

Abstract
There have been reports showing a protective role of nonsteroidal anti‐inflammatory drugs (NSAIDs) against gastrointestinal cancers. CpG island hyper methylation (CIHM) of tumor suppressor genes is a major event in carcinogenesis. We investigated the CIHM status of non‐cancerous gastric mucosa in chronic NSAID users and non‐users and assessed the effect of NSAIDs on CIHM. Gastric mucosa samples were obtained from 51 chronic NSAID users and 180 non‐users. CIHM of p14(ARF), p16(INK4a), death‐associated protein kinase (DAP‐kinase), and E‐cadherin (CDH1) genes were determined by methylation‐specific PCR. CIHM high was defined as two or more CpG islands methylated. CIHM of p14, p16, CDH1, and CIHM high were lower in chronic NSAID users than in non‐users (p14: non‐users vs users = 32.2%vs 9.8%, P = 0.003; p16: non‐users vs users = 35.0%vs 15.7%, P = 0.02; CDH1: non‐users vs users = 36.1%vs 9.8%, P = 0.0009; CIHM high: non‐users vs users = 44.4%vs 17.6%, P = 0.0009). NSAID use was also associated with decreased number of CIHM by anova (R = –0.32, P < 0.0001). Multivariate logistic regression analysis with adjustment for sex, age, Helicobacter pylori infection, and NSAID use revealed that NSAID use was inversely correlated with all four CIHM and CIHM high as an independent factor (p14: odds ratio [OR] = 0.17, 95% confidence interval [CI] = 0.06–0.48; p16: OR = 0.32, 95% CI = 0.14–0.75; DAP‐kinase: OR = 0.45, 95% CI = 0.22–0.92; CDH1: OR = 0.18, 95% CI = 0.06–0.48; CIHM high: OR = 0.21, 95% CI = 0.09–0.49). No association was found between CIHM status and the duration or dose of NSAIDs. Chronic NSAID use suppresses CIHM in human gastric mucosa. NSAIDs may have a suppressive role against methylation‐related gastric carcinogenesis. (Cancer Sci 2009; 100: 1192–1197)
Abbreviations:
- CIHM
CpG island hyper methylation
- H. pylori
Helicobacter pylori
- MSP
methylation‐specifc PCR
- NSAID
nonsteroidal anti‐inflammatory drug
NSAIDs are widely used for prevention of ischemic heart or cerebral diseases or for the treatment of chronic inflammation and various musculoskeletal disorders. Their usage, however, remains limited because of their potential risk for gastrointestinal damage.( 1 , 2 , 3 , 4 )
However, the hypothesis that NSAIDs prevents gastrointestinal cancers is also gaining support. Mechanisms involving the inhibitory effect of aspirin and other NSAIDs on the cyclooxygenase‐2 (COX‐2) enzyme's production of various prostaglandins, resulting in modulation of inflammation and immunoresponse, induction of cell apoptosis, and inhibition of angiogenesis, have been suggested( 5 ) as the antitumorgenetic effects.
Epidemiologic studies have also addressed association between NSAID use and reduced risk of cancers in the colorectal( 6 , 7 , 8 , 9 ) esophagus( 10 , 11 , 12 , 13 , 14 ) and stomach.( 10 , 11 , 12 ) Recent study has shown that chronic users of celecoxib, a selective COX‐2 inhibitor, showed more significant regression of gastric intestinal metaplasia than non‐users after H. pylori eradication therapy,( 15 ) suggesting that the use of NSAIDs may suppress the early processes of carcinogenesis.
CIHM is shown to be an important mechanism in gene silencing. In the stomach, CIHM of several tumor suppressor genes have been documented in gastric cancer tissues, precancerous lesions, and H. pylori‐infected non‐neoplastic mucosa.( 16 , 17 , 18 , 19 , 20 ) Among several genes, p14(ARF), p16(INK4a), death‐associated protein kinase DAP‐kinase, and E‐cadherin (CDH1) have been frequently methylated in non‐neoplastic gastric mucosa, in relation to age,( 20 ) H. pylori infection( 18 , 21 , 22 ) and histological degree of gastritis,( 22 ) and gastric cancer occurrence.( 23 , 24 , 25 ) Therefore, epigenetic silencing of these genes may be involved in early steps of carcinogenesis.
Recently, we have reported that chronic aspirin users show lower prevalence of CDH1 methylation in non‐neoplastic gastric mucosa than non‐users.( 26 ) Since DNA methylation play a important role in the early steps of gastric carcinogenesis, it is speculated that the chronic use of NSAIDs might influence methylation status.
This is a consequent study of this recent preliminary report. To clarify whether the chronic use of various NSAIDs might also prevent methylation‐related gastric carcinogenesis through suppression of CIHM of multiple CpG promoters, we examined the prevalence of CIHM in four candidate promoters (p14, p16, DAP‐kinase, and CDH1) in the non‐neoplastic gastric mucosa of chronic users of various NSAIDs and non‐users and assessed its influence on DNA methylation.
