All chronically instrumented rats (TM, TM+I3C and TM+I3C+Cap) showed a granulomatous inflammation with epitheloid macrophages and multi-nuclear giant cells as well as a deposition of an amorphous acellular material, characteristic for a foreign body reaction, at the insertion site of the catheter (Fig. 3A). There were no differences regarding this reaction between rats with and without I3C treatment. In addition to these changes, TM+I3C rats clearly showed histological signs resembling classical polyarteritis nodosa, including fibrinoid necrosis of the media, associated with transmural infiltration of lymphocytes, macrophages and plasma cells as well as a few neutrophils and eosinophils. Additionally, prominent perivascular granulation tissues with haemorrhages and intravascular thrombi were present. This chronic necrotizing vasculitis was predominantly found in medium-sized mesenteric arteries, such as the cranial mesenteric artery and/or celiac artery, with nodules up to 3 mm in diameter (Fig. 3B). Together with the celiac artery, the arteries of the pancreas were affected (Fig. 3C). Furthermore, a necrotizing vasculitis in the testes (not shown) and kidneys (Fig. 3D) of these animals was observed. Additionally, tubular protein casts and intracytoplasmatic protein resorption droplets were seen in the kidneys of these rats, as is typical for hypertension (Fig. 3D). Signs of microscopic polyarteritis, especially in the glomeruli, were not observed. Pulmonary and skeletal muscles arteries (hind limb) were not affected (not shown). The coronary and cerebral arteries showed thickening of the media as a sign of hypertensive vasculopathy but no inflammation typical for polyarteritis nodosa (not shown).
Fig 3.

Histological evidence of polyarteritis nodosa in cyp1a1ren-2 transgenic rats. Representative tissue sections of I3C-treated rats with transmitters (TM+I3C) (A) Foreign body reaction at the insertion site of the catheter (haematoxylin and eosin staining). (B) Vasculitis of a mesenteric vessel with dissection of the arterial wall and intraluminal thrombotic material (haematoxylin and eosin staining). (C) Pancreas: arterial nature of the affected vessel confirmed by Elastica van Gieson staining. (D) Fibrinoid necrosis of the endothelial wall of an intraparenchymal medium-sized renal artery; protein casts in the tubuli and intratubular protein droplets are stained orange (AFOG staining). Bar = 100 μm.
Arterial wall infiltration by T cells and leucocytes in polyarteritis nodosa in cyp1a1 ren-2 transgenic rats
Immunohistochemical staining with anti-CD3 monoclonal antibody demonstrated the presence of T lymphocytes in the inflammatory areas (Fig. 4A) of the mesenteric arteries of TM+I3C rats. Additionally, chloracetate esterase-positive polymorphic leucocytes (Fig. 4B), activated fibroblasts and monocytes as components of the inflammation were observed.
Fig 4.
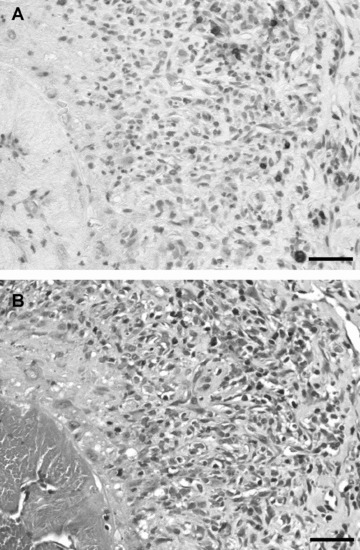
Identification of CD3-positive cells. (A) Immunohistochemistry of mesenteric polyarteritis lesions with anti-CD3 (DAB staining). Positively stained cells are labelled brown. (B) Chloracetate esterase reaction of mesenteric polyarteritis lesions for the identification of polymorphic leucocytes (red). Bar = 100 μm.
Polyarteritis nodosa occurred only in the group of TM+I3C rats (five out of six rats were positive for polyarteritis nodosa; Fig. 5C) and was absent in the groups of Con rats (Fig. 5A) and I3C rats (Fig. 5B). In an independent experiment (groups: Con, I3C: 0.125% and TM+I3C, n= 5 each), all TM+I3C rats, but none of the Con or I3C rats, developed polyarteritis nodosa (see Supporting Information). Thus, in our model, polyarteritis nodosa was reproducibly induced by I3C treatment combined with chronic instrumentation (TM+I3C: 10 out of 11 rats).
