

Abstract
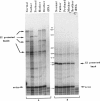
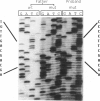
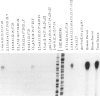
Maple syrup urine disease in humans results from inherited defects in branched chain alpha-ketoacid dehydrogenase, a mitochondrial multienzyme complex. A variety of genetic changes may produce this phenotype by affecting the function of any of the three complex-specific subunits. The varied clinical expression observed in patients may be partially explained by the defects in the involved subunit. Here we report localization of the gene for the branched chain acyltransferase component of the complex to human chromosome 1 and describe a proband who is a compound heterozygote at this locus. One allele, inherited from the father, produces transcripts with 124 nucleotides deleted from the coding region. The deletion is not found in the branched chain acyltransferase gene, implying that the deleted transcripts arise by an error in transcript processing. Cells from the patient's mother contain 50% of the normal amount of mRNA for the subunit, and the proband has inherited this nonexpressing allele from her. As a result, the proband produces no acyltransferase protein and therefore has greatly impaired complex activity. A phenotypically normal sibling is shown to be genetically similar to the mother having inherited the mother's nonexpressing allele and the father's normal allele.
Full text
PDF








Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Danner D. J., Litwer S., Herring W. J., Pruckler J. Construction and nucleotide sequence of a cDNA encoding the full-length preprotein for human branched chain acyltransferase. J Biol Chem. 1989 May 5;264(13):7742–7746. [PubMed] [Google Scholar]
- Feinberg A. P., Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983 Jul 1;132(1):6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- Fekete G., Plattner R., Crabb D. W., Zhang B., Harris R. A., Heerema N., Palmer C. G. Localization of the human gene for the El alpha subunit of branched chain keto acid dehydrogenase (BCKDHA) to chromosome 19q13.1----q13.2. Cytogenet Cell Genet. 1989;50(4):236–237. doi: 10.1159/000132768. [DOI] [PubMed] [Google Scholar]
- Gibbs R. A., Nguyen P. N., McBride L. J., Koepf S. M., Caskey C. T. Identification of mutations leading to the Lesch-Nyhan syndrome by automated direct DNA sequencing of in vitro amplified cDNA. Proc Natl Acad Sci U S A. 1989 Mar;86(6):1919–1923. doi: 10.1073/pnas.86.6.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie L. L., Argan C., Taneja A. T., Hodges R. S., Freeman K. B., Shore G. C. A synthetic signal peptide blocks import of precursor proteins destined for the mitochondrial inner membrane or matrix. J Biol Chem. 1985 Dec 25;260(30):16045–16048. [PubMed] [Google Scholar]
- Gough N. M. Rapid and quantitative preparation of cytoplasmic RNA from small numbers of cells. Anal Biochem. 1988 Aug 15;173(1):93–95. doi: 10.1016/0003-2697(88)90164-9. [DOI] [PubMed] [Google Scholar]
- Harris R. A., Kuntz M. J., Simpson R. Inhibition of branched-chain alpha-keto acid dehydrogenase kinase by alpha-chloroisocaproate. Methods Enzymol. 1988;166:114–123. doi: 10.1016/s0076-6879(88)66017-4. [DOI] [PubMed] [Google Scholar]
- MENKES J. H., HURST P. L., CRAIG J. M. A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics. 1954 Nov;14(5):462–467. [PubMed] [Google Scholar]
- Miller R. H., Eisenstein R. S., Harper A. E. Effects of dietary protein intake on branched-chain keto acid dehydrogenase activity of the rat. Immunochemical analysis of the enzyme complex. J Biol Chem. 1988 Mar 5;263(7):3454–3461. [PubMed] [Google Scholar]
- Ono H., Tuboi S. The cytosolic factor required for import of precursors of mitochondrial proteins into mitochondria. J Biol Chem. 1988 Mar 5;263(7):3188–3193. [PubMed] [Google Scholar]
- Otulakowski G., Robinson B. H., Willard H. F. Gene for lipoamide dehydrogenase maps to human chromosome 7. Somat Cell Mol Genet. 1988 Jul;14(4):411–414. doi: 10.1007/BF01534650. [DOI] [PubMed] [Google Scholar]
- Pons G., Raefsky-Estrin C., Carothers D. J., Pepin R. A., Javed A. A., Jesse B. W., Ganapathi M. K., Samols D., Patel M. S. Cloning and cDNA sequence of the dihydrolipoamide dehydrogenase component human alpha-ketoacid dehydrogenase complexes. Proc Natl Acad Sci U S A. 1988 Mar;85(5):1422–1426. doi: 10.1073/pnas.85.5.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore G. C., Rachubinski R. A., Argan C., Rozen R., Pouchelet M., Lusty C. J., Raymond Y. Synthesis and intracellular transport of mitochondrial matrix proteins in rat liver: studies in vivo and in vitro. Methods Enzymol. 1983;97:396–408. doi: 10.1016/0076-6879(83)97151-3. [DOI] [PubMed] [Google Scholar]
- Urlaub G., Mitchell P. J., Ciudad C. J., Chasin L. A. Nonsense mutations in the dihydrofolate reductase gene affect RNA processing. Mol Cell Biol. 1989 Jul;9(7):2868–2880. doi: 10.1128/mcb.9.7.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Smith R. L. Lowry determination of protein in the presence of Triton X-100. Anal Biochem. 1975 Feb;63(2):414–417. doi: 10.1016/0003-2697(75)90363-2. [DOI] [PubMed] [Google Scholar]
- Weber J. L., Kwitek A. E., May P. E. Dinucleotide repeat polymorphism at the D1S102 locus. Nucleic Acids Res. 1990 Apr 25;18(8):2199–2199. [PMC free article] [PubMed] [Google Scholar]
- Weber J. L., May P. E. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet. 1989 Mar;44(3):388–396. [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Edenberg H. J., Crabb D. W., Harris R. A. Evidence for both a regulatory mutation and a structural mutation in a family with maple syrup urine disease. J Clin Invest. 1989 Apr;83(4):1425–1429. doi: 10.1172/JCI114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Healy P. J., Zhao Y., Crabb D. W., Harris R. A. Premature translation termination of the pre-E1 alpha subunit of the branched chain alpha-ketoacid dehydrogenase as a cause of maple syrup urine disease in Polled Hereford calves. J Biol Chem. 1990 Feb 15;265(5):2425–2427. [PubMed] [Google Scholar]