Materials and Methods
Chronic NSAID users and non‐users. Enrolled were 231 subjects who were attending the Endoscopy Center of Fujita Health University Hospital from January 2005 to May 2008. All subjects underwent upper gastroscopy for various indications. The reasons for gastroscopy included health check, secondary complete check‐up of stomach cancer following to barium X‐ray examination, follow‐up examination of ulcer diseases, or for the complaint of abdominal discomfort. Among 231 subjects, 51 chronic NSAID users were identified as having chronic use of NSAIDs for at least 3 months or more. The other 180 subjects had no history of chronic use of NSAIDs in the past, or occasional use of NSAIDs within 12 months. This cohort was partly recruited from recent study investigating the association between the promoter CIHM and severity of chronic gastritis,( 22 ) host genetic factors,( 27 ) and chronic aspirin use.( 26 ) Those who had a history of continuous or occasional use of NSAIDs in a period of less than 3 months were excluded from this study. Patients who had severe systemic disease or malignancy in the stomach or other organ were also excluded. The Ethics Committee of the Fujita Health University School of Medicine approved the protocol, and prior, written informed consent was obtained from all participating subjects.
Tissue sample, DNA extraction, and detection of H. pylori infection. Biopsy specimens were taken from the antrum as well as the upper corpus along the greater curvature from grossly non‐pathological mucosa in all patients. The specimens taken from the antrum were cut into two pieces, and one part of specimens was immediately frozen and stored at –80°C until use. Genomic DNA was extracted directly from these frozen specimens using the standard phenol/chloroform method. Other specimens were fixed in 10% buffered formalin and embedded in paraffin for microscopic histological examination. H. pylori infection status was assessed by histological analysis of both the biopsy specimens from the greater curvature of the gastric antrum and upper corpus using antibodies to H. pylori to avoid false‐negative results for H. pylori infection as much as possible.( 28 , 29 ) NSAID users were ordered to stop using their drugs 1 week before and after gastroscopy to avoid gastrointestinal bleeding.
Bisulfite modification and MSP. To detect CIHM, we chose four candidate promoter CpG islands, which have been shown to be frequently methylated in gastric mucosa in relation to age, gender,( 20 ) and H. pylori infection:( 18 , 21 , 22 ) p14, p16, DAP‐kinase, and CDH1. For the examination of DNA methylation, genomic DNA was treated with sodium bisulfite using the BislFast DNA Modification Kit for Methylated DNA Detection (Toyobo, Osaka, Japan). MSP was carried out with the following primers: p14 methylated forward (p14 MF): 5′‐gtgttaaagggcggcgtagc‐3′, p14 methylated reverse (p14 MR): 5′‐aaaaccctcactcgcgacga‐3′, which amplified 122‐bp product; p14 unmethylated forward (p14 UF): 5′‐tttttggtgttaaagggtggtgtagt‐3′, p14 unmethylated reverse (p14 UR): 5′‐cacaaaaaccctcactcacaacaa‐3′, which amplified 132‐bp product;( 30 ) p16 methylated forward (p16 MF): 5′‐ttattagagggtggggcggatcgc‐3′, p16 methylated reverse (p16 MR): 5′‐gaccccgaaccgcgaccgtaa‐3′, which amplified 150‐bp product; p16 unmethylated forward (p16 UF): 5′‐ttattagagggtggggtggattgt‐3′, p16 unmethylated reverse: (p16 UR): 5′‐caaccccaaaccacaaccataa‐3′, which amplified 151‐bp product;( 31 ) DAP‐kinase methylated forward (DAP‐kinase MF): 5′‐ggatagtcggatcgagttaacgtc‐3′, DAP‐kinase methylated reverse (DAP‐kinase MR): 5′‐ccctcccaaacgccga‐3′, which amplified 98‐bp product; DAP‐kinase unmethylated forward (DAP‐kinase UF): 5′‐ggaggatagttggattgagttaatgtt‐3′, DAP‐kinase unmethylated reverse (DAP‐kinase UR): 5′‐caaatccctcccaaacaccaa‐3′, which amplified 106‐bp product;( 32 ) CDH1 methylated forward (CDH1 MF): 5′‐ ttaggttagagggttatcgcgt‐3′, CDH1 methylated reverse (CDH1 MR): 5′‐taactaaaaattcacctaccgac‐3′, which amplified 115‐bp product; CDH1 unmethylated forward (CDH1 UF): 5′‐taattttaggttagagggttattgt‐3′, CDH1 unmethylated reverse (CDH1 UR): 5′‐cacaaccaatcaacaacaca‐3′, which amplified 97‐bp product.( 31 ) An annealing temperature and times were determined using DNA from the peripheral blood of a young individual without H. pylori infection and DNA methylated with SssI methylase (New England BioLabs, Beverly, MA, USA). MSP was carried out in a volume of 20 µL containing 0.1 µg of bislufite‐modificated DNA. The DNA was denatured at 95°C for 5 min, followed by 33–35 cycles at 95°C for 30 s and 57–69°C according to primers for 1 min, and 72°C for 1 min with a final extension at 72 for 5 min. MSP reactions were done using EX Taq HS (Takara Bio, Shiga, Japan). PCR products (10 µL) were separated by electrophoresis in 2.5% agarose gels, and visualized by UV illumination using ethdium bromide staining. CIHM was defined as the presence of a positive methylation band, showing signals approximately equivalent to or greater than that of the size marker (10 ng/µL: 100 bp DNA Ladder; Takara Bio), irrespective of the presence of unmethylated bands. Samples giving faint positive signals were analyzed a further two times and only those samples with consistent positive methylation bands were considered as positive. In addition, we measured the fluorescence intensities of methylated bands for 50 randomly selected CIHM samples using a digital densitometer (Lane Analyzer; ATTO, Tokyo, Japan), and confirmed that the fluorescence intensities of all 50 methylated bands were approximately equivalent to or greater than that of the size marker (data not shown). CIHM high was also defined as two or more CpG islands methylated.
