

Abstract
Primordial organisms of the putative RNA world would have required polymerase ribozymes able to replicate RNA. Known ribozymes with polymerase activity best approximating that needed for RNA replication contain at their catalytic core the class I RNA ligase, an artificial ribozyme with a catalytic rate among the fastest of known ribozymes. Here we present the 3.0 angstrom crystal structure of this ligase. The architecture resembles a tripod, its three legs converging near the ligation junction. Interacting with this tripod scaffold through a series of 10 minor-groove interactions (including two A-minor triads) is the unpaired segment that contributes to and organizes the active site. A cytosine nucleobase and two backbone phosphates abut the ligation junction; their location suggests a model for catalysis resembling that of proteinaceous polymerases.
The RNA world hypothesis proposes that early life forms lacked DNA and coded proteins, depending instead on RNA for both chemical catalysis and information storage (1). Central to this RNA world would have been polymerase ribozymes able to replicate RNA. Among the efforts to generate ribozymes with such ability, the most productive have started with the class I RNA ligase ribozyme (2-4). This ribozyme was originally isolated from a large pool of random sequences (5, 6). It has since been improved by mutation and selection and has served as a platform for modeling ribozyme evolution in vitro (6-8). Because it rapidly promotes a reaction with chemistry identical to that catalyzed by proteinaceous enzymes that replicate RNA (Fig. 1A) (6), the ligase has provided the catalytic engine for more sophisticated RNA enzymes that use the information from an external RNA template and nucleoside triphosphates to synthesize short strands of RNA (2, 3, 9). Although more efficient with some templates than with others, this primer-extension reaction is general in that all templates tested support detectable extension (2-4). To understand the structural basis behind RNA-catalyzed RNA polymerization, we have solved the crystal structure of the class I ligase ribozyme.
Fig. 1.
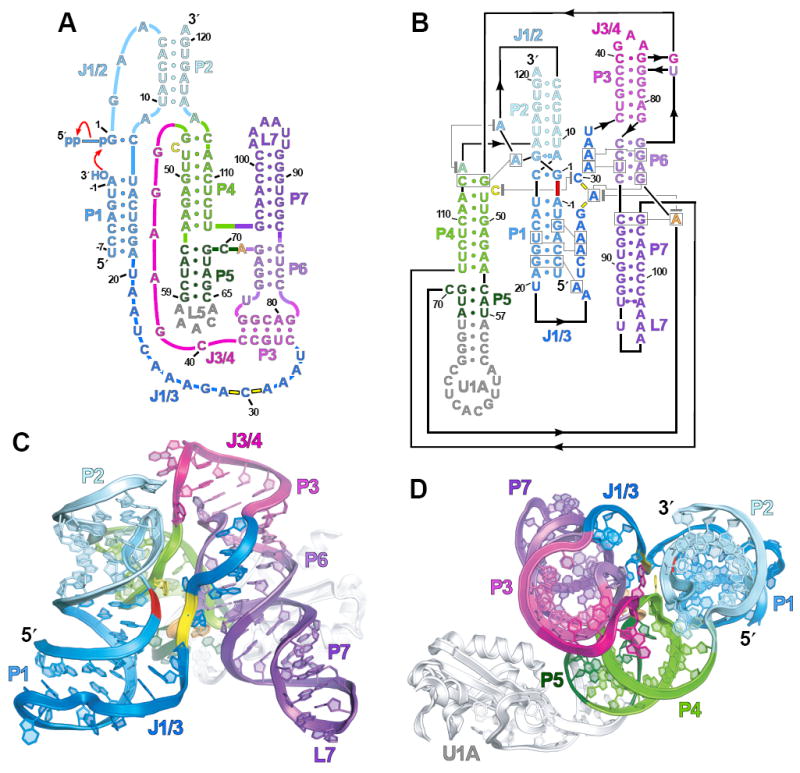
Global architecture of the ligase ribozyme. (A) Secondary structure and reaction scheme of a ligase variant with decreased Mg2+ dependence (10). It is depicted undergoing ligation, using the classical secondary-structure representation (15). Red arrows indicate attack by the substrate 3′-hydroxyl on the ribozyme α-phosphate with concomitant loss of pyrophosphate. (B) Revised secondary structure of the crystallization construct, reflecting the coaxial stacking and relative domain orientation. Indicated is the ligation junction (thick red dash), backbone phosphates at the active site (yellow dashes), base triples (boxed residues connected with gray lines), and stacking interactions (residues vertically aligned or connected with gray lines terminating in gray bars). Residues numbered as in (A); those in gray were added to facilitate crystallization. Base-pair geometries indicated using nomenclature of (27). (C) Ribbon representation of ligase structure, as if peering into the active site (yellow) and ligation junction (red). (D) Top-down view, rotated ~90° relative to (C).