Fig 5.

Captopril prevented the development of polyarteritis nodosa. Representative tissue sections (same magnification) of the mesenteric vessels of cyp1a1ren-2 rats (haematoxylin and eosin staining). (A) control rats, (B) I3C-treated rats (I3C), (C) I3C-treated rats with transmitter (TM + I3C), (D) transmitter-implanted rats (TM), (E) sham-operated rats treated with I3C (Sham TM + I3C) and (F) co-application of I3C and captopril to transmitter-implanted rats (TM + I3C + Cap). Bar = 250 μm.
Telemetric devices are frequently used in hypertension research. To exclude that polyarteritis nodosa was related to the device, six rats received a catheter without induction of the renin–angiotensin system (TM). None of these rats developed polyarteritis nodosa (Fig. 5D) nor did the rats that were sham-operated (Sham TM+I3C; Fig. 5E).
To investigate whether the development of polyarteritis nodosa is influenced by the renin–angiotensin system, six rats received catheters and were treated with I3C as well as with captopril (TM+I3C+Cap). The oral administration of captopril at a final dose of 25 mg/100 ml of drinking water completely prevented the development of polyarteritis nodosa (Figs 5F and 6 (bottom)). At the same time, five out of six TM+I3C rats developed severe polyarteritis nodosa (Fig. 5C).
Fig 6.

Prorenin levels during the development of polyarteritis nodosa. Prorenin levels in I3C-treated rats that developed polyarteritis nodosa in a period of 10 weeks of treatment (PAN+) or remained unaffected (PAN–: without captopril; PAN– Cap: with captopril) at 3 and 6 weeks of treatment. I3C: circles (n= 6), ShamTM+I3C: crosses (n= 3), TM+I3C: triangles (n= 6) and TM+I3C+Cap: diamonds (n= 6).
Absence of ANCA and ANA
Blood serum was obtained from the rats in all groups at the end of the experiment. All sera were negative for ANCA and ANA.
Prorenin is necessary but not sufficient to induce polyarteritis
To test whether any differences in prorenin levels in I3C-treated rats may have influenced the development of polyarteritis nodosa, prorenin levels were compared between the following groups: rats positive for polyarteritis nodosa (PAN+: TM+I3C, n= 5) versus non-affected rats without captopril treatment (PAN– : I3C, Sham TM+I3C and one negative TM+I3C rat, n= 10) and with captopril treatment [PAN– Cap: TM+I3C+Cap, n= 6; at day 21 (Fig. 7A) and 42 (Fig. 7B)]. Prorenin levels were compared at days 21 and 42 of the experiment, representing time points before and at the beginning of statistically significant weight loss in these rats. There was no statistically significant difference in plasma prorenin levels between PAN+ and PAN– rats without captopril (day 21: median for PAN–: 11.3 μg Ang I/ml/hr versus PAN+: 11.2 μg Ang I/ml/hr, n.s.; day 42: PAN–: 14.9 μg Ang I/ml/hr versus PAN+: 27.8 μg Ang I/ml/hr, n.s.). Prorenin levels at days 21 and 42 were significantly higher in captopril-treated rats than in PAN– rats (median for PAN– Cap day 21: 30.2 μg Ang I/ml/hr; day 42: 36.8 μg Ang I/ml/hr, P < 0.05, respectively). Thus, prorenin levels were higher in thecaptopril-treated group than in I3C-treated rats with transmitters who developed polyarteritis nodosa.
Fig 7.

Prevention of polyarteritis nodosa by captopril. Top: prominent nodular enlargement of mesenteric vessels (yellow arrow) of an I3C-treated rat with transmitter. The mesentery is devoid of any fat. Bottom: macroscopic normal vasculature of an I3C-treated rat with transmitter (black arrow) treated with captopril.