Statistical analysis. Statistical analysis was using the χ2‐test for the comparison of CIHM between NSAID users and non‐users. Association between the number of methylated CpG islands and NSAID use was assessed by anova. The strength of association between CIHM and various clinicopathological factors was assessed by calculating the odds ratio (OR) and 95% confidence interval (CI). A probability value of less than 0.05 was considered statistically significant in all analyses.
Results
Characteristics of subjects. Age and sex distributions, H. pylori infection status, and prevalence of active peptic ulcer disease among chronic NSAID users and non‐users are shown in Table 1. Age and prevalence of active peptic ulcer disease were significantly higher among aspirin users, while sex distribution and H. pylori infection status were not different among these two groups. Clinicopathological characteristics of chronic NSAID users are shown in Table 2. Most NSAID users consisted of users of aspirin (33 subjects, 64.7%) and loxoprofen sodium (12 subjects, 23.5%). NSAID use was occasional in four (7.8%) loxoprofen sodium users, and for all others, usage was continuous. The reasons for NSAID use were prevention of ischemic heart or cerebral diseases in 31 (60.8%) subjects (they were all 100 mg or 200 mg/day aspirin users), and for control of muscular pain or arthralgia in 18 (35.3%) subjects. In 39 (76.5%) subjects, detailed information on previous periods of chronic NSAIDs use was available.
Table 1.
Age and sex distribution, H. pylori infection status, and prevalence of active peptic ulcers in NSAID users and non‐users
| Variables | NSAID non‐users | NSAID users | P‐values |
|---|---|---|---|
| Number of subjects | 180 | 51 | |
| Age (mean ± SD) † | 59.9 ± 13.0 | 68.0 ± 11.0 | < 0.0001 |
| Male : female ‡ | 105:75 | 29:22 | 0.85 |
| H. pylori positive | |||
| Subjects, n (%) ‡ | 99 (55.0) | 29 (56.9) | 0.81 |
| Active peptic ulcer, n (%) ‡ | 19 (10.6) | 18 (35.3) | P = 0.0001 |
Student's t‐test,
χ2‐test.
H. pylori, Helicobacter pylori; NSAID, nonsterioidal anti‐inflammatory drug.
Table 2.
Characteristics of 51 NSAID users
| Variables (n) | Number of subjects |
|---|---|
| Aspirin (n = 33) | |
| 80 mg/day | 1 |
| 100 mg/day | 29 |
| 200 mg/day | 2 |
| 330 mg/day | 1 |
| Loxoprofen sodium (n= 12) | |
| 60 mg/day | 6 |
| 120 mg/day | 2 |
| 180 mg/day | 2 |
| Unknown dose | 2 |
| Lornoxicam (n= 1) | |
| 8 mg/day | 1 |
| Dicrofenac sodium (n= 2) | |
| Unknown dose | 2 |
| Etodolac (n= 1) | |
| 400 mg/day | 1 |
| Other NSAIDs (n= 2) | |
| Unknown dose | 2 |
| Purpose of NSAID use | |
| Prevention of ischemic heart or cerebral diseases | 31 |
| Control of muscular pain or arthralgia | 18 |
| Unknown | 2 |
| Past periods of NSAID use | |
| 3 months~ <1 year | 12 |
| 1 year~ <2 years | 10 |
| 2 year~ | 17 |
| Unknown | 12 |
Note: NSAID use was occasional in only four of the 12 Loxoprofen sodium users, and all others were continuous users.
NSAID, nonsterioidal anti‐inflammatory drug.
CIHM in chronic NSAID users and non‐users. All 231 gastric mucosa samples were available for MSP analysis. In all subjects, CIHM was found in 63 (27.2%) for p14, 71 (30.7%) for p16, 91 (39.4%) for DAP‐kinase, and 70 subjects (30.3%) for CDH1. CIHM high was also found in 89 subjects (38.5%).
In the comparison of frequency of CIHM between chronic NSAID users and non‐users, we found that rates of CIHM of p14, p16, CDH1, and CIHM high were significantly lower in chronic NSAID users than in non‐users (p14: non‐users vs users = 32.2%vs 9.8%, P = 0.003; p16: non‐users vs users = 35.0%vs 15.7%, P = 0.02; CDH1: non‐users vs users = 36.1%vs 9.8%, P = 0.0009; CIHM high: non‐users vs users = 44.4%vs 17.6%, P = 0.0009; Fig. 1). We also investigated the association between the number of methylated CpG islands NSAID use. In all subjects, none, one, two, three, or all four CIHM were found in 75 (32.5%), 67 (29.0%), 49 (21.2%), and 40 subjects (17.3%), respectively, while the prevalence of NSAID users in the four groups were 31 (41.3%) for none, 11 (16.4%) for one, six (10.2%) for two, and one subject (2.5%) for three or all four CIHM. This association was significant by anova (R = –0.32, P < 0.0001; Fig. 2).
Figure 1.
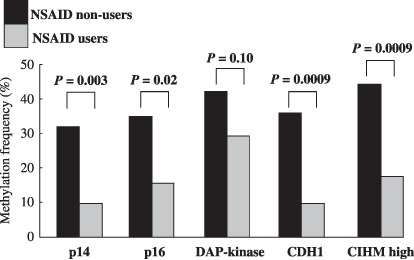
Rates of CpG island hyper methylation (CIHM) of four CpG islands and CIHM high in the gastric mucosa of nonsteroidal anti‐inflammatory drug (NSAID) users and non‐users. CIHM of p14, p16, E‐cadherin (CDH1), and CIHM high were significantly lower in NSAID users than in non‐users. CIHM high was defined as two or more CpG islands methylated. DAP‐kinase, death‐associated protein kinase.