The ligase sequence variant we crystallized was the product of three successive in vitro selection experiments, the last of which mutagenized segments not participating in known base pairs (termed “joining regions”) and selected variants that folded and reacted within milliseconds (5, 6, 10). This experiment produced an improved variant that, unlike its predecessor, yielded useful crystals (data to 3.0 Å, tables S1 to S3, figs. S1 and S2) (11). This variant is more tolerant of low Mg2+ concentrations; it reacts 15 times faster than the predecessor in 1 mM Mg2+ (10) but only slightly faster than the predecessor in high Mg2+ [predecessor reaction rate, 800/min in 60 mM Mg2+, pH 9 (12)]. As with the predecessor, its reaction is pH-dependent, slowing to 2.2/min in our crystallization conditions (10 mM Mg2+, pH 6.0).
To promote crystallization, we replaced loop 5 (L5) with the U1A-binding loop, and grew crystals of the ligase-U1A complex (Fig. 1B) (11, 13). A parallel effort used a phage-display system to generate antibodies for cocrystallization (14), which yielded crystals with data to 3.1 Å (11). The ligase structure in this second crystal form, solved independently, confirmed the structure presented here (all-atom root mean square deviation = 1.48 Å) (fig. S3).
The global structure features three coaxially stacked domains: P1-P2, P3-P6-P7 and P4-P5 (Fig. 1, B and C), consistent with the previously predicted topology (15), but with the three domains placed at relative angles of 58° to 71°, converging near the ligation junction so as to resemble a tripod (Fig. 1D). Because the tripod legs protrude into solvent, the fraction of surface area occluded from solvent is less than that of similarly sized RNAs (fig. S4).
Positioning these domains are tertiary interactions at the top of the tripod (Fig. 2A). G45 stacks on U76, the joining residue of the P6-P3 pseudoknot. This interaction pulls the 5′ strands of P4 and P6 close to—and nearly parallel with—J1/2, facilitating a contact between the G45 2′-hydroxyl and a C5 nonbridging oxygen, two groups with confirmed function (10). Preceding G45 is an unexpected Watson-Crick pair, G44:C40, which we confirmed biochemically (fig. S5) (11). This pair extends the P3 helical stack and closes a 3-nucleotide (nt) loop resembling a GNRA tetraloop (in which N is A, C, G, or U; R is A or G). In addition to this loop, two other regions (L7 and part of J1/3) resemble previously defined substructures (figs. S6 and S7).
Fig. 2.

Tertiary contacts involving the three longest joining regions. (A) Interactions bridging the three domains. (B) The path of J1/3. (C) Hydrogen bonds of the two A-minor triads (fig. S8).
Interacting with the tripod scaffold is J1/3, which docks into the P1 and P6 minor grooves, passing from one to the other near the ligation junction (Figs. 1C and 2B). Of the 10 minor-groove interactions, eight involve adenosines of J1/3. Residues A25–A26–A27 dock into the fourth and fifth base pairs of P1 (Fig. 2C and fig. S9), which corresponds to the primer-template duplex used by the polymerase. Each interaction could form irrespective of the P1 base-pair identity. Of particular note are the hydrogen bonds involving the 2′-hydroxyls of U16 and G–3. In the polymerase, 2′-deoxy substitution is more detrimental at the position analogous to U16 than at any other template residue, and 2′-deoxy substitution at the position analogous to G–3 is among the most detrimental primer substitutions (16). Hence A25–A26–A27 make defined, yet sequence-independent, contacts that help explain the ability of the polymerase to utilize primer-template helices of any sequence (2-4).