I3C treatment did not increase plasma active renin concentrations within the first 6 weeks (Con: 252 ± 125 ng Ang I/ml/hr, I3C: 113 ± 38 ng Ang I/ml/hr, TM: 199 ± 93 ng Ang I/ml/hr, Sham TM+I3C: 198 ± 30 ng Ang I/ml/hr and TM+I3C: 191 ± 63 ng Ang I/ml/hr, n.s.). Captopril treatment led to a slight increase in active renin concentration compared with I3C treatment alone (TM+I3C+Cap: 323 ± 133 ng Ang I/ml/hr versus I3C: 113 ± 38 ng Ang I/ml/hr, P < 0.05).
Discussion
The pathogenesis of the hepatitis B-negative form of polyarteritis nodosa is currently unknown. Here, we demonstrate that necrotizing vasculitis resembling classical polyarteritis nodosa can be reproducibly induced by the activation of the renin–angiotensin system under defined conditions (the PAN-cyp1a1ren-2 transgenic model). Furthermore, we show that polyarteritis nodosa in this model can be prevented by the angiotensin-converting enzyme inhibitor captopril.
In transgenic models of angiotensin-induced end-organ damage such as dTGR (human angiotensin and human renin gene transgenic) or TGR(mRen2)27, severe phenotypes were observed with proteinuria, fibrosis of the heart, myocardial infarction and early death [13–15]. The predominantly affected vessels are the renal and coronary arteries. With high doses of I3C, cyp1a1ren-2 transgenic rats develop malignant hypertension with glomerulosclerosis and cardiac microinfarctions after 2 weeks of induction. Under these conditions, fibrinoid necrosis of the mesenteric vessels occurred, which was seen as a premature state of hypertension-induced endangiitis obliterans [6]. In the present study, lower doses of I3C were applied, leading to only moderate non-malignant hypertension and thus allowing us to study the rats for a longer period of time than in the previous studies. The coronary and cerebral arteries only showed thickening of the media as a sign of hypertensive vasculopathy. The mesenteric lesions seen in the present study are typical for polyarteritis nodosa rather than for hypertension-induced end-organ damage. These lesions require several weeks to develop and may have escaped detection in shorter-lasting studies. As in the human macroscopic form of polyarteritis nodosa, affected vessels were medium-sized arteries and the sera were negative for ANCA and ANA. At present, no information is available, however, about cryoglobulins, which also are negative in human macroscopic polyarteritis nodosa.
The activation of the renin–angiotensin system and the subsequent development of arterial hypertension are frequent sequelae of polyarteritis nodosa. In this context, renin is believed to be released, because of the vasculitis and a subsequent stenosis of the renal arteries [2, 4]. The present study shows that the activation of the renin–angiotensin system-in addition to its known role as a sequela of polyarteritis nodosa-may also act as a causal factor in the pathogenesis of the disease. In our model, polyarteritis nodosa was preceded by increased plasma prorenin levels and the activation of local renin–angiotensin systems.
Prorenin may induce polyarteritis nodosa by increasing Ang I formation, which subsequently increases Ang II and aldosterone levels . On the other hand, prorenin has been shown to exert angiotensin-independent effects by binding to the (pro)renin receptor [16]. These effects include the activation of ERK1/2 and activation of promyelotic zinc finger protein (PLZF)/p85-alpha [17], which promotes proliferation and inhibits apoptosis. Also, prorenin stimulates transforming growth factor (TGF)-β[18], which is a potent pro-inflammatory and pro-fibrotic mediator. In the present study, polyarteritis nodosa was prevented by captopril. As captopril increases prorenin and active renin levels but prevents the formation of Ang II, our study excludes angiotensin-independent effects of prorenin and supports angiotensin-dependent effects of prorenin in the pathogenesis of polyarteritis nodosa.