Figure 2.
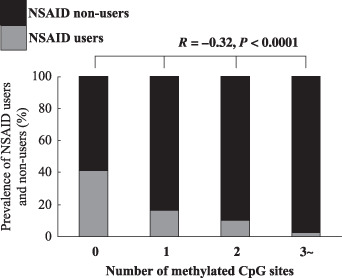
Association between the number of methylated CpG islands and nonsteroidal anti‐inflammatory drug (NSAID) use. The number of methylated CpG islands was inversely associated with NSAID use. Statistical analysis was performed by anova.
Univariate and multivariate logistic regression analysis for relationship between CIHM and clinicopathological factors. Since CIHM in non‐neoplastic gastric mucosa would be influenced by clinicopathological factors such as age, sex,( 20 ) and H. pylori infection,( 18 , 21 , 22 ) univariate (Table 3) and multivariate logistic regression analysis (Table 4) were conducted. Univariate analysis showed that H. pylori infection was closely associated with CIHM of p16 (OR = 3.69, 95% CI = 1.97–6.91, P < 0.0001), CDH1 (OR = 3.58, 95% CI = 1.91–6.70, P < 0.0001), and CIHM high (OR = 3.12, 95% CI = 1.77–5.51, P < 0.0001), while NSAID use was inversely correlated with CIHM of p14 (OR = 0.23, 95% CI = 0.09–0.61, P = 0.003), p16 (OR = 0.35, 95% CI = 0.15–0.78, P = 0.02), CDH1 (OR = 0.19, 95% CI = 0.07–0.51, P = 0.0009), and CIHM high (OR = 0.27, 95% CI = 0.12–0.58, P = 0.0009) (Table 3).
Table 3.
Univariate analysis for relationship between CIHM and clinicopathological factors
| Genes | p14 | Univariate | p16 | Univariate | DAP‐kinase | Univariate | CDH1 | Univariate | CIHM high | Univariate | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CIHM | UM | M | OR (95% CI) | P‐ values | UM | M | OR (95% CI) | P‐ values | UM | M | OR (95% CI) | P‐ values | UM | M | OR (95% CI) | P‐ values | (–) | (+) | OR (95% CI) | P |
| Variables | ||||||||||||||||||||
| Gender (n) | ||||||||||||||||||||
| Female | 70 | 27 | Reference | 66 | 31 | Reference | 58 | 39 | Reference | 67 | 30 | Reference | 56 | 41 | Reference | |||||
| Male | 98 | 36 | 0.95 (0.53–1.71) | 0.87 | 94 | 40 | 0.91 (0.52–1.59) | 0.73 | 82 | 52 | 0.94 (0.55–1.69) | 0.83 | 94 | 40 | 0.95 (0.54–1.68) | 0.86 | 86 | 48 | 0.76 (0.45–1.30) | 0.32 |
| Age (Mean ± SD) | 60.9 ± 13.4 | 63.4 ± 11.8 | 1.02 (0.99–1.04) | 0.11 | 62.3 ± 13.6 | 60.4 ± 11.7 | 0.99 (0.97–1.01) | 0.31 | 60.5 ± 13.0 | 63.6 ± 13.0 | 1.02 (0.99–1.04) | 0.08 | 62.5 ± 13.3 | 60.0 ± 12.3 | 0.99 (0.97–1.01) | 0.19 | 61.7 ± 13.2 | 61.7 ± 12.9 | 1.00 (0.98–1.02) | 0.99 |
| H. pylori infection (n) | ||||||||||||||||||||
| Negative | 78 | 25 | Reference | 86 | 17 | Reference | 69 | 34 | Reference | 86 | 17 | Reference | 78 | 25 | Reference | |||||
| Positive | 90 | 38 | 1.32 (0.73–2.37) | 0.36 | 74 | 54 | 3.69 (1.97–6.91) | <0.0001 | 71 | 57 | 1.63 (0.95–2.79) | 0.08 | 75 | 53 | 3.58 (1.91–6.70) | <0.0001 | 64 | 64 | 3.12 (1.77–5.51) | <0.0001 |
| NSAID use (n) | ||||||||||||||||||||
| Non‐users | 122 | 58 | Reference | 117 | 63 | Reference | 104 | 76 | Reference | 115 | 65 | Reference | 100 | 80 | Reference | |||||
| Users | 46 | 5 | 0.23 (0.09–0.61) | 0.003 | 43 | 8 | 0.35 (0.15–0.78) | 0.02 | 36 | 15 | 0.57 (0.29–1.12) | 0.10 | 46 | 5 | 0.19 (0.07–0.51) | 0.0009 | 42 | 9 | 0.27 (0.12–0.58) | 0.0009 |
CIHM high was defined as two or more CpG islands methylated.
CDH1, E‐cadherin; CI, confidence interval; CIHM, CpG island hyper methylation; DAP‐kinase, death‐associated protein kinase; M, methylated; NSAID, nonsteroidal anti‐inflammatory drug; OR, odds ratio; UM, unmethylated.
Table 4.