At the other end of J1/3, A31–A32–A33 dock into the P6 minor groove, passing from one helical strand to the other through a succession of hydrogen bonds identical to that of A25–A26–A27 (Fig. 2, B and C). We call this recurring motif the A-minor triad and note another instance in the small subunit of the bacterial ribosome (fig. S8). The P6 A-minor triad helps form a Mg2+-binding site (Figs. 2B and 3A). Direct metal coordination by the A31 and A32 nonbridging (pro-RP) phosphate oxygens brings these oxygens ~3.1 Å from one another, inducing a 90° kink that positions C30 out of the helical docking register of A31–A33. Outer-sphere contacts involving N7 of A32, N7 and N2 of A33, and O4 of U34 further stabilize this interaction—roles that, in concert with their packing into P6, explain both the absolute conservation of these nucleotides in active ribozyme isolates (2, 4, 8, 10, 17) and the deleterious effects of chemically modifying them (10).
Fig. 3.

Architecture of the active site. (A) The active site, as viewed from the ligation junction, removing P1–P2 for clarity. (B) Interactions near G1:C12, which is analogous to the NTPtemplate pair during polymerization (2-4, 9). Meshes are simulated-annealing OMIT maps in which active-site nucleotides (gray, contoured at 2σ) or the hydrated metal cluster (aqua, 4σ) were excluded from map calculations. (C) Stereograph of the active site. Black dashes indicate hydrogen bonds; magenta dashes indicate proximity between A29 and C30 phosphate oxygens and the ligation junction (red). Mesh represents a simulated-annealing OMIT map (4.5 σ) in which the hydrated metal was excluded from map calculations. (D) Mean interference values (±SD) from three α-phosphorothiolate NAIM experiments. The secondary structure is aligned above. Strong interference was truncated at the detection limit, 6.0 (10, 22). Missing positions are those modified to facilitate crystallization (hashes) or too close to the termini to measure.
Between the two A-minor triads lies the active site (Figs. 2B and 3, A to C). Forming the “floor” of the active site is A71, an absolutely conserved residue at the center of the four-way junction linking the P4-P5 and P3-P6-P7 domains (Fig. 1, A and B). A71 forms an imperfect type I A-minor interaction (18) with C86:G105, the first base pair of P7 (Fig. 3A and fig. S9). Chemical modification of A71 or loss of the C86 2′-hydroxyl impairs catalysis (10). A29 stacks on the A71 floor, itself forming an A-minor base triple (fig. S9). Residues A29 and C30 are hence enclosed between the A71 A-minor interaction and the metal-stabilized backbone kink (Fig. 3C). The consequences of this enclosure are twofold. First, C30 is extruded from the minor groove of P6, the rotation of its base constrained by A73 so as to form a cross-strand stack with C47 (Fig. 3, A to C). C47 is likewise extruded from helix P4, the plane of its base roughly perpendicular to the adjoining base pairs, with its N4 exocyclic amine positioned 3.1 Å away from the ligation junction. Second, the A71 interaction prevents G28 from stacking below A29. The consequent rotation of G28 places the phosphates of A29 and C30 in close proximity to one another (~5 Å between phosphorus centers), and facing P1. The nonbridging oxygens at these residues are 4.5 to 5.3 Å from the 3′-hydroxyl and phosphate oxygens of the ligation junction.
We therefore propose that C47, as positioned by C30, and the backbone phosphates of A29 and C30 comprise the ligase active site. Both cytidines are conserved among active isolates (2, 4, 8, 10, 17), although their contributions to activity differ (fig. S10). The C30U substitution decreases activity about fivefold, perhaps from disrupting the hydrogen bond between the C30 N4 and the C47 O4′ (Fig. 3B), whereas, the C47U substitution diminishes activity >104-fold, consistent with a more direct role in catalysis.
With only minor perturbation, the A29 and C30 phosphates could provide a binding site for a catalytic metal ion, as observed at active sites for some natural ribozymes (19, 20) and the L1 ligase, an artificial ribozyme that promotes a reaction resembling that of our ligase (21). Although we observed no electron density for such a metal ion in the crystal structure of the product, a metal might be bound more tightly before catalysis. To test for a functional role for these and other backbone phosphate oxygens, we performed nucleotide-analog interference mapping (NAIM) (10, 22), randomly incorporating RP -phosphorothioate substitutions and identifying those that interfered with activity (Fig. 3D). Substitution at residues A29 through A32 resulted in maximal interference. Substitution at A31 and A32 would disrupt the metal-stabilized kink needed to form the C30 to C47 stack at the active site (Fig. 3C). That substitution at A29 or C30 similarly abrogated function supports the hypothesis that these other phosphates coordinate at least one magnesium ion that is catalytically critical in the transition state but not bound tightly in the crystallized product.