To test if a foreign body reaction to the telemetric device was involved in the development of polyarteritis in cyp1a1ren-2 transgenic rats, telemetric devices were implanted in the abdomen without puncturing the aorta. When these rats were treated with I3C, they developed neither a para-aortal granulomatous inflammation nor polyarteritis nodosa. Further studies are needed to elucidate the potential roles of inflammation and/or direct endothelial damage, i.e. induced by malignant hypertension, in conjunction with increased prorenin levels, for the development of polyarteritis nodosa. In any case, our data suggest that several pathogenic factors acted in concert to cause panarteritis nodosa in our model. The exact role of each individual factor remains to be determined. In rat models with an activated renin–angiotensin system, instrumentation in the abdomen with injury to the aorta might provide a basis for the development of necrotizing vasculitis.
Polyarteritis nodosa also develops infrequently in old stroke-prone spontaneously hypertensive rats (SHRSP). Saito et al. [19] found that polyarteritis nodosa in SHRSP mesenteric arteries was closely related to systolic blood pressures higher than 200 mmHg, suggesting that hypertension per se or hypertension-induced increased vascular shear stress may be able to cause polyarteritis nodosa. However, this model is limited as the disease develops inconsistently, only in old SHRSP, and the primary events leading to hypertension as well as to polyarteritis nodosa in this model are unknown. Thus, at the old age of 15.5 months, only 60% of SHRSP had polyarteritis nodosa. In contrast, in the present study, polyarteritis nodosa was present in 91% of young rats after 10 weeks of I3C treatment. By this time, the mean arterial blood pressure was increased only to approximately 170 mmHg. Clinical symptoms such as weight loss became statistically significant already after 6 weeks of I3C treatment. In view of the short duration of hypertension and the relatively low blood pressure levels achieved, the present study renders a primary role of hypertension per se in the pathogenesis of polyarteritis nodosa unlikely, but a permissive role cannot be ruled out. In particular, the combination of oscillatory shear-induced redox signalling with Ang II activation of redox-sensitive molecules may play a pathophysiological role in vascular injury and inflammation [20].
Necrotizing vasculitis in rats has been observed under treatment with various vasoactive compounds such as theophylline [21], endothelin receptor antagonists, fenoldopam [22] and selective phosphodiesterase type 3 (PDE3) [23] as well as PDE4 inhibitors [24]. These compounds have in common that they can induce systemic hypotension and it has been proposed that this may lead to decreased oxygen delivery, necrosis, enhanced endothelial permeability, protein exudation and immigration of inflammatory cells [25]. It should be noted that all these components also stimulate renin release [26–29]. In view of the present results, it is conceivable that renin release with the subsequent elevation of Ang II and aldosterone may have played an important role in the development of necrotizing vasculitis, induced by the above-mentioned compounds.
ANG II as well as aldosterone in combination with high-salt diet can induce oxidative stress and inflammation by multiple actions on VSMCs, monocytes, macrophages and endothelial cells [30]. Up-regulation of the local vascular renin–angiotensin– aldosterone system during monocyte differentiation to macrophages may amplify monocyte migration into the vessel wall [31]. Recently, it has been demonstrated that T cells possess a functional renin–angiotensin system [32] and are capable of producing and delivering Ang II to sites of inflammation. In the present study, vascular lesions were infiltrated with polymorphic leucocytes and T cells. As Ang II induces the recruitment of inflammatory cells [33] and promotes the secretion of the chemokine CCL5/RANTES by natural killer (NK) and T cells [34], overexpression of prorenin in an already pro-inflammatory status might induce a vicious circle, which results in massive T-cell infiltration of the vascular tissue.
Perspectives
In the present model of cyp1a1ren-2 transgenic rat vascular lesions, resembling classical polyarteritis nodosa, can be induced under defined conditions by only moderately activating the renin–angiotensin system. Furthermore, the development of polyarteritis nodosa can be prevented by an angiotensin-converting enzyme inhibitor. This model now allows us to investigate the causal factors and molecular mechanisms of the inflammatory processes that induce, maintain or aggravate the disease in a well-controlled manner.
Our findings may also have implications for therapy. The fact that captopril was able to prevent polyarteritis nodosa suggests that angiotensin-converting enzyme inhibitors may have benefits beyond blood pressure lowering in hypertensive patients with polyarteritis nodosa.