Multivariate logistic regression analysis for relationship between CIHM and clinicopathological factors
| CIHM | p14 | p16 | DAP‐kinase | CDH1 | CIHM high | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Multivariate OR (95% CI) | P‐values | Multivariate OR (95% CI) | P‐values | Multivariate OR (95% CI) | P‐values | Multivariate OR (95% CI) | P‐values | Multivariate OR (95% CI) | P‐values | |
| Variables | ||||||||||
| Gender | ||||||||||
| Female | Reference | Reference | Reference | Reference | Reference | |||||
| Male | 0.85 (0.46–1.58) | 0.61 | 0.77 (0.42–1.41) | 0.39 | 0.85 (0.49–1.47) | 0.56 | 0.80 (0.43–1.49) | 0.49 | 0.61 (0.34–1.10) | 0.10 |
| Age | 1.03 (1.01–1.06) | 0.01 | 0.99 (0.97–1.02) | 0.64 | 1.03 (1.00–1.05) | 0.02 | 0.99 (0.97–1.02) | 0.55 | 1.01 (0.99–1.03) | 0.35 |
| H. pylori infection | ||||||||||
| Negative | Reference | Reference | Reference | Reference | Reference | |||||
| Positive | 1.36 (0.73–2.51) | 0.34 | 4.07 (2.13–7.77) | <0.0001 | 1.69 (0.97–2.93) | 0.06 | 4.09 (2.12–7.89) | <0.0001 | 3.62 (1.99–6.61) | <0.0001 |
| NSAID use | ||||||||||
| Non‐users | Reference | Reference | Reference | Reference | Reference | |||||
| Users | 0.17 (0.06–0.48) | 0.0006 | 0.32 (0.14–0.75) | 0.009 | 0.45 (0.22–0.92) | 0.03 | 0.18 (0.06–0.48) | 0.0007 | 0.21 (0.09–0.49) | 0.0003 |
Note: CIHM high was defined as two or more CpG islands methylated.
CDH1, E‐cadherin; CIHM, CpG island hyper methylation; CI, confidence interval; DAP‐kinase, death‐associated protein kinase; H. pylori, Helicobacter pylori; NSAID, nonsteroidal anti‐inflammatory drug; OR, odds ratio.
After multivariate logistic regression analysis with adjustment for sex, age, H. pylori infection, and NSAID use, it was revealed that NSAID use was inversely correlated with all four CIHM and CIHM high as a independent factor (p14: OR = 0.17, 95% CI = 0.06–0.48, P = 0.0006; p16: OR = 0.32, 95% CI = 0.14–0.75, P = 0.009; DAP‐kinase: OR = 0.45, 95% CI = 0.22–0.92, P = 0.03; CDH1: OR = 0.18, 95% CI = 0.06–0.48, P = 0.0007; CIHM high: OR = 0.21, 95% CI = 0.09–0.49, P = 0.0003; Table 5).
Table 5.
Association between CHIM and past duration of NSAID use in 39 chronic NSAID users
| CIHM (n) | Past duration of NSAIDs(Mean ± SD): years | Multivariate analysis | |
|---|---|---|---|
| OR (95% CI) | P‐values | ||
| p14 | |||
| UM (34) | 1.46 ± 1.04 | Reference | |
| M (5) | 3.09 ± 3.36 | 1.68 (0.77–3.66) | 0.20 |
| p16 | |||
| UM (34) | 1.49 ± 0.91 | Reference | |
| M (5) | 2.88 ± 3.77 | 1.46 (0.83–2.58) | 0.19 |
| DAP‐kinase | |||
| UM (27) | 1.51 ± 0.91 | Reference | |
| M (12) | 2.03 ± 2.49 | 1.22 (0.76–1.95) | 0.41 |
| CDH1 | |||
| UM (35) | 1.49 ± 1.01 | Reference | |
| M (4) | 3.28 ± 3.92 | 1.65 (0.84–3.25) | 0.14 |
| CIHM high | |||
| (–) (33) | 1.42 ± 0.93 | Reference | |
| (+) (6) | 3.17 ± 3.19 | 1.72 (0.88–3.38) | 0.12 |
Note: All data were adjusted for age, sex, and Helicobacter pylori infection status. CIHM high was defined as two or more CpG islands methylated.
CDH1, E‐cadherin; CI, confidence interval; CIHM, CpG island hyper methylation; DAP‐kinase, death‐associated protein kinase; M, methylated; NSAID, nonsteroidal anti‐inflammatory drug; OR, odds ratio; UM, unmethylated.
Multivariate logistic regression analysis for relationship between CHIM and past duration of NSAID use in chronic NSAID users. In 39 subjects for whom information on previous NSAID use was available, we investigated the association between CHIM and past duration of NSAID use. Although past duration of NSAID use was relatively longer in methylated subjects for all CIHM and CHIM high, differences were not significant by multivariate logistic regression analysis with adjustment for sex, age, and H. pylori infection (Table 5). We also investigated whether CIHM status would be affected by different NSAIDs as well as dosage, but no association was found between CIHM status and such factors (data not shown).
Discussion
This is a consequent study of our recent preliminary report showing a suppressive role of chronic aspirin use against CIHM of CDH1 promoter in gastric mucosa.( 26 ) We investigated the prevalence of CIHM of four candidate promoters (p14, p16, DAP‐kinase, and CDH1) in the non‐neoplastic gastric mucosa of chronic users of various NSAIDs and non‐users.
Chronic use of various NSAIDs is associated with lower frequency of CIHM in human gastric mucosa compared to non‐users. In particular, rates of CIHM of p14, p16, CDH1, and CIHM high were significantly lower in chronic NSAID users than in non‐users. NSAID use was also associated with a decreased number of CIHM by anova. Furthermore, multivariate logistic regression analysis also revealed that NSAID use was inversely correlated with all four CIHM and CIHM high as an independent factor.