We propose a preliminary model for catalysis by the class I ligase and its polymerase derivatives that resembles the mechanism of proteinaceous enzymes. Proteinaceous nucleic acid polymerases require a pair of aspartic acid–bound divalent metal ions supplemented by a general acid that stabilizes and protonates the pyrophosphate-leaving group (Fig. 4A) (23, 24). In our model, the substrate α-phosphate and backbone phosphates of A29 and C30 jointly bind a catalytic magnesium ion (Fig. 4B). This metal activates the primer 3′-hydroxyl for nucleophilic attack and stabilizes the transition-state geometry, akin to Metal A of proteinaceous polymerases (23). In addition, because free NTPs bind divalent cations, we suggest that the G1 triphosphate (or the incoming NTP) enters the active site complexed with a second metal, which, after binding, would remain coordinated by oxygens on the β- and γ-phosphates. At the transition state, this second metal helps stabilize the developing negative charge on the pyrophosphate-leaving group. This stabilization is aided by the exocyclic amine of C47, which hydrogen bonds to the (α,β) bridging oxygen.
Fig. 4.

Transition-state stabilization by polymerases built from either protein or RNA. (A) Catalysis by proteinaceous polymerases (23, 24). Indicated are bonds formed or broken during the transition state (red arrows), coordination of catalytic metal ions, MA and MB (gray solid lines), and an active-site acid (A•••H). (B) Model for catalysis by the ligase ribozyme. Notation as in (A), with the addition of a hydrogen bond between C47 N4 and the leaving group (dashed gray line). Some magnesium ligands are not specified; for those that are, relative orientations are unknown. A proposed contact to the reactive phosphate pro-RP oxygen (28) and two speculative contacts implied by NAIM are in blue. Features not supported (or refuted) by structural or biochemical evidence are in gray.
Our model, which postulates a hydrogen bond to the leaving oxygen in the transition state, differs from that of proteinaceous polymerases, which involves proton transfer to this oxygen in the transition state (24). Although nucleobases can act as general acids at ribozyme active sites (25), we disfavor ascribing such a function to C47. With increasing pH, the ribozyme ligation rate increases log-linearly with a slope of 1.0 (pH 5.7 to 8.5), consistent with the net loss of one proton, presumably that of the nucleophile, when proceeding from the ground state to the transition state (12). If general-acid catalysis by C47 were dominant at the transition state, the pH-rate profile would likely deviate from linearity over this pH range. Moreover, if C47 were a general acid, the functional group donating the proton would differ from that of the active-site cytidine of the hepatitis delta virus (HDV) ribozyme, wherein the N3 imine is thought to act as the proton donor (25). For the ligase, methylating N3 has little effect on catalysis, which rules out direct participation of N3 but not N4 (10).
By identifying the residues at the active site, the ligase crystal structure will facilitate directed examination of the catalytic mechanism of RNA-catalyzed RNA polymerization. Our model also provides insights into how known polymerase ribozymes recognize primer-template duplexes, the feature most in need of improvement for developing a self-replicating polymerase ribozyme (4, 26), and one that now can be targeted more explicitly in design and selection experiments.
Supplementary Material
Acknowledgments
We thank A. Ferré-D’Amaré, J. Doudna, R. Batey, and K. Zhou for the generous donation of reagents and protocols; K. Rajashankar for assistance in phasing; E. Duguid, V. Malashkevich, C. Drennan, T. Schwartz, U. RajBhandary, J. Cochrane, M. Stahley, S. Strobel, M. Robertson, W. Scott, A. Ricardo, and J. Chen for advice, encouragement, and helpful discussions; and members of the Bartel laboratory for support and comments. Supported by NIH grant GM61835. X-ray data were collected with the Northeastern Collaborative Access Team, at the Advanced Photo Source, Argonne National Laboratory (11). D.M.S. was an NSF predoctoral fellow; S.C.B was a Howard Hughes Medical Institute (HHMI) predoctoral Fellow. D.B. and J.A.P. are HHMI investigators. Coordinates for the U1A-bound and Fab-bound ribozymes are deposited in the Protein Data Bank (3HHN and 3IVK, respectively).