Acknowledgments
We thank Doreen Nierath, Brigitte Sturm and Anke Wolter for excellent technical assistance and Barbara M. Broeker and Monika Ehlers for helpful discussion. The study was supported by a grant from the Federal Ministry of Education and Research (BMBF/NBL3; FKZ:01ZZ0403). John J. Mullins is the recipient of a Wellcome Trust Principal Research Fellowship.
Supporting Information
Fig. S1 Weight loss in rats with polyarteritis nodosa.White circles: untreated control rats, black circles: I3C-treatedrats and black triangles: I3C-treated rats with transmitter(n ∇ 5 each).
Fig. S2 Prorenin levels during the development ofpolyarteritis nodosa. Prorenin levels of rats that which developedpolyarteritis nodosa in a period of 10 weeks of I3C treatment(PAN+) or remained unaffeced(PAN–) at time points 3 and 8 weeks of I3Ctreatment. I3C: circles (n ∇ 5) and TM + I3C:triangles (n ∇ 5).
Fig. S3 Vasculitis of mesenterial arteries.Representative tissue sections of mesenteric vessels oftransmitter-implanted rats with I3C treatment. Vasculitis andperivasculitis of a mesenterial artery branch (A). Thearterial nature of the vessel was confirmed by Elastica van Giesonstain (B). Segmental fibrinoid necrosis with thrombosistypical for panarteritis nodosa (C). Arterial vessel withsubsided inflammation and fibrous obliteration of the lumen(D). (A) and (C) Haematoxylin and eosin stain;(B) and (D) Elastica van Gieson stain. Bar ∇400 μm.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Guillevin L, Mahr A, Callard P, et al. Hepatitis B virus-associated polyarteritis nodosa: clinical characteristics, outcome, and impact of treatment in 115 patients. Medicine. 2005;84:313–22. doi: 10.1097/01.md.0000180792.80212.5e. [DOI] [PubMed] [Google Scholar]
- 2.Bourgarit A, Le Toumelin P, Pagnoux C, et al. Deaths occurring during the first year after treatment onset for polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: a retrospective analysis of causes and factors predictive of mortality based on 595 patients. Medicine. 2005;84:323–30. doi: 10.1097/01.md.0000180793.80212.17. [DOI] [PubMed] [Google Scholar]
- 3.Cohen L, Guillevin L, Meyrier A, et al. Malignant arterial hypertension in periarteritis nodosa. Incidence, clinicobiologic parameters and prognosis based on a series of 165 cases. Arch Mal Coeur Vaiss. 1986;79:773–8. [PubMed] [Google Scholar]
- 4.Graham PC, Lindop GB. The renin-secreting cell in polyarteritis-an immunocytochemical study. Histopathology. 1990;16:339–45. doi: 10.1111/j.1365-2559.1990.tb01137.x. [DOI] [PubMed] [Google Scholar]
- 5.Pintar TJ, Zimmerman S. Hyperreninemic hypertension secondary to a subcapsular perinephric hematoma in a patient with polyarteritis nodosa. Am J Kidney Dis. 1998;32:503–7. doi: 10.1053/ajkd.1998.v32.pm9740170. [DOI] [PubMed] [Google Scholar]
- 6.Kantachuvesiri S, Fleming S, Peters J, et al. Controlled hypertension: a transgenic toggle switch reveals differential mechanisms underlying vascular disease. J Biol Chem. 2001;276:36727–33. doi: 10.1074/jbc.M103296200. [DOI] [PubMed] [Google Scholar]
- 7.Peters B, Grisk O, Becher B, et al. Dose-dependent titration of prorenin and blood pressure in Cyp1a1ren-2 transgenic rats: absence of prorenin-induced glomerulosclerosis. J Hypertens. 2008;26:102–9. doi: 10.1097/HJH.0b013e3282f0ab66. [DOI] [PubMed] [Google Scholar]
- 8.Opay AL, Mouton CR, Mullins JJ, et al. Cyclooxygenase-2 inhibition normalizes arterial blood pressure in CYP1A1-REN2 transgenic rats with inducible ANG II-dependent malignant hypertension. Am J Physiol Renal Physiol. 2006;291:F612–8. doi: 10.1152/ajprenal.00032.2006. [DOI] [PubMed] [Google Scholar]