CIHM has been reported in many kinds of cancers as an important mechanism of gene silencing. Issa et al.( 33 ) first reported that CIHM of the estrogen receptor (ER) promoter increased as a function of age and suggested that ER was a tumor suppressor gene in the colon. Subsequently, age‐related CIHM of other tumor suppressor genes has been reported in the colon( 34 , 35 , 36 , 37 ) and stomach.( 16 , 17 , 18 , 19 , 20 ) CIHM in non‐cancerous gastric mucosa has been shown to be associated with degree of H. pylori‐related gastritis and gastric cancer occurrence;( 22 , 23 , 24 , 25 ) thus, this epigenetic change has been considered to be a early step in gastric carcinogenesis. Reversal or prevention of the CIHM of tumor suppressor genes in the stomach, resulting in their increased expression, could contribute to a delay and decrease in the risk of cancer. Our current data support a series of epidemiologic studies reporting the suppressive role of NSAIDs in carcinogenesis in the digestive tract,( 6 , 7 , 8 , 9 , 10 , 11 , 12 ) and indicate the novel evidence that use of various NSAIDs may reduce the risk of methylation‐related carcinogenesis in human gastric mucosa.
The theory of methylation reduction by NSAIDs is not a new one. Pereira et al. first reported that administration of celecoxib, and celecoxib and α‐difluoromethylornithine (DFMO), significantly reduced the number of methylated sites in the ER‐α gene at both 7 and 28 days, resulting in an increase of ER‐α gene mRNA expression in colon tumors induced in rats by azoxymethane.( 38 )
Although we did not investigate the mRNA expression of tumor suppressor genes in human gastric mucosa, it is possible that NSAIDs may uphold mRNA levels by suppressing methylation, and that they may reduce the risk of gastric cancer in the long term.
Concerning the possible mechanisms of CIHM, inflammation and cell proliferation have considered to be promoting factors. Cell proliferation itself has been reported to accelerate de novo DNA methylation.( 37 , 39 ) Inflammation has also been known to promote de novo methylation through repressing the expression of many genes.( 39 , 40 , 41 , 42 ) In our study, CIHM of p16, CDH1, and CIHM high were associated with H. pylori infection by both univariate and multivariate analysis, which is in agreement with previous studies and our recent study.( 18 , 21 , 22 ) H. pylori‐related inflammation in methylation induction seems to be clearly evident in the stomach. On the other hand, NSAIDs blocks COX‐2 and H. pylori‐associated enhanced epithelial prostaglandin production and associated hyperproliferation.( 43 , 44 ) Since COX‐2 expression has been documented in gastric cancer( 45 , 46 ) and H. pylori‐infected chronic gastritis and premalignant lesions,( 47 ) this inhibitory effect against cell proliferation may be a main, possible explanation of our results.
Recently, a novel animal study disclosed that celecoxib could significantly decrease gastric tumor volume, but had no significant effect on COX‐2 and prostaglandin signals,( 48 ) implying that NSAIDs also have an antitumorigenic effect which is involved in the COX‐2‐independent pathway, such as nuclear factor–kappa B (NF‐κB), and I‐kappa B (IκB).( 48 , 49 , 50 , 51 ) Further study would be necessary to trace the exact mechanism of aspirin in counteracting gastric carcinogenesis, especially in a stage as early as the precancerous stage.
We also hypothesized that CIHM status would be influenced by past duration of NSAID use and the different agents as well as their dosage. In this study, however, contrary to our expectation, multivariate logistic regression analysis revealed that no association was found between CIHM status and past duration. Also, no association was found between CIHM status and different agents and their doses. This result may be due to the small number of subjects for whom information on past use of NSAIDs was available. Future study will be needed of a larger sample to resolve this issue.
In conclusion, we have shown that chronic NSAID use is significantly associated with a reduced risk of CHIM in human gastric mucosa. The suppressive role of various NSAIDs in CHIM of multiple promoters in human gastric mucosa provided a novel finding. However, our result is not meant to imply that such alterations in CHIM status in human gastric mucosa lead to direct manifestation of the NSAIDs as chemopreventive agents, because the detailed mechanism by which NSAIDs reduce the CHIM of multiple promoters is unclear. Moreover, whether this epigenetic alteration would actually reduce the development of gastric cancer is unclear. In addition, since our result has been obtained by retrospective observational study, evaluation of the effect of NSAIDs on CHIM by comparison of the methylation status before and after NSAID use would be also needed as part of longitudinal research.
Furthermore, there has been strong evidence that use of NSAIDs is accompanied by risk for gastrointestinal damage and bleeding.( 1 , 2 , 3 , 4 ) Careful consideration of the clinical significance of NSAIDs as chemopreventive agents is required in regards to the stomach.