Footnotes
www.sciencemag.org/cgi/content/full/326/ISSUE/PAGE/DC1
Materials and Methods
SOM Text
Figs. S1 to S12
Tables S1 to S3
References
References and Notes
- 1.Joyce GF, Orgel LE. In: The RNA World. Gesteland RF, Cech TR, Atkins JF, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1999. pp. 49–77. [Google Scholar]
- 2.Johnston WK, Unrau PJ, Lawrence MS, Glasner ME, Bartel DP. Science. 2001;292:1319. doi: 10.1126/science.1060786. [DOI] [PubMed] [Google Scholar]
- 3.Lawrence MS, Bartel DP. RNA. 2005;11:1173. doi: 10.1261/rna.2110905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zaher HS, Unrau PJ. RNA. 2007;13:1017. doi: 10.1261/rna.548807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartel DP, Szostak JW. Science. 1993;261:1411. doi: 10.1126/science.7690155. [DOI] [PubMed] [Google Scholar]
- 6.Ekland EH, Szostak JW, Bartel DP. Science. 1995;269:364. doi: 10.1126/science.7618102. [DOI] [PubMed] [Google Scholar]
- 7.Wright MC, Joyce GF. Science. 1997;276:614. doi: 10.1126/science.276.5312.614. [DOI] [PubMed] [Google Scholar]
- 8.Joyce GF. Annu Rev Biochem. 2004;73:791. doi: 10.1146/annurev.biochem.73.011303.073717. [DOI] [PubMed] [Google Scholar]
- 9.Ekland EH, Bartel DP. Nature. 1996;382:373. doi: 10.1038/382373a0. [DOI] [PubMed] [Google Scholar]
- 10.Bagby SC, Bergman NH, Shechner DM, Yen CC, Bartel DP. RNA. published online 26 November 2009 (10.1261/rna.191250) [Google Scholar]
- 11.Materials and methods and supporting text are available as supporting materials on Science Online
- 12.Glasner ME, Bergman NH, Bartel DP. Biochemistry. 2002;41:8103. doi: 10.1021/bi012179b. [DOI] [PubMed] [Google Scholar]
- 13.Ferré-D’Amaré AR, Doudna JA. J Mol Biol. 2000;295:541. doi: 10.1006/jmbi.1999.3398. [DOI] [PubMed] [Google Scholar]
- 14.Ye JD, et al. Proc Natl Acad Sci U S A. 2008;105:82. doi: 10.1073/pnas.0709082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergman NH, Lau NC, Lehnert V, Westhof E, Bartel DP. RNA. 2004;10:176. doi: 10.1261/rna.5177504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müller UF, Bartel DP. Chem Biol. 2003;10:799. doi: 10.1016/s1074-5521(03)00171-6. [DOI] [PubMed] [Google Scholar]
- 17.Ekland EH, Bartel DP. Nucleic Acids Res. 1995;23:3231. doi: 10.1093/nar/23.16.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. Proc Natl Acad Sci U S A. 2001;98:4899. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stahley MR, Strobel SA. Science. 2005;309:1587. doi: 10.1126/science.1114994. [DOI] [PubMed] [Google Scholar]
- 20.Toor N, Keating KS, Taylor SD, Pyle AM. Science. 2008;320:77. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robertson MP, Scott WG. Science. 2007;315:1549. doi: 10.1126/science.1136231. [DOI] [PubMed] [Google Scholar]
- 22.Strobel SA. Curr Opin Struct Biol. 1999;9:346. doi: 10.1016/S0959-440X(99)80046-3. [DOI] [PubMed] [Google Scholar]
- 23.Sträter N, Lipscomb WN, Klabunde T, Krebs B. Angew Chem Int Ed Engl. 1996;35:2024. [Google Scholar]
- 24.Castro C, et al. Nat Struct Mol Biol. 2009;16:212. doi: 10.1038/nsmb.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bevilacqua PC, Yajima R. Curr Opin Chem Biol. 2006;10:455. doi: 10.1016/j.cbpa.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 26.Lawrence MS, Bartel DP. Biochemistry. 2003;42:8748. doi: 10.1021/bi034228l. [DOI] [PubMed] [Google Scholar]
- 27.Leontis NB, Westhof E. RNA. 2001;7:499. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glasner ME, Yen CC, Ekland EH, Bartel DP. Biochemistry. 2000;39:15556. doi: 10.1021/bi002174z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.