- 9.Patterson ME, Mullins JJ, Mitchell KD. Renoprotective effects of neuronal NOS-derived nitric oxide and cyclooxygenase-2 metabolites in transgenic rats with inducible malignant hypertension. Am J Physiol Renal Physiol. 2008;294:F205–11. doi: 10.1152/ajprenal.00150.2007. [DOI] [PubMed] [Google Scholar]
- 10.Touyz RM, Yao G, Viel E, et al. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens. 2004;22:1141–9. doi: 10.1097/00004872-200406000-00015. [DOI] [PubMed] [Google Scholar]
- 11.Grisk O, Kloting I, Exner J, et al. Long-term arterial pressure in spontaneously hypertensive rats is set by the kidney. J Hypertens. 2002;20:131–8. doi: 10.1097/00004872-200201000-00019. [DOI] [PubMed] [Google Scholar]
- 12.Peters J, Wanka H, Peters B, et al. A renin transcript lacking exon 1 encodes for a non-secretory intracellular renin that increases aldosterone production in transgenic rats. J Cell Mol Med. 2008;12:1229–37. doi: 10.1111/j.1582-4934.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luft FC, Mervaala E, Muller DN, et al. Hypertension-induced end-organ damage: a new transgenic approach to an old problem. Hypertension. 1999;33:212–8. doi: 10.1161/01.hyp.33.1.212. [DOI] [PubMed] [Google Scholar]
- 14.Pilz B, Shagdarsuren E, Wellner M, et al. Aliskiren, a human renin inhibitor, ameliorates cardiac and renal damage in double-transgenic rats. Hypertension. 2005;46:569–76. doi: 10.1161/01.HYP.0000179573.91016.3f. [DOI] [PubMed] [Google Scholar]
- 15.Mullins JJ, Peters J, Ganten D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 1990;344:541–4. doi: 10.1038/344541a0. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen G, Delarue F, Burckle C, et al. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–27. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schefe JH, Menk M, Reinemund J, et al. A novel signal transduction cascade involving direct physical interaction of the renin/prorenin receptor with the transcription factor promyelocytic zinc finger protein. Circ Res. 2006;99:1355–66. doi: 10.1161/01.RES.0000251700.00994.0d. [DOI] [PubMed] [Google Scholar]
- 18.Huang Y, Wongamorntham S, Kasting J, et al. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int. 2006;69:105–13. doi: 10.1038/sj.ki.5000011. [DOI] [PubMed] [Google Scholar]
- 19.Saito N, Kawamura H. The incidence and development of periarteritis nodosa in testicular arterioles and mesenteric arteries of spontaneously hypertensive rats. Hypertens Res. 1999;22:105–12. doi: 10.1291/hypres.22.105. [DOI] [PubMed] [Google Scholar]
- 20.Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res. 2006;71:247–58. doi: 10.1016/j.cardiores.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Collins JJ, Elwell MR, Lamb JC, et al. Subchronic toxicity of orally administered (gavage and dosed-feed) theophylline in Fischer 344 rats and B6C3F1 mice. Fundam Appl Toxicol. 1988;11:472–84. doi: 10.1016/0272-0590(88)90111-x. [DOI] [PubMed] [Google Scholar]
- 22.Yuhas EM, Morgan DG, Arena E, et al. Arterial medial necrosis and hemorrhage induced in rats by intravenous infusion of fenoldopam mesylate, a dopaminergic vasodilator. Am J Pathol. 1985;119:83–91. [PMC free article] [PubMed] [Google Scholar]
- 23.Sandusky GE, Means JR. Acute and subchronic toxicology of LY-195115 in rats and dogs. Toxicol Lett. 1987;38:177–86. doi: 10.1016/0378-4274(87)90126-3. [DOI] [PubMed] [Google Scholar]