References
- 1. Hawkey CJ. Non‐steroidal anti‐inflammatory drug gastropathy. Gastroenterology 2000; 119: 521–35. [DOI] [PubMed] [Google Scholar]
- 2. Allison MC, Howatson AG, Torrance CJ, Lee FD, Russel RI. Gastrointestinal damage associated with the use of non‐steroidal anti‐inflammatory drugs. N Eng J Med 1992; 327: 749–54. [DOI] [PubMed] [Google Scholar]
- 3. Wolf MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of non‐steroidal anti‐inflammatory drugs. N Eng J Med 1999; 340: 1888–999. [DOI] [PubMed] [Google Scholar]
- 4. Rich M, Scheiman JM. Nonsteroidal anti‐inflammatory drug gastropathy at the millennium: mechanisms and prevention. Semin Arthritis Rheum 2000; 30: 167–79. [DOI] [PubMed] [Google Scholar]
- 5. Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti‐inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst 2002; 94: 252–66. [DOI] [PubMed] [Google Scholar]
- 6. Kune GA, Kune S, Watson LF. Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res 1988; 48: 4399–404. [PubMed] [Google Scholar]
- 7. Rosenberg L, Palmer JR, Zauber AG, Warshauer ME, Stolley PD, Shapiro S. A hypothesis: nonsteroidal anti‐inflammatory drugs reduce the incidence of large‐bowel cancer. J Natl Cancer Inst 1991; 83: 355–8. [DOI] [PubMed] [Google Scholar]
- 8. Thun MJ, Namboodiri MM, Heath CW Jr. Aspirin use and reduced risk of fatal colon cancer. N Engl J Med 1991; 325: 1593–6. [DOI] [PubMed] [Google Scholar]
- 9. Giovannucci E, Egan KM, Hunter DJ et al . Aspirin and the risk of colorectal cancer in women. N Engl J Med 1995; 333: 609–14. [DOI] [PubMed] [Google Scholar]
- 10. Langman MJ, Cheng KK, Gilman EA, Lancashire RJ. Effect of anti‐inflammatory drugs on overall risk of common cancer: Case control study in general practice research database. BMJ 2000; 320: 1642–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW Jr. Aspirin use and risk of fatal cancer. Cancer Res 1993; 15 (53): 1322–7. [PubMed] [Google Scholar]
- 12. Coogan PF, Rosenberg L, Palmer JR et al . Nonsteroidal anti‐inflammatory drugs and risk of digestive cancers at sites other than the large bowel. Cancer Epidemiol Biomarkers Prev 2000; 1: 119–23. [PubMed] [Google Scholar]
- 13. Farrow DC, Vaughan TL, Hansten PD et al . Use of aspirin and other nonsteroidal anti‐inflammatory drugs and risk of esophageal and gastric cancer. Cancer Epidemiol Biomarkers Prev 1998; 2: 97–102. [PubMed] [Google Scholar]
- 14. Funkhouser EM, Sharp GB. Aspirin and reduced risk of esophageal carcinoma. Cancer 1995; 76: 1116–19. [DOI] [PubMed] [Google Scholar]
- 15. Yang HB, Cheng HC, Sheu BS, Hung KH, Liou MF, Wu JJ. Chronic celecoxib users more often show regression of gastric intestinal metaplasia after Helicobacter pylori eradication. Aliment Pharmacol Ther 2007; 15 (25): 455–61. [DOI] [PubMed] [Google Scholar]
- 16. Toyota M, Ahuja N, Suzuki H et al . Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res 1999; 59: 5438–42. [PubMed] [Google Scholar]
- 17. Kang GH, Shim YH, Jung HY, Kim WH, Ro JY, Rhyu MG. CpG island methylation in premalignant stages of gastric carcinoma. Cancer Res 2001; 61: 2847–51. [PubMed] [Google Scholar]
- 18. Chan AO, Lam SK, Wong BC et al . Promoter methylation of E‐cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut 2003; 52: 502–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Waki T, Tamura G, Sato M, Terashima M, Nishizuka S, Motoyama T. Promoter methylation status of DAP‐kinase and RUNX3 genes in neoplastic and non‐neoplastic gastric epithelia. Cancer Sci 2003; 94: 360–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kang GH, Lee HJ, Hwang KS, Lee S, Kim JH, Kim JS. Aberrant CpG island hypermethylation of chronic gastritis, in relation to aging, gender, intestinal metaplasia, and chronic inflammation. Am J Pathol 2003; 163: 1551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maekita T, Nakazawa K, Mihara M et al . High levels of aberrant DNA methylation in Helicobacter pylori‐infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res 2006; 12: 989–95. [DOI] [PubMed] [Google Scholar]
- 22. Tahara T, Arisawa T, Shibata T et al . Increased number of methylated CpG islands correlates with Helicobacter pylori infection, histological and serological severity of chronic gastritis. Eur J Gastroenterol Hepatol 2009. (e‐pub ahead of print). [DOI] [PubMed]
- 23. Tahara T, Arisawa T, Shibata T et al . Risk prediction of gastric cancer by analysis of aberrant DNA methylation in non‐neoplastic gastric epithelium. Digestion 2007; 75: 54–61. [DOI] [PubMed] [Google Scholar]
- 24. Kaise M, Yamasaki T, Yonezawa J, Miwa J, Ohta Y, Tajiri H. CpG island hypermethylation of tumor‐suppressor genes in H. pylori‐infected non‐neoplastic gastric mucosa is linked with gastric cancer risk. Helicobacter 2008; 13: 35–41. [DOI] [PubMed] [Google Scholar]
- 25. Nakajima T, Maekita T, Oda I et al . Higher methylation levels in gastric mucosae significantly correlate with higher risk of gastric cancers. Cancer Epidemiol Biomarkers Prev 2006; 15: 2317–21. [DOI] [PubMed] [Google Scholar]
- 26. Tahara T, Shibata T, Nakamura M et al . Chronic aspirin use suppresses CDH1 methylation in human gastric mucosa. Dig Dis Sci (e‐pub ahead of print). [DOI] [PubMed]
- 27. Tahara T, Shibata T, Nakamura M et al . MTHFR 677 T carrier influences the methylation status of H. pylori infected gastric mucosa in older subjects. Dig Dis Sci 2008. (e‐pub ahead of print). [DOI] [PubMed]
- 28. Sipponen P, Kekki M, Haapakoski J, Ihamaki T, Siurala M. Gastric cancer risk in chronic gastritis: statistical calculations of cross‐sectional data. Int J Cancer 1985; 35: 173–7. [DOI] [PubMed] [Google Scholar]
- 29. Enomoto H, Watanabe H, Nishikura K, Umezawa H, Asakura H. Topographic distribution of Helicobacter pylori in the resected stomach. Eur J Gastroenterol Hepatol 1998; 10: 473–8. [DOI] [PubMed] [Google Scholar]
- 30. Esteller M, Tortola S, Toyota M et al . Hypermethylation‐associated inactivation of p14 (ARF) is independent of p16 (INK4a) methylation and p53 mutational status. Cancer Res 2000; 60: 129–33. [PubMed] [Google Scholar]
- 31. Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 1996; 93: 9821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Katzenellenbogen RA, Baylin SB, Herman JG. Hypermethylation of the DAP‐kinase CpG island is a common alteration in B‐cell malignancies. Blood 1999; 93: 4347–53. [PubMed] [Google Scholar]
- 33. Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen rececptor CpG island links ageing and neoplasma in human colon. Nature Genet 1994; 7: 536–40. [DOI] [PubMed] [Google Scholar]
- 34. Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998; 8: 5489–94. [PubMed] [Google Scholar]
- 35. Issa JP. Aging, DNA methylation and cancer. Crit Rev Oncol Hematol 1999; 32: 31–43. [DOI] [PubMed] [Google Scholar]
- 36. Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev 2004; 23: 9–39. [DOI] [PubMed] [Google Scholar]
- 37. Issa PJ, Ahuja N, Toyota M, Bronner P, Brentnall TA. Accelerated age‐related CpG island methylation in ulcerative colitis. Cancer Res 2001; 61: 3573–7. [PubMed] [Google Scholar]
- 38. Pereira MA, Tao L, Wang W et al . Modulation by celecoxib and difluoromethylornithine of the methylation of DNA and the estrogen receptor‐alpha gene in rat colon tumors. Carcinogenesis 2004; 25: 1917–23. [DOI] [PubMed] [Google Scholar]
- 39. Velicescu M, Weisenberger DJ, Gonzales FA, Tsai YC, Nguyen CT, Jones PA. Cell division is required for de novo methylation of CpG islands in bladder cancer cells. Cancer Res 2002; 62: 2378–84. [PubMed] [Google Scholar]
- 40. Song JZ, Stirzaker C, Harrison J, Melki JR, Clark SJ. Hypermethylation trigger of the glutathione‐S‐transferase gene (GSTP1) in prostate cancer cells. Oncogene 2002; 21: 1048–61. [DOI] [PubMed] [Google Scholar]
- 41. De Smet C, Loriot A, Boon T. Promoter‐dependent mechanism leading to selective hypomethylation within the 5′ region of gene MAGE‐A1 in tumor cells. Mol Cell Biol 2004; 24: 4781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ushijima T, Okochi‐Takada E. Aberrant methylations in cancer cells: where do they come from? Cancer Sci 2005; 96: 206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhu GH, Yang XL, Lai KC et al . Nonsteroidal antiinflammatory drugs could reverse Helicobacter pylori‐induced apoptosis and proliferation in gastric epithelial cells. Dig Dis Sci 1998; 43: 1957–63. [DOI] [PubMed] [Google Scholar]
- 44. Hudson N, Balsitis M, Filipowicz F, Hawkey CJ. Effect of Helicobacter pylori colonisation on gastric mucosal eicosanoid synthesis in patients taking non‐steroidal anti‐inflammatory drugs. Gut 1993; 34: 748–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ristimäki A, Honkanen N, Jänkälä H, Sipponen P, Härkönen M. Expression of cyclooxygenase‐2 in human gastric carcinoma. Cancer Res 1997; 57: 1276–80. [PubMed] [Google Scholar]
- 46. Uefuji K, Ichikura T, Mochizuki H, Shinomiya N. Expression of cyclooxygenase‐2 protein in gastric adenocarcinoma. J Surg Oncol 1998; 69: 168–72. [DOI] [PubMed] [Google Scholar]
- 47. Sung JJ, Leung WK, Go MY et al . Cyclooxygenase‐2 expression in Helicobactor pylori associated premalignant and malignant gastric lesions. Am J Pathol 2000; 157: 729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hu PJ, Yu J, Zeng ZR et al . Chemoprevention of gastric cancer by celecoxib in rats. Gut 2004; 53: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karin M, Cao Y, Greten FR, Li ZW. NF‐kappa B in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2002; 2: 301–10. [DOI] [PubMed] [Google Scholar]
- 50. Lin A, Karin M. NF‐kappa B in cancer: a marked target. Semin Cancer Biol 2003; 13: 107–14. [DOI] [PubMed] [Google Scholar]
- 51. Yin MJ, Yamamoto Y, Gaynor RB. The anti‐inflammatory agents aspirin and salicylate inhibit the activity of I (kappa) B kinase‐beta. Nature 1998; 396: 77–80. [DOI] [PubMed] [Google Scholar]