- 24.Mecklenburg L, Heuser A, Juengling T, et al. Mesenteritis precedes vasculitis in the rat mesentery after subacute administration of a phosphodiesterase type 4 inhibitor. Toxicol Lett. 2006;163:54–64. doi: 10.1016/j.toxlet.2005.09.037. [DOI] [PubMed] [Google Scholar]
- 25.Robertson DG, Reily MD, Albassam M, et al. Metabonomic assessment of vasculitis in rats. Cardiovasc Toxicol. 2001;1:7–19. doi: 10.1385/ct:1:1:07. [DOI] [PubMed] [Google Scholar]
- 26.Reid IA, Stockigt JR, Goldfien A, et al. Stimulation of renin secretion in dogs by theophylline. Eur J Pharmacol. 1972;17:325–32. doi: 10.1016/0014-2999(72)90112-4. [DOI] [PubMed] [Google Scholar]
- 27.Pedersen EB, Thomsen IM, Fjordside LS. Effect of BQ-123, an endothelin antagonist, on renal hemodynamics, tubular function, vasoactive hormones, and blood pressure in healthy humans: a dose response study. Am J Hypertens. 2005;18:1578–85. doi: 10.1016/j.amjhyper.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Velasco M, Luchsinger A. Dopamine: pharmacologic and therapeutic aspects. Am J Ther. 1998;5:37–43. [PubMed] [Google Scholar]
- 29.Friis UG, Jensen BL, Sethi S, et al. Control of renin secretion from rat juxtaglomerular cells by cAMP-specific phosphodiesterases. Circ Res. 2002;90:996–1003. doi: 10.1161/01.res.0000017622.25365.71. [DOI] [PubMed] [Google Scholar]
- 30.Brown NJ. Aldosterone and vascular inflammation. Hypertension. 2008;51:161–7. doi: 10.1161/HYPERTENSIONAHA.107.095489. [DOI] [PubMed] [Google Scholar]
- 31.Okamura A, Rakugi H, Ohishi M, et al. Upregulation of renin-angiotensin system during differentiation of monocytes to macrophages. J Hypertens. 1999;17:537–45. doi: 10.1097/00004872-199917040-00012. [DOI] [PubMed] [Google Scholar]
- 32.Jurewicz M, McDermott DH, Sechler JM, et al. Human T and natural killer cells possess a functional renin-angiotensin system: further mechanisms of angiotensin II-induced inflammation. J Am Soc Nephrol. 2007;18:1093–102. doi: 10.1681/ASN.2006070707. [DOI] [PubMed] [Google Scholar]
- 33.Mateo T, Abu Nabah YN, Abu TM, et al. Angiotensin II-induced mononuclear leukocyte interactions with arteriolar and venular endothelium are mediated by the release of different CC chemokines. J Immunol. 2006;176:5577–86. doi: 10.4049/jimmunol.176.9.5577. [DOI] [PubMed] [Google Scholar]
- 34.Wolf G, Ziyadeh FN, Thaiss F, et al. Angiotensin II stimulates expression of the chemokine RANTES in rat glomerular endothelial cells. Role of the angiotensin type 2 receptor. J Clin Invest. 1997;100:1047–58. doi: 10.1172/JCI119615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Weight loss in rats with polyarteritis nodosa.White circles: untreated control rats, black circles: I3C-treatedrats and black triangles: I3C-treated rats with transmitter(n ∇ 5 each).
Fig. S2 Prorenin levels during the development ofpolyarteritis nodosa. Prorenin levels of rats that which developedpolyarteritis nodosa in a period of 10 weeks of I3C treatment(PAN+) or remained unaffeced(PAN–) at time points 3 and 8 weeks of I3Ctreatment. I3C: circles (n ∇ 5) and TM + I3C:triangles (n ∇ 5).
Fig. S3 Vasculitis of mesenterial arteries.Representative tissue sections of mesenteric vessels oftransmitter-implanted rats with I3C treatment. Vasculitis andperivasculitis of a mesenterial artery branch (A). Thearterial nature of the vessel was confirmed by Elastica van Giesonstain (B). Segmental fibrinoid necrosis with thrombosistypical for panarteritis nodosa (C). Arterial vessel withsubsided inflammation and fibrous obliteration of the lumen(D). (A) and (C) Haematoxylin and eosin stain;(B) and (D) Elastica van Gieson stain. Bar ∇400 